
Abstract
Microfibrillated cellulose (MFC) is continuously gaining attention due to its outstanding mechanical properties, in particular high strength-to-weight ratio. Recently, more and more studies target the production of porous materials, such as foams, out of this natural resource. Commonly, an energy-consuming freeze–drying method is utilized for producing pure MFC porous structures from water-based suspensions, which renders these products particularly unattractive for industry. Although alternatives for foam production have been proposed, using either modified MFC or with various additives, the freeze–drying step is still one of the most critical bottle-neck of MFC foam production upscaling. A novel straightforward freeze–thawing–drying procedure assisted by the common additive urea was herein proposed. Such method allows the production of mechanically stable, lightweight MFC structures under low-cost ambient conditions drying. The influence of the cellulose fibril characteristics, the suspension formulation and the process parameters on the final foam properties have been studied in terms of porosity, density and mechanical properties.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In comparison to dense bulk materials, foams combine the advantages of having a light structure, an increased surface-to-weight ratio and being permeable for fluids or gases in the case of open-cell structures. The application field of porous structures is broad including e.g. thermal and acoustic isolation materials, catalysts supports, packaging materials, binders or absorbents for environmental protection and cleaning purposes (Gibson and Ashby 1999). Beside foam production by sophisticated and expensive cellulose regeneration processes (Ming Chih 1960), different procedures for generating foams out of cellulose fibers have been proposed since the 70s. In 1970, Norman and Frank patented a process to foam cellulose fibers by whipping air into aqueous suspensions assisted by the use of surfactants. The foamed suspension was dewatered over a screen by applying vacuum, thus avoiding structure collapse before drying at 204 °C (Friedberg Norman and Adams Frank 1970). Similarly, Alimadadi and Uesaka describe a process to foam thermomechanical pulp suspensions with sodium dodecyl sulfate to achieve foam densities as low as 5 kg/m3 (Alimadadi and Uesaka 2016). Recently, it was shown that the addition of 10 wt% of microfibrillated cellulose (MFC) to a wood fiber suspension improved the properties of foam-formed paper obtained by a surfactant-based process including a freeze–drying step (Korehei et al. 2016). Disadvantageously, aqueous MFC suspensions cannot be dewatered fast enough, causing a progressive collapse of the porous structure upon oven or room temperature drying and subsequent fibril re-aggregation (hornification) (Weise 1998). In 2008, Paakko et al. first pioneered aerogels by freeze–drying of aqueous nanofibrillar cellulose gels (Paakko et al.). Similarly, ultra-high porosity foams were prepared from cellulose nanofiber suspensions by Sehaqui et al. (2010) and Srinivasa et al. (2015). Furthermore, functionalized ultra-light weight cellulose foams (ρ ≤ 17.3 mg/cm3) with high porosity (≥99%) have been developed using a modification-freeze–drying pathway in water in the presence of methyltrimethoxysilane resulting in hydrophobic and oleophilic porous structures (Zhang et al. 2014, 2015).
Prior to freeze–drying and in the case of aqueous systems, a freeze-casting or ice-templating sequence can be employed in order to form the cellulose scaffold. This process can be described as a “segregation-induced templating of a second phase by a solidifying solvent” (Deville 2008, 2013). Although this technique is governed by numerous parameters and complex interactions which influence the final structure of the foam (Li et al. 2012; Rempel and Worster 1999), it is employed in various fields like biotechnologies, ceramics or polymers (O’Brien et al. 2004; Pawelec et al. 2014; Zhang et al. 2005). Recently, MFC and cellulose nanocrystals (CNC) porous structures through ice-templating were reported in literature: In these works, MFC and CNC were added as reinforcement agent to the polymers polylactic acid, polyvinyl alcohol, xylan, clays or dissolved cellulose (Blaker et al. 2010; Butylina et al. 2016; Dash et al. 2012; Donius et al. 2014; Koehnke et al. 2012, 2014).
The influence of different conditions, like freezing rate upon ice-templating, on the pore size distribution of the cellulose foam were investigated by Martoïa et al. (2016). Quenching of aqueous 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) -oxidized and enzymatic MFC suspensions at −13 °C resulted in foams with pore sizes in the range of 50–300 µm. In contrast, quenching at −80 °C led to pores sizes in the range 10–20 µm. This effect was explained in terms of competition of the ice-crystals nucleation and the ice-crystals growth. High freezing rates at low temperatures favor the generation of numerous ice nuclei resulting in smaller ice-crystals and, consequently, smaller pore sizes are templated.
Other alternatives to freeze–drying of aqueous suspensions have been proposed in literature. By solvent-exchange procedures, various solvents like methanol, ethanol, propanol, isopropanol, acetone, acetonitrile, tert-butanol, DMF, DMSO, dioxane, THF, ethylene glycol, or glycerol, gradually replace the ice/water in CNC suspensions upon immersion and subsequent evaporation (Azerraf et al. 2015). Another approach is to use TEMPO-oxidized MFC in the presence of the toxic surfactant octylamin, which enables the production of light foams upon careful removal of water in a Büchner funnel under vacuum and ambient conditions (Cervin et al. 2013; Johansson et al. 2016).
In this work, we describe a novel approach to produce porous structures with unmodified aq. MFC/urea suspensions—without freeze–drying. Our investigations follow the approach of previous studies, in which urea was shown to promote dissolution of cellulose, by interacting with the hydrophobic part of the cellulose because of its amphiphilic properties (Xiong et al. 2014). Furthermore, regenerated cellulose scaffolds were fabricated by an ice-templating process from urea/sodium hydroxide solutions (Flauder et al. 2014). Finally, the property of urea to work as a seeding agent for ice nucleation in aqueous solutions (Hazra et al. 2006) motivated this work on the properties of frozen MFC/urea suspensions.
The procedure is based on an ice-templating step of the MFC/urea suspensions at temperatures down to −45 °C, subsequent thawing and dewatering of the stabilized porous MFC structures under ambient conditions and, finally, drying of the MFC foam at 105 °C. Produced foams were characterized by X-ray micro-tomography and their mechanical properties were assessed through compression loading tests. The dried foams are mechanically stable when placed in water or common organic solvents, thus allowing for a straightforward post-functionalization.
Experimental
Material
Never-dried elemental chlorine free (ECF) fibers from bleached softwood pulp were obtained from the company Stendal, Berlin, Germany. Urea (>99%) was purchased from Merck, Darmstadt, Germany.
MFC production and characterization
MFC suspensions were produced and characterized according to Josset et al. (2014). In brief, ECF fibers in aqueous suspension were ground at a concentration of 2 wt% with an ultra-fine friction grinder “Supermasscolloider” (MKZA10-20J CE, Masuko Sangyo Co., Ltd., Kawaguchi/Saitama, Japan). Three different grinding energy inputs (3, 6 and 9 kWh/kg referred to dry MFC content) were applied. After grinding, the MFC suspensions were dewatered on a sieve under pressure to reach a solid content of about 5 wt%.
The morphology of the MFC was monitored by scanning electron microscopy (Nova NanoSEM 230 instrument, FEI, Hillsboro, Oregon, USA) at an accelerating voltage of 5 keV. A vacuum-dried droplet of 0.01 wt% MFC suspension was deposited on a mica sheet and sputtered with 7 nm platinum (BAL-TEC MED 020 Modular High Vacuum Coating Systems, BAL-TEC AG, Liechtenstein).
The dynamic viscosity was monitored with a rheometer RheolabQC from Anton Paar, Switzerland, on 2 wt% MFC suspensions for 30 s at a shear rate of 1000 s−1 and during progressive reduction down to 6.46 s−1. The specific surface area (SSA) of the MFC was determined after 4 h degassing at 105 °C on a “3 Flex” apparatus (Micromeritics, USA). The results rely on the Brunauer–Emmett–Teller (BET) theory based on N2 physisorption on aerogels prepared by critical CO2 drying, as described elsewhere (Josset et al. 2014).
Foam production, experimental setup and nomenclature
A full factorial design of experiment was performed as detailed in the Table 1. It forms 144 parameters sets that were realized in triplicates. The suspension mixtures (35 g) were prepared in 50 mL polypropylene tubes (diameter: 30 mm, length: 115 mm) and mixed for 15 s with a high-speed mixer (T 10 basic ULTRA-TURRAX®, IKA, Germany). In the case of the “fast freezing” process, samples precooled at 2 °C were placed directly in a climate chamber (HT 7033, Vötsch Industrietechnik GmbH, Germany) at −45 °C, whereas samples subjected to the “slow freezing” process were chilled at a rate of −2.57 °C/h from room temperature down to −25 °C. Finally, samples were dried either through room temperature drying (RT) or vacuum freeze–drying (FD). One replicate of each set was thawed, dried at room temperature for 48 h and subsequently dried at 105 °C for 18 h. As reference foams, a second replicate was vacuum freeze dried (Lyovac freeze–dryer, SRK system technik GmbH, Germany) in order to preserve the templated porous structure existing in the ice matrix.
The name of each individual sample is generated from the values of five parameters: grinding energy, concentration, urea mass ratio, freezing process and drying conditions, e.g. 3-0.5-0:1-f-FD.
Preparation of samples with large volume
Based on the results of the small-scale study, large volume samples (0.8 kg) were prepared with slurries containing 2 wt% MFC (3, 6 and 9 kWh/kg) and the same amount (2 wt%) of urea, following the slow freezing process. Therefore, rectangular PVC tubes (4 × 4 × 50 = 800 cm3, wall thickness: 2 mm) were filled with the slurries and frozen. After thawing overnight and dewatering at room temperature for 1 week, the foams were finally dried at 105 °C for 18 h. These foams are referred as F3 urea , F6 urea and F9 urea accordingly to the energy consumed for the MFC production and the content of urea. SEM was performed on dried foam samples following a similar procedure than for the MFC.
Density, porosity and urea content
For the determination of the foam densities and urea contents, 10 samples of each material (dimensions: 2.5 × 2.5 × 1.0 cm3) were intensively rinsed with deionized water, subsequently dried at 105 °C overnight and weighted. To ensure the successful washing of urea, the nitrogen content of five samples was determined by elemental analysis (Truspec Micro Analyzer, LECO Corporation, USA).
X-ray micro-tomography
The computer tomography was performed on foam samples with dimensions of 0.5 × 0.5 × 1.0 cm3 at 25 °C and 50% relative humidity at the Empa Center for X-ray Analytics (ZRA 2016). The foam parallelepipeds were cut from the center of the large volume samples F3 urea , F6 urea , and F9 urea . A custom made micro-tomography setup was equipped with a micro-focus X-ray tube (Viscom XT9160-TXD) and a 40 × 40 cm2 flat panel detector (Perkin Elmer XRD 1621 CN2 ES).
Since these foams are very weakly absorbers of X-rays, the X-ray source had to be operated at a low voltage of 50 kV and 190 µA. 1441 projections were acquired per tomogram. Due to the rather low contrast of the projections and the remaining fluctuations in the X-ray source flux, the projections were pre-processed and filtered to reach a high signal-to-noise ratio in the 3D volume. Pre-processing and tomographic reconstruction starting from the projections were performed with in-house developed software tools. The tomographic reconstruction part of the software consists of an implementation of the Feldkamp–Davis–Kress (FDK) cone beam filtered back-projection algorithm (Feldkamp et al. 1984), optimized for fast reconstruction of large tomographic datasets exploiting GPU processing, developed and maintained at Empa’s Center for X-ray Analytics. The voxel sizes were 3.7 μm (F3 urea ) and 4.4 μm (F6 urea and F9 urea ), respectively.
Tomographic data analysis
Processing and analysis of the data were performed by the software ImageJ, version 1.51d (Schneider et al. 2012). Each experimental 3D image (tomogram) of the scanned layer of a foam specimen was converted, by 3D image segmentation (see “Tomographic data analysis” section of the Supplementary Materials for details), into two distinct 3D binary images (also called “3D binary masks”), each identifying the voxels belonging to either of the two material phases of the scanned volumes: the foam cell walls (“wall mask”) and the air contained in its pore space (“air mask”).
For all 3D binary images of each specimen only an inner sub-volume of 0.31 × 0.31 × 0.31 cm3, positioned in the center of the specimen, was selected and actually used for the analysis in order to avoid artifact in the foam structure, close to the boundaries, created during the samples preparation.
The cropped 3D air mask was used to analyze the size distribution of the pore space of the foam. Given (1) the high degree of interconnections between several cells, the specimens being opened-cell foams, and (2) the high degree of cell shape variation, we adopted the continuous size distribution analysis approach developed for the quantitative characterization of porous materials with highly interconnected pores (Torquato 2002).
The actual realization of such analysis was performed with the algorithm developed by Münch and Holzer (2008) and implemented in the ImageJ plugin “Pore size distribution” of the Empa Bundle of ImageJ Plugins for Image Analysis “EBIPIA” (Münch 2014), used with the option for a continuous pore size distribution (cPSD) calculation. The algorithm, summarized in the “Tomographic data analysis” of the Supplementary Materials, allows estimating, for each value of a critical pore diameter, D c , adopted as a definition of pore size, the cumulative pore space volume fraction (volume of pore space divided by total volume of the analyzed samples) due to pores with size larger than D c , which is related with the complementary cumulative distribution function (cCDF or \(P(D_{c} > D )\), where \(P\left( \ldots \right)\) indicates the probability of the event within the parentheses) of D c when it is considered as a random variable with statistical ensemble given by the pore space.
The implementation of the algorithm also allows obtaining a 3D image where each voxel is assigned a value equal to pore space local size, D c . Such image is used for a graphical representation of the spatial distribution of D c .
The cropped 3D cell wall mask was used to analyze the cell wall “thickness” distribution according to an approach similar to the cPSD one and originally proposed by Hildebrand and Rüegsegger for analyzing bone microstructures (1997). Such cell wall continuous thickness distribution analysis was performed with the “Thickness” algorithm implemented in “BoneJ (version 1.4.1)”, a ImageJ plugin for analyzing cellular solids, e.g., bone microstructures (Doube et al. 2010; Dougherty and Kunzelmann 2007). This plugin also allows obtaining a spatial distribution representation of the local cell wall thickness, in the form of a colored 3D image.
Mechanical properties
For each dry MFC foam, compression tests were performed on 10 samples prepared by sawing parallelepiped with dimensions of 2.5 × 2.5 × 1.0 ± 0.025 × 0.025 × 0.025 cm3 using a universal testing machine type Zwick 1484 (Zwick GmbH & Co. KG, Germany), equipped with a 500 N load cell and crosshead displacement sensor, both with an accuracy of measurement of <1% (class I). The compression speed in the vertical (z) direction was 1 mm/min, down to a 50% compressive strain. E-modulus, yield stress and absorbed energy were calculated from the compressive stress-compressive strain curves as described in the work of Sehaqui et al. (2011).
Results and discussion
Characterization of the starting material MFC
Morphology of MFC
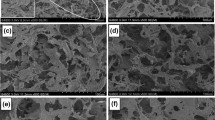
SEM micrographs of three different MFC grades with applied grinding energies of 3, 6 and 9 kWh/kg (MFC3, MFC6, MFC9) are presented in Fig. 1. As expected, the MFC3 obtained with the lowest grinding energy still contains large non disintegrated fiber debris even though fibrils with diameters of about 50–150 nm are also visible. The proportion of debris was visibly reduced with increasing the energy input (MFC6, MFC9) but the size of the fibrils remained in the same range.

SEM micrographs at two increasing magnification levels (left–right columns) of MFC3 (a, b), MFC6 (c, d) and MFC9 (e, f)
Viscosity and specific surface area (SSA) of MFC
The degree of fibrillation of the MFC samples was further assessed by viscosity measurement of the suspensions (2 wt%) and determination of the specific surface area (SSA) of dry fibrils. The results of the viscosity measurements are presented in Fig. 2. The values of the viscosity at different shear rates for MFC6, MFC9 suspensions were respectively 3 and 5 times higher than the viscosity of the MFC3 suspension. The specific surface areas of MFC3, MFC6, and MFC9 were 147, 198, and 228 m2/g, respectively. Those values represent increases in the SSA of 34% (MFC6) and 55% (MFC9) in respect to the least ground material MFC3. The increase of viscosity and SSA with the grinding energy is in agreement with an enhanced degree of disintegration (Josset et al. 2014; Kangas et al. 2014).

Dynamic viscosities of MFC3, MFC6, and MFC9 suspensions (2 wt%), respectively
Freezing–thawing–drying parameters
Freezing, thawing and drying of suspensions with MFC concentrations below or equal to 1 wt% did not lead to any highly porous structures. After thawing, the ice-templated structures shrank and flattened in a non-homogeneous manner. This is in accordance with the observations of Martoïa et al. (2016). Foams, which were produced out of aqueous MFC suspensions followed by freeze–drying, were subject to strong shrinkage below a critical concentration of 1 wt%. This was explained by the reduction of inter-fibrils bounds at low MFC concentrations, leading to depletion: the fiber network cannot withstand the ice crystal growth, in particular at the boundaries of the sample holder.
At higher MFC concentrations, the fast freezing procedure led upon RT drying indeed to 3D structures, but these presented strong deformations or collapses, as visible in the Fig. 3. This figure depicts foams originated from 2 wt% suspensions of MFC9, without the additive urea (9-2-0:1-f-FD, 9-2-0:1-f-RT), and with different amounts of urea (9-2-0.5:1-f-FD, 9-2-1:1-f-FD, 9-2-0.5:1-f-RT, 9-2-1:1-f-RT). The suspensions were frozen at high cooling rate and freeze- dried or thawed and dried under ambient conditions. In the present case, freeze–drying was used to visualize the effect of urea on the MFC network in the frozen structure. The urea-free foam (9-2-0:1-f-FD) exhibited strong depletion; the transport of the fibrils to the center of the tube during ice formation occurred. The liquid/solid drift started at the tube wall and propagated to the center. This drift of the fibrils was significantly reduced by adding urea to the MFC suspensions. Finally, complete porous structures filling up the whole tube volume were achieved (Fig. 3a: 9-2-0.5:1-f-FD, 9-2-1:1-f-FD). Obviously, urea influenced the formation of the foam structure by either changing the ice crystal grow process and/or by hindering fibril agglomeration. Nonetheless, these foams were still inhomogeneous, containing a dense texture of agglomerated fibrils.

Foams produced from 2 wt% suspensions of MFC9 and 0, 1 and 2 wt% urea (l. to r.) at high cooling rate and subsequent freeze–drying (a: 9-2-0:1-f-FD, 9-2-0.5:1-f-FD, 9-2-1:1-f-FD) and ambient drying (b: 9-2-0:1-f-RT, 9-2-0.5:1-f-RT, 9-2-1:1-f-RT)
Thawing of frozen samples (Fig. 3b: 9-2-0:1-f-RT, 9-2-0.5:1-f-RT, 9-2-1:1-f-RT) under ambient conditions led to liquid water and subsequent dewatering induced a collapse of the mechanical instable foam structure independent of adding urea or not (Fig. 3b).
Beside urea, also the cooling rate and the solid–liquid drift velocity influenced the foam homogeneity during freezing. In order to investigate these parameters, suspensions of 2 wt% of MFC9 with and without urea were prepared and frozen with a slower cooling rate of −2.57 °C/h down to −25 °C. In analogy to the previous samples (Fig. 3), drying was performed under freeze–drying (9-2-0:1-s-FD, 9-2-0.5:1-s-FD, 9-2-1:1-s-FD) or ambient conditions (9-2-0:1-s-RT, 9-2-0.5:1-s-RT, 9-2-1:1-s-RT). Remarkably, by reducing the cooling rate and the drift velocity, the fibrils formed a more homogenous porous structure (9-2-0:1-s-FD in Fig. 4a). By increasing the urea content, the homogeneity of the foam structure was improved (9-2-0.5:1-s-FD, 9-2-1:1-s-FD in Fig. 4a). As expected, water generated by thawing and dewatering led to shrinkage (9-2-0:1-s-RT in Fig. 4b). Differently to the previous foams (9-2-0.5:1-f-RT, 9-2-1:1-f-RT in Fig. 3b), visually homogeneous foams (9-2-0.5:1-s-RT, 9-2-1:1-s-RT in Fig. 4b) could be obtained under the influence of urea. Here, the stability and homogeneity of the porous structure could benefit from the low cooling rate.

a Freeze-dried samples 9-2-0:1-s-FD, 9-2-0.5:1-s-FD, and 9-2-1:1-s-FD (l. to r.), b under ambient conditions dried samples 9-2-0:1-s-RT, 9-2-0.5:1-s-RT, and 9-2-1:1-s-RT (l. to r.)—all samples are frozen at low cooling rate. c Freeze-dried samples (3-2-1:1-s-FD, 6-2-1:1-s-FD, and 9-2-1:1-s-FD (l. to r.), d under ambient conditions dried samples 3-2-1:1-s-RT, 6-2-1:1-s-RT, and 9-2-1:1-s-RT (l. to r.)—samples are prepared with MFC3, MFC6, and MFC9, respectively, and were frozen at low cooling rate
Finally, the influence of degree of fibrillation on the foam forming process was investigated. Therefore, suspensions with MFC3, MFC6, and MFC9, respectively, at 2 wt% MFC content and urea content of 1:1 were frozen at low cooling rate and, subsequently, freeze-dried (3-2-1:1-s-FD, 6-2-1:1-s-FD, 9-2-1:1-s-FD) or dried under ambient condition (3-2-1:1-s-RT, 6-2-1:1-s-RT, 9-2-1:1-s-RT). Under the applied conditions, all suspensions could be converted into almost homogenous foams with shape retention independently from the degree of fibrillation (Fig. 4c, d). Exclusively, the foam with MFC3 exhibits slight depletion after thawing and drying (Fig. 4d: 3-2-1:1-s-RT).
Summarizing the aforementioned set-up, a set of most promising parameters could be determined that enables the production of foams with high homogeneity:
MFC3, MFC6 and MFC9 were all suitable as starting material. The concentration of MFC leading to the most stable cellular solids was 2 wt%. The optimal mass ratio urea/MFC was found to be 1:1. A cooling rate of −2.57 °C/h down to −25 °C is suitable. And finally, thawing and drying under ambient conditions lead to homogeneous foams.
Based on these parameters, MFC3, MFC6, and MFC9 were used to produce large foam samples in 800 cm3 containers. These foams still containing a given amount of urea were comprehensively characterized afterwards (F3 urea , F6 urea , and F9 urea ).
Monitoring the temperature profile of MFC suspensions upon freezing
Figure 5 shows the monitored temperature progress inside the climate chamber and in the center of one profile filled with MFC9 suspension mixed with urea. This program was chosen to minimize the temperature gradient inside the samples and at the walls of the container. After a temperature equilibrium phase between the climate chamber and the sample down to 0.6 °C, the temperature was reduced with a very slow ramp of −2.57 °C/h. Super-cooling was observed, with recalescent nucleation occurring at approx. −6 °C, and the suspension started to freeze at approx. −2 °C. After a phase transition of roughly 3 h, the temperature of the frozen suspension decreased again to reach the final temperature of the program of −25 °C after 4.75 h.

Temperatures profile inside the climate chamber (black curve) and in the center of a 0.8 L sample of MFC9 and urea (red curve)
SEM characterization of the foams
In Fig. 6, SEM micrographs of the three cellular structures of F3 urea , F6 urea , and F9 urea , are provided at two magnifications (100× and 1000×). The MFC network in all cases underwent compression by ice crystals, templating the MFC into sheets arranging into a cellular fashion distribution. The cell size reached few hundreds of micrometers and the wall thickness was in the range of a few tens of micrometers. Furthermore, at the surface of the sheets single fibrils and or bundles of fibrils spanning along the sheets can be distinguished. In detail, F3 urea exhibited spherical pits with an average diameter of about 100 μm. Spherical pits in foams F6 urea and F9 urea were roughly two to three times larger in size. Seemingly, the reduced volume of the F3 urea could be a consequence of the shrinkage during thawing and drying which could also be observed for 3-2-1:1-s-RT.

SEM micrographs of the foams: F3 urea (a, b), F6 urea (c, d), F9 urea (e, f)
Density, porosity and urea content
The foams F3 urea , F6 urea , and F9 urea , respectively, were prepared from ternary mixtures of water-based MFC and urea, containing cMFC = 2 wt% and curea = 2 wt%. Based on the following approximated values of the densities (ρwater = 1000 kg/m3, ρcellulose = 1500 kg/m3, and ρurea = 1320 kg/m3), and under the assumption that between 0 and 100% of urea can be leached out with water after thawing or drying, the theoretical value of the density of the foams F3, F6, and, F9, \(\rho_{foam}^{theo.}\), without depletion, should vary between the values of \(\rho_{foam}^{min}\) (0% urea) and \(\rho_{foam}^{max}\) (100% urea) (Eqs. 1–3).
Out of this, the calculated values were respectively \(\rho_{foam}^{min}\) = 20 kg/m3 (Eq. 2) and \(\rho_{foam}^{max}\) = 41 kg/m3 (Eq. 3).
Porosity, P, is defined as the complement to the ratio of the density of the foam (ρfoam) to the density of cellulose/urea mixture (ρs) (Gibson and Ashby 1999):
The value of the density, ρs, varies between the value of the density of cellulose, ρcellulose, and the value of the density (ρ mins ) of the mixture of cellulose/urea which is evaluated by using the mixing rule (Eq. 5 with xurea representing the mass fraction of urea):
With a cellulose/urea mass ratio of 1:1, the highest fraction of urea remaining in the foam was xurea = 0.5, leading to a theoretical minimum density of the solid material of ρ mins = 1420 kg/m3. Hence, the theoretical porosity of foams can be estimated to be between 97.3 and 98.6% which is in agreement to the value for a foam generated by Lee and Deng from a 2 wt% MFC suspension by freeze–drying (2011).
Table 2 summarizes the experimental results obtained for the density, the porosity and the urea content of the foams F3, F6, and F9 (10 specimens with a volume of 2.5 × 2.5 × 1.0 cm3 for each foam) after freezing–thawing–drying (unwashed) and subsequent leaching out of urea (washed).
As already observed, the foam F3 urea tends to deplete resulting in a density of 36.4 kg/m3 which is 20% higher than the density of the foams F6 urea and F9 urea (30.5 and 30.2 kg/m3). The low standard deviations of the density of all foams are also an indication of their homogenous porous structure templated by the freeze–thawing–drying process. According to Eq. (5), the density of cellulose is 1500 kg/m3 and of cellulose:urea = 2:1 is 1420 kg/m3. However, due to the very low specific densities of the foam compared to the bulk material, the differences in the porosity values for F3 urea , F6 urea , and F9 urea are negligible (F3 urea : 97.4–97.6%; F6 urea and F9 urea : 97.9–98.0%).
In order to determine the parameters of the urea-free foams F3, F6, and F9, samples were intensively leached with deionized water. The mass fractions of nitrogen determined by elemental analysis of the neat MFC and pure urea were respectively 0.19 and 46.7%. Equations (6) and (7) show the relationship between the weight fraction of urea (xurea) and the nitrogen contribution ([N], wt%) in the foams.
Based on the MFC mass conservation through the leaching step (Eq. 8), the weight fraction of urea and the foam density before (x 0urea , ρ 0foam ) and after leaching (x 1urea , ρ 1foam ) are related as described by the Eq. (9).
After leaching, the nitrogen mass fractions for F3, F6, and F9 were 0.72 (±0.04) %, 0.50 (±0.03) %, and 0.31 (±0.01) %, respectively. Almost the complete urea content used for templating was efficiently removed. By neglecting the remaining traces of urea, the values of the density and the porosity of the foams were calculated to be 33.9 kg/m3 (F3), 29.1 kg/m3 (F6) and 29.3 kg/m3 (F9). Similarly, the urea content of the different foams before leaching can be evaluated: 7.9, 5.6 and 4.0%, respectively. The higher the cellulose mass per volume, the higher the urea content which was trapped. Since the original suspension contained 50% urea (1:1 weight ratio to MFC), these results indicated that 84–92% of the urea were directly washed out until thawing and drying. Finally, a simple washing procedure could remove more than 97% of the initial urea.
Tomography
Figure 7 depicts a 2D cross-section, orthogonal to the rotation axis of the micro-tomography setup and typically called “slice”, from the reconstructed tomogram of F9 urea . Such slice demonstrates the sufficient contrast obtained between the cellulose sheets (bright filaments) and air (dark area in between), enabling a facile segmentation.

Reconstructed slice of the tomogram of F9 urea
As described in the “Tomographic data analysis” section, the information on pore size and wall thickness was computed in 3D by two algorithms, the EBIPIA’s continuous pore size distribution (“cPSD”) and BoneJ’s “Thickness”, respectively. For the tomograms of the three foams, each algorithm produced a new 3D image, attributing to each voxel a value equal to the critical diameter of the largest sphere still fitting either in the pore volume (cPSD) or in the wall (BoneJ/Thickness) and being centered in the voxel (cPSD) or just containing the voxel itself (BoneJ/Thickness). These two 3D images are thus spatial distribution maps of the pore space and cell wall size/thickness, respectively. For each foam, one slice of the 3D map at the same position is provided in Fig. 8.

Slices extracted from the 3D (color) maps produced with the algorithms cPSD (left) and BoneJ/thickness (right) from the three tomograms (F3 urea : a, b; F6 urea : c, d; F9 urea : e, f)
Concerning the pore size, colored slices in Fig. 8 visually confirm the SEM measurements: the foam produced with the least processed MFC3 shrank more, leading to smaller pores. For F3 urea , the largest D c observed in the slice is around 275 μm, but most of the values are lower than 200 μm (cold hues of the color scale), whereas F6 urea and F9 urea are similarly presenting much more frequently D c values larger than 200 μm (warm hues of the color scale). However, the wall thickness does not present large differences between the three structures.
The quantified results from the cPSD analysis are reported in the Fig. 9. The volume fraction represents the amount of the pore voxels having a given critical diameter. The probability for a voxel to have a critical diameter larger than a given value, P(D c > D), is indicated as well and represents the volume fraction of the total pore space comprised by pores having a critical diameter larger than a given value. For F3 urea , the 50% cut-off critical diameter was obtained for 120 μm, whereas this value almost doubled to 240–250 μm for the other foams. For F6 urea and F9 urea , less than 20% of the void was comprised by pores having smaller critical diameters than 120 μm. Martoïa et al. obtained foams with pores in the range 50–300 μm, when quenching the suspensions at −13 °C, and this interval was reduced to tenths of micrometers while diminishing the temperature down to −80 °C (2016).

Volume fraction of the total pore space comprised in spheres with a critical diameter Dc (l.) and probability that any region of the pore space can be covered by a sphere with (critical) diameter Dc larger than a given value (r.)
The BoneJ’s algorithm “Thickness” computed for each tomogram the average of the wall thickness and the results are between 11 and 14 μm. Cell walls with lower average thickness may exist within the samples. However, due to the voxel size, comprised between 3.7 μm (F3 urea ) and 4.4 μm (F6 urea and F9 urea ), and given that the effective spatial resolution of the tomograms is approximately double the voxel size, such thinner walls could not be completely analyzed. Still, the obtained wall thickness average values derive from the analysis of the majority of the cell wall volume and are in agreement with the SEM observations. Martoïa et al. obtained lower values (few microns) based on tomography analysis; this might be related to the lower MFC concentration used in their work (2016).
Compression tests
In Fig. 10 typically S-shaped curves of the compressive stress–strain measurements of the foams F3 urea , F6 urea , and F9 urea and their mechanical properties E-modulus, yield stress, and absorbed energy are depicted. For open-cell foam deformations, three different micromechanical regions were typically defined: a linear elastic region, a plastic deformation region and a densification region (Gibson and Ashby 1999). In the linear elastic region, the foams F3 urea , F6 urea , and F9 urea behaved as an elastic solid up to a strain of approximately five percent. Here, the MFC walls contribute to the load-bearing, which depends on the bending and stretching of the cellulose material. Within this strain range, the foams could fully recover upon force release. Especially, the slope of the initial linear region corresponds to the Young’s modulus (E-modulus) which was found to be 2.4 MPa for F3 urea , 2.9 MPa for F6 urea , and 4.1 MPa for F9 urea , respectively. These values correlated almost with the increasing degree of the fibrillation of the MFCs and thus with the increasing energy provided during grinding. Probably, less fibrillated bulk-material MFC3 of F3 urea formed more heterogeneous sheet structures compared to highly fibrillated bulk-material MFC9 of F9 urea . In turn, this led to the lower mechanical stability of the foam F3 urea under compression. But in general, a lower E-modulus for F3 urea was expected.

Compressive stress–strain curves (a: F3 urea , b: F6 urea , c: F9 urea ). E-modulus, yield stress and absorbed energy (d: F3 urea , e: F6 urea , f: F9 urea ). For the foam produced with the MFC3, the dashed lines correspond to the corrected values for a density of 30 kg/m3 instead of 36 kg/m3
With increasing compression stress, a regime of irreversible plastic deformations (collapse plateau) was reached. The transition from elasticity to collapse is characterized by the compressive yield stress which is the intersection of the slope of linear regime and the plateau. The foams F3 urea , F6 urea , and F9 urea showed a yield stress of 66, 71, and 91 kPa, respectively. The extent of the collapse plateau remains limited to the strain range of 5–10% for the foams F3 urea , F6 urea , and F9 urea , which is in agreement to the literature (Sehaqui et al. 2010). The collapse of the foam under compression can be considered as an irreversible buckling and braking of the brittle MFC sheets.
The value of the absorbed energy per unit volume is the area under the stress–strain curve before densification and it is an indicator for the energy absorption property of the foam. Since the densification occurred in the three cases at a moderate rate, the absorbed energy was estimated by the integration between 0 and 50% compressive strain. In the case of the MFC foams, an almost linearly increasing amount of deformation energy could be absorbed: 39 kJ/m3 (F3 urea ), 40 kJ/m3 (F6 urea ), and 49 kJ/m3 (F9 urea ) (Fig. 10).
The apparent deviation of the values of the yield stress and absorbed energy of foam F3 urea is assigned to the higher density of the foam (Table 2). Therefore accordingly to the literature, the values of the E-modulus and the yield stress of a foam material at different densities can by determined by using allometric scaling (Eq. 10) (Gibson and Ashby 1999; Sehaqui et al. 2010).
In these equations, ES and ρS are the Young’s modulus and density of the scaffold, respectively, E* and ρ* are the Young’s modulus and density of the overall foam, respectively, and \(\sigma^{*}\) and \(\sigma_{s}\) indicate the respective yield stress values.
These equations are valid for foams produced out of one MFC but having different densities values, e.g. ρ* = ρ1 and ρ* = ρ2, so that they can be rewritten as Eq. (11).
The last equation allows the allometric scaling of the E-modulus and the yield stress of foam F3 urea assuming a density of 30 kg/m3 (of F6 urea and F9 urea ) instead of 36 kg/m3: E corr. F3 = 1.6 MPa and σ corr. F3 = 49.8 kPa. The correction of the absorbed energy was based on the assumption that the stress–strain curves would be simply vertically shifted by the difference between σF3 = 65.6 kPa and σ corr.F3 = 49.8 kPa, leading to an estimated value of ca. 32 kJ/m3. Close results are obtained using scaling factors similar to Eq. (11).
With the allometric scaling, the expected and almost linear correlations of the E-modulus, yield stress and absorbed energy with the degree of fibrillation could be demonstrated. The higher the degree of fibrillation, and the higher the spatial homogeneity of fibril density and fibril orientation in the wall-forming bulk material, the higher the values of E-modulus, yield stress and absorbed energy for MFC foams.
With strain values over 10%, a densification of the foam structure occurs. The pores of the foam collapse under compression, adjacent MFC walls progressively get closer and finally touch each other. The foams responded with higher stress values within small increase in compression.
Sehaqui et al. investigated the mechanical properties of foams (ρ = 30 kg/m3) prepared with softwood MFC, obtained by vacuum freeze drying. For these porous structures, the obtained values for the E-modulus and the yield stress were respectively 800 and 73 kPa (2010). Due to the very high cooling rate favoring the nucleation rather than crystal growth and the careful removal of the water through freeze–drying, aerogels produced through this procedure are mostly built out of thin fibrils aggregates resulting in extremely soft porous structures. In comparison to the foams of this study, the cellulose sheets forming the porous structure were built through slow ice-templating: fibrils were pushed together by the growth of larger ice crystals, which aggregated to form dense and bulky sheets.
An overview of the data sets of E-moduli for other MFC based foams or aerogels as a function of the density is provided in Fig. 11, adapted from Donius et al. with the values of the E-modulus of F3 urea , F6 urea , and F9 urea (2014). In the work of Donius et al. clays (montmorillonite, MTM) and MFC were mixed in different ratios and frozen using either an isotropic, i.e. an immersion in liquid N2, or a unidirectional process using a so called “coldfinger”. More than 100 times higher E-modulus values were recorded along the freezing direction, in comparison to the homogeneous freezing. However, the E-modulus measurements performed perpendicularly to the freezing direction did not exceed 5 times the values of the isotropic material. Using a similar unidirectional freezing process, Lee and Deng found, for foams prepared out of a 2 wt% MFC suspension, a yield stress in the freezing direction of about 30.7 kPa, a value 40, 56 and 67% inferior to the ones obtained for F3 urea , F6 urea , and F9 urea , respectively (2011).

Adapted from Donius et al. (2014)
E-modulus—density diagram for different cellulose based foams and expanded polystyrene. The values for F3 urea , F6 urea , and F9 urea are depicted by a small area and lines
The E-modulus values obtained in this study show that the ice-templating process with urea followed by ambient drying leads to three dimensional structures as resistant as or even slightly stronger against deformation than the strongest MTM-MFC composites, which already have properties close to those of expanded polystyrene.
Conclusions
This study showed that a very simple freeze–thawing process followed by a drying under ambient conditions can be used to produce homogeneous MFC foam thanks to the addition of urea. It is noteworthy to emphasize that first satisfactory foams were prepared in a kitchen freezer, indicating that just minimum equipment is required.
The role of the additive urea in the freezing–thawing–drying procedure is not completely understood. But, because of its amphiphilic character, which allows the interaction with water and cellulose, and its property to influence the ice nucleation, it can be assumed, that urea has a positive impact on the nucleation rate in the super-cooled MFC slurries, preventing the formation of large ice-crystals growing from the boundaries into the MFC network.
A similar effect was observed as well with the fast freezing process but the resulting structures after RT drying were not homogeneous. Interestingly, about 90% of the urea was eliminated with the thawed water, so that recycling of this additive could be envisaged.
In agreement with former results, we confirmed that the grinding energy used for the MFC production is an efficient mean to tune its properties, like the viscosity or the BET surface area. Moreover, this energy could be well correlated to the mechanical properties of the foams, the most ground material leading to the highest performances in terms of E-modulus, yield stress and absorbed energy, with values rivaling with those of expended polystyrene. In turn, the foams produced out of the least ground material were more subject to shrinkage, resulting in smaller pores and a higher density, whereas the other foams almost maintained their initial shape and presented very close pore size distributions. However, under the presented experimental conditions, this process failed with MFC concentrations less or equal to 1 wt% so that this cannot compete with the freeze–drying procedure for the preparation of aerogels.
Further work should focus on the impact of the freezing program since there are still large possibilities between the two used in this work. Moreover, the exact role of urea and its influence on the ice-crystals nucleation and growth should be elucidated.
References
Alimadadi M, Uesaka T (2016) 3D-oriented fiber networks made by foam forming. Cellulose 23:661–671. doi:10.1007/s10570-015-0811-z
Azerraf C, Braslavsky I, Lapidot S, Roth Shalev S, Shoseyov O, Slattegard R, Yashunsky V (2015) Porous structure used for article, consists of unidirectionally oriented partially interconnected sheets comprising nanocrystalline cellulose and/or microfibrillar cellulose as cellulose-based material. WO2015114630-A1
Blaker JJ, Lee KY, Mantalaris A, Bismarck A (2010) Ice-microsphere templating to produce highly porous nanocomposite PLA matrix scaffolds with pores selectively lined by bacterial cellulose nano-whiskers. Compos Sci Technol 70:1879–1888. doi:10.1016/j.compscitech.2010.05.028
Butylina S, Geng S, Oksman K (2016) Properties of as-prepared and freeze–dried hydrogels made from poly(vinyl alcohol) and cellulose nanocrystals using freeze–thaw technique. Eur Polym J. doi:10.1016/j.eurpolymj.2016.06.028
Cervin NT, Andersson L, Ng JBS, Olin P, Bergstrom L, Wagberg L (2013) Lightweight and strong cellulose materials made from aqueous foams stabilized by nanofibrillated cellulose. Biomacromol 14:503–511. doi:10.1021/bm301755u
Dash R, Li Y, Ragauskas AJ (2012) Cellulose nanowhisker foams by freeze casting. Carbohyd Polym 88:789–792. doi:10.1016/j.carbpol.2011.12.035
Deville S (2008) Freeze-casting of porous ceramics: a review of current achievements and issues. Adv Eng Mater 10:155–169. doi:10.1002/adem.200700270
Deville S (2013) Ice-templating, freeze casting: beyond materials processing. J Mater Res 28:2202–2219. doi:10.1557/jmr.2013.105
Donius AE, Liu A, Berglund LA, Wegst UGK (2014) Superior mechanical performance of highly porous, anisotropic nanocellulose-montmorillonite aerogels prepared by freeze casting. J Mech Behav Biomed Mater 37:88–99. doi:10.1016/j.jmbbm.2014.05.012
Doube M et al (2010) BoneJ: free and extensible bone image analysis. ImageJ Bone 47:1076–1079. doi:10.1016/j.bone.2010.08.023
Dougherty R, Kunzelmann K-H (2007) Computing local thickness of 3D structures with ImageJ. Microsc Microanal 13:1678–1679. doi:10.1017/S1431927607074430
Feldkamp LA, Davis LC, Kress JW (1984) Practical cone-beam algorithm. J Opt Soc Am A 1:612–619. doi:10.1364/JOSAA.1.000612
Flauder S, Heinze T, Müller F (2014) Cellulose scaffolds with an aligned and open porosity fabricated via ice-templating. Cellulose 21:97–103. doi:10.1007/s10570-013-0119-9
Friedberg Norman D, Adams Frank S (1970) Method for drying a wet foam containing cellulosic fibers. US Patent US 3542640 A, 1970/11/24
Gibson LJ, Ashby MF (1999) Cellular solids: structure and properties. In: Clarke DR, Suresh S, Ward IM (eds) Cambridge solid state science series, 2nd edn (with corrections). Cambridge: Cambridge University Press. ISBN 9780521499118
Hazra A, Paul S, De UK, Kar S, Goswami K (2006) Study of ice nucleation/hydrate crystallisation over urea. Nova Science Publishers, New York
Hildebrand T, Rüegsegger P (1997) A new method for the model-independent assessment of thickness in three-dimensional images. J Microsc 185:67–75. doi:10.1046/j.1365-2818.1997.1340694.x
Johansson E, Tchang Cervin N, Gordeyeva K, Bergström L, Wagberg L-E (2016) CNF cellular solid material. WO Patent WO 2016/068771 A1, 2016/05/06
Josset S, Orsolini P, Siqueira G, Tejado A, Tingaut P, Zimmermann T (2014) Energy consumption of the nanofibrillation of bleached pulp, wheat straw and recycled newspaper through a grinding process. Nord Pulp Pap Res J 29:167–175
Kangas H, Lahtinen P, Sneck A, Saariaho A-M, Laitinen O, Hellen E (2014) Characterization of fibrillated celluloses. A short review and evaluation of characteristics with a combination of methods. Nordic Pulp Paper Res J 29:129–143
Koehnke T, Lin A, Elder T, Theliander H, Ragauskas AJ (2012) Nanoreinforced xylan-cellulose composite foams by freeze-casting. Green Chem 14:1864–1869. doi:10.1039/c2gc35413f
Koehnke T, Elder T, Theliander H, Ragauskas AJ (2014) Ice templated and cross-linked xylan/nanocrystalline cellulose hydrogels. Carbohyd Polym 100:24–30. doi:10.1016/j.carbpol.2013.03.060
Korehei R, Jahangiri P, Nikbakht A, Martinez M, Olson J (2016) Effects of drying strategies and microfibrillated cellulose fiber content on the properties of foam-formed paper. J Wood Chem Technol 36:235–249. doi:10.1080/02773813.2015.1116012
Lee J (2011) Deng YL (2011) The morphology and mechanical properties of layer structured cellulose microfibril foams from ice-templating methods. Soft Matter 7:11547
Li WL, Lu K, Walz JY (2012) Freeze casting of porous materials: review of critical factors in microstructure evolution. Int Mater Rev 57:37–60. doi:10.1179/1743280411y.0000000011
Martoïa F, Cochereau T, Dumont PJJ, Orgéas L, Terrien M, Belgacem MN (2016) Cellulose nanofibril foams: links between ice-templating conditions, microstructures and mechanical properties. Mater Des. doi:10.1016/j.matdes.2016.04.088
Ming Chih C (1960) Manufacture of regenerated cellulose sponge material. US Patent US 2927034 A, 1960/03/01
Münch B (2014) Empa bundle of ImageJ Plugins for image analysis (EBIPIA). http://wiki.imagej.net/Xlib
Münch B, Holzer L (2008) Contradicting geometrical concepts in pore size analysis attained with electron microscopy and mercury intrusion. J Am Ceram Soc 91:4059–4067. doi:10.1111/j.1551-2916.2008.02736.x
O’Brien FJ, Harley BA, Yannas IV, Gibson L (2004) Influence of freezing rate on pore structure in freeze-dried collagen-GAG scaffolds. Biomaterials 25:1077–1086. doi:10.1016/s0142-9612(03)00630-6
Paakko M et al (2008) Long and entangled native cellulose I nanofibers allow flexible aerogels and hierarchically porous templates for functionalities. Soft Matter 4:2492–2499. doi:10.1039/B810371B
Pawelec KM, Husmann A, Best SM, Cameron RE (2014) Ice-templated structures for biomedical tissue repair: from physics to final scaffolds. Appl Phys Rev. doi:10.1063/1.4871083
Rempel AW, Worster MG (1999) The interaction between a particle and an advancing solidification front. J Cryst Growth 205:427–440. doi:10.1016/S0022-0248(99)00290-0
Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi:10.1038/nmeth.2089
Sehaqui H, Salajkova M, Zhou Q, Berglund LA (2010) Mechanical performance tailoring of tough ultra-high porosity foams prepared from cellulose I nanofiber suspensions. Soft Matter 6:1824–1832. doi:10.1039/b927505c
Sehaqui H, Zhou Q, Berglund LA (2011) High-porosity aerogels of high specific surface area prepared from nanofibrillated cellulose (NFC). Compos Sci Technol 71:1593–1599. doi:10.1016/j.compscitech.2011.07.003
Srinivasa P, Kulachenko A, Aulin C (2015) Experimental characterisation of nanofibrillated cellulose foams. Cellulose 22:3739–3753. doi:10.1007/s10570-015-0753-5
Torquato S (2002) Sections 2.6 and 12.5.4. In: Random heterogeneous materials—microstructure and macroscopic properties. Springer: New York
Weise U (1998) Hornification—mechanisms and terminology. Paperi Ja Puu Paper Timber 80:110–115
Xiong B, Zhao P, Hu K, Zhang L, Cheng G (2014) Dissolution of cellulose in aqueous NaOH/urea solution: role of urea. Cellulose 21:1183–1192. doi:10.1007/s10570-014-0221-7
Zhang HF, Hussain I, Brust M, Butler MF, Rannard SP, Cooper AI (2005) Aligned two- and three-dimensional structures by directional freezing of polymers and nanoparticles. Nat Mater 4:787–793. doi:10.1038/nmat1487
Zhang Z, Sèbe G, Rentsch D, Zimmermann T, Tingaut P (2014) Ultralightweight and flexible silylated nanocellulose sponges for the selective removal of oil from water. Chem Mater 26:2659–2668. doi:10.1021/cm5004164
Zhang Z, Tingaut P, Rentsch D, Zimmermann T, Sebe G (2015) Controlled silylation of nanofibrillated cellulose in water: reinforcement of a model polydimethylsiloxane network. Chemsuschem 8:2681–2690. doi:10.1002/cssc.201500525
ZRA (2016) Accessed June 2017. http://www.empa.ch/web/empa/center-for-x-ray-analytics
Acknowledgments
The authors would like to thank Esther Strub (Applied Wood Materials, Swiss Federal Laboratories for Materials Science and Technology (Empa), Duebendorf, Switzerland.) and Anja Huch (Applied Wood Materials, Swiss Federal Laboratories for Materials Science and Technology (Empa), Duebendorf, Switzerland.) for the SEM microscopy, Daniel Heer (Applied Wood Materials, Swiss Federal Laboratories for Materials Science and Technology (Empa), Duebendorf, Switzerland.) and Hans Michel (Mechanical Systems Engineering, Swiss Federal Laboratories for Materials Science and Technology (Empa), Duebendorf, Switzerland.) for the compression tests, as well as the company Stendal (Berlin, Germany) for providing the ECF fibers. Part of this work has been performed by the use of the Empa Platform for Image Analysis (http://empa.ch/web/s499/software-/-imaging-platform) at Empa’s Center for X-ray Analytics.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Josset, S., Hansen, L., Orsolini, P. et al. Microfibrillated cellulose foams obtained by a straightforward freeze–thawing–drying procedure. Cellulose 24, 3825–3842 (2017). https://doi.org/10.1007/s10570-017-1377-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-017-1377-8