
Abstract
Cellulose microfibrils are recalcitrant toward dissolution, thus it is difficult to extract and characterize them without modifying their native state. To study the molecular level behavior of microfibrils over 100 sugar residues, we construct a coarse-grained model of solvated cellulose Iβ microfibril using one bead per sugar residue. We derive the coarse-grained force field from atomistic simulation of a 36 chain, 40-residue microfibril by requiring consistency between the chain configuration, intermolecular packing and hydrogen bonding of the two levels of modeling. Coarse-grained force sites are placed at the geometric center of each glucose ring. Intermolecular van der Waals and hydrogen bonding interactions are added sequentially until the microfibril crystal structure in the atomistic simulation is achieved. This requires hydrogen bond potentials for pairs that hydrogen bond in cellulose Iβ, as well as those that can hydrogen bond in other structures, but not in cellulose Iβ. Microfibrils longer than 100 nm form kinks along their longitudinal direction, with an average periodicity of 70 nm. The behavior of kinked regions is similar with a bending angle of approximately 20°. These kinked regions might be linked to observations of periodic disorder from small angle neutron scattering and acid hydrolysis.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Cellulose is an unbranched polysaccharide of contiguous β-(1,4)-linked d-glucopyranose residues, and is the primary structural component in plant cell walls. As the most abundant biological material on earth, cellulose has gained increasing attention due to its potential for conversion to useful products including biofuels and direct energy (Ragauskas et al. 2006). Cellulose chains in native plants tend to organize into microfibrils, which sometimes bundle into macrofibrils and larger organized structures. Microfibrils in higher plants have diameters between 2 and 8 nm (Newman 1999), while larger microfibrils occur in cellulosic algae (Koyama et al. 1997). Although no direct proof is available, and smaller numbers of chains have been suggested (Fernandes et al. 2011; McCann et al. 1990; Thomas et al. 2013), it is widely accepted that the cellulose microfibril in plants contains 36 chains. Cellulose has been studied for more than 16 decades, yet we lack agreement on some microfibril properties. Direct imaging suggests that native cellulose microfibrils have some flexibility: they contain bends and turns along the extending direction (Kennedy et al. 2007). Their cause is still unclear: both the fibril–fibril interaction and the interaction between cellulose and other components can contribute to this behavior. Another area of contention is the presence of disorder in the microfibril. The existence of disordered regions is clear from X-ray scattering, which does not reveal its location. Two possibilities have been proposed. The disordered regions may be located on the surface of the microfibril, as supported by the correlation between surface hydroxyl group percentage and cellulose crystallinity (O’Sullivan 1997), or they may be periodic, as demonstrated combining small-angle neutron scattering and acid hydrolysis on ramie cellulose (Nishiyama et al. 2003), and suggested by the 50–400 nm nanowhiskers that result from acid hydrolysis on native cellulose (Araki et al. 1998; Bondeson et al. 2006; Nelson and Tripp 1953; Nickerson and Habrle 1947). In order to progress further on these issues, molecular level information is advantageous.
The insolubility of cellulose makes it difficult to extract from cell walls for experimental characterization of structure. Removing other cell wall components requires chemical treatments (such as strong alkali and heating), which may alter the properties of the extracted cellulose (Albersheim et al. 2010; Moran et al. 2008). Molecular dynamics (MD) simulation offers an alternative to chemical extraction, but is subject to a force field that is simultaneously accurate and computationally efficient. Simulation of cellulose microfibrils is often accomplished using atomistic force fields, such as CHARMM35 (Guvench et al. 2008, 2009; Matthews et al. 2006, 2011, 2012), GLYCAM06 (Kirschner et al. 2008; Matthews et al. 2011, 2012), GROMOS96 45A4 (Bergenstrahle et al. 2007, 2009; Bergenstråhle et al. 2008; Matthews et al. 2012; Lins and Hunenberger 2005), OPLS (Damm et al. 1997; Kony et al. 2002; Paavilainen et al. 2011), and PCFF (Hanus and Mazeau 2006; Mazeau and Heux 2003; Mazeau 2005; Mazeau and Rivet 2008). Cellulose in primary cell walls has been reported in two molecular weight fractions: 500–1,000 and 5,000–8,000 glucopyranose residues (Albersheim et al. 2010; Blaschek et al. 1982). The length of cellulose in secondary cell walls is even higher (Albersheim et al. 2010; Blaschek et al. 1982). In contrast, computational requirements limit atomistic simulations to a single, 36-chains microfibril, in most cases with 40 residues. Thus, methods with lower computational cost are required to simulate microfibrils of native lengths.
In this paper, we report a coarse-grained (CG) force field that is used to simulate 36 chain microfibrils of up to 400 residues. A CG model is a mesoscopic model in which force sites contain multiple atoms. Each force site is treated as one particle, and the interactions are only defined between the CG force sites (Girard and Muller-Plathe 2004). This significantly reduces the number of force and the computational cost. CG simulation has been widely applied to simulate biomolecular systems, including lipids (Ayton et al. 2007; Izvekov and Voth 2005; Marrink et al. 2004, 2007; Shelley et al. 2001; Stevens 2004), proteins (Ayton et al. 2007; Bahar and Rader 2005; Bond et al. 2007; Doruker et al. 2002; Monticelli et al. 2008; Tozzini 2005; Shih et al. 2006), carbohydrates (Ayton et al. 2007; Lopez et al. 2009; Liu et al. 2007; Molinero and Goddard 2004), and polymers (Depa et al. 2011). Coarse-grained models of cellulose have been developed and used to study processes (such as the enzymatic pathways of degradation process) or systems (such as microfibrils with disordered regions) involving long microfibrils (Bu et al. 2009; Glass et al. 2012; Hynninen et al. 2011; Srinivas et al. 2011; Queyroy et al. 2004; Wohlert and Berglund 2011).
Two levels of coarse-graining are reported: three beads per residue (Bu et al. 2009; Hynninen et al. 2011; Wohlert and Berglund 2011) and one bead per residue (Glass et al. 2012; Srinivas et al. 2011; Queyroy et al. 2004). In the three-bead models, force sites were assigned based on the MARTINI coarse-grained force field (Lopez et al. 2009) (each site contains two carbon atoms with oxygen and hydrogen atoms adjacent to them) or the M3B model (Molinero and Goddard 2004) (force sites are placed on C1, C4, and C6 atoms). The coarse-grained models used atomistic simulation of a solvated Iβ microfibril as the target system. Several of these models (Bu et al. 2009; Wohlert and Berglund 2011) were applied to study the interaction between the carbohydrate-binding domain and cellulose surfaces.
The first single-bead cellulose CG model was developed based on an atomistic simulation of octaose, the 8-ring oligomer of cellulose (Queyroy et al. 2004). The authors placed CG force sites on the glycosidic oxygen atoms, and the model was used to prepare a relaxed system of bulk amorphous cellulose. More recently, a solvent-free single-bead CG model was used to study the transition of cellulose fibrils from crystalline to amorphous structures (Srinivas et al. 2011), and the residue-scale REACH (Realistic Extension Algorithm via Covariance Hessian) coarse-grained force field was used to calculate mechanical properties such as Young’s modulus and persistence length (Glass et al. 2012).
Compared to single-bead models, three-bead models have more molecular detail. For example, each bead in a three-bead model contains one hydroxyl group, which dictates the directionality of hydrogen bonds (Bu et al. 2009; Wohlert and Berglund 2011). Single-bead models lose this directionality, but are required to study larger systems. In this report, we present a solvated single-bead model for cellulose. The bonded potentials are obtained using the iterative Boltzmann Inversion method (Reith et al. 2003), and the van der Waals potential is assigned from the pair distribution function of amorphous cellulose chains. We solve the directionality problem by including bonded potentials between pairs of CG beads that either do or can form a hydrogen bond. We use the model to simulate fibrils with 100–400 residues. For fibrils longer than 100 nm, we observe bends along the longitudinal direction. In the longest fibril we tested (400 residues), multiple bends are present. This behavior has not been previously reported, and may provide insights about the periodicity and the behavior of the disordered regions in cellulose microfibrils in future studies.
Methodology
Simulation details
We used an atomistic target simulation provided by collaborators (Zhao et al. 2013) in which they used the CHARMM (Brooks et al. 1983) simulation package with a 36 chain 40-residue microfibril (Fig. 1) in a TIP3P (Jorgensen et al. 1983) water box. A solvation shell (minimum of 10-Å) prevents the microfibril from interacting with its periodic images. The carbohydrates are represented with the C35 force field (Guvench et al. 2008, 2009), and long-range electrostatics are handled using the particle-mesh Ewald algorithm (Darden et al. 1993) with a 1-Å charge grid size. Non-bonded interactions are truncated at 10 Å. The SHAKE algorithm (Ryckaert et al. 1977) maintains constant lengths for bonds involving hydrogen atoms. After performing minimization, the system undergoes stepwise heating of 10 ns at 100, 200 and 300 K. A 20-ps equilibration is performed at 300 K. The 9-ns production run was performed in the NVE ensemble using a 1 fs time step. Atomic coordinates form the last 3-ns production were output every 0.5 ps for later analysis.

Cellulose microfibril at the atomistic level. In this paper, the dimension of microfibrils is denoted by the number of chains on each side surface and the number of glucose units along the longitudinal direction. In case of atomistic simulation, the microfibril has a diamond shape cross-section consisting of 36 chains with 6 chains on each surface, and each chain contains 40 glucose units. This is referred as the 6 × 6 × 40 microfibril
We use the LAMMPS (Plimpton 1995) package to perform the CG simulations. The initial system contains a microfibril solvated in a 10 Å water shell. In order to more accurately describe interactions between the microfibril and water in the first solvation shell, we describe water molecules using a coarse-grained explicit force field instead of using an implicit solvent model. A maximum of 3,000 iterations of conjugate gradient minimization is performed to relieve large strains. All CG simulations are performed in the NVT ensemble using a 1 fs time step. Here we require a 1 fs as our time step because some of the potentials are sharp. For example, the bond potential of the coarse-grained beads (Table 1) and that of the atomistic bond potentials reported in C35 force field have the same order of magnitude. In this case, using a small time step will prevent forces from becoming too high during the simulation. The temperature is maintained at 300 K using a Nose–Hoover thermostat. We allow the system to equilibrate for at least 5 ns before collecting the production trajectories. The cut-off distances for all non-bonded interactions are set to 15 Å.
Coarse-grained force field development
We derive the CG force field based on the atomistic simulation described above. The objectives in deriving this force field are to realize the largest computational gain while maintaining microfibril structure, and to keep the force field general enough that branches may be easily added for simulation of xyloglucan or other polysaccharides. We choose one force site per residue and compensate for the loss of directional hydrogen bonds by introducing interactions for pairs that can or do form hydrogen bonds. These interactions resemble bonded interactions that occur between specific pairs, but are allowed to break and reform during the simulation. We refer to these potentials as hydrogen bonds, although it is important to note that only one of them corresponds to an actual hydrogen bond in the native cellulose Iβ crystal structure. The others are required to reproduce the glucose residue packing pattern. Atomistic simulations suggest that some do form hydrogen bonds at high temperature (Matthews et al. 2011). It is not problematic to enforce a specific separation between pairs that do not hydrogen bond, as the atomistic simulations clearly show a preferred separation distance without such bonding. To achieve generality for later simulation of branched polysaccharides, we develop our force field starting with CG bond stretching, angle, and torsional potentials, and non-bonded pair interactions. The hydrogen bonds are added consecutively, and their impact on the microfibril structure is noted at each addition.
The force field parameters are obtained by Boltzmann inversion of the corresponding intramolecular and intermolecular probability distributions. In order to obtain the distributions, we first determine the location of the force sites. Two potential placements of the CG bead center are the glycosidic oxygen (O4 model) or the center of the d-glucose rings (ring center model) (Fig. 2) (Queyroy et al. 2004). To determine the best choice, we consider the probability distributions of intramolecular bonding (bond lengths, bending angles, and torsional angles) and intermolecular pair interactions (intermolecular pair distribution function) (Fig. 3a–c) to look for obvious difficulties in using them as CG potential targets. The two models have similar bond length and bending angle distributions, but the torsional angle distribution is much softer in the ring center model. Choosing a softer torsion potential minimizes the risk of artificially reducing conformational freedom of the glucan chains. More importantly, the pair distribution function of the ring center model (Fig. 3e) is less structured than the O4 model (Fig. 3d), so that we can decompose the pair distribution function into individual hydrogen bonds and more accurately describe the preferred separations with intermolecular potentials. Therefore, we place the force site at the d-glucose ring center, which benefits computational cost and accuracy.

Coarse-grained mapping of cellulose chains with beads centered on a O4 (the glycosidic oxygen) or b ring center

Intramolecular probability distributions of O4 (dashed line) and ring center (solid line) models: a bond length, b bending angle, c torsional angle. Intermolecular pair distribution functions of d O4 model and e ring center model
The CG force field incorporates bonded and non-bonded potentials. Bonded potentials include bond stretching, bending, and torsional transitions. The target intramolecular distributions (Fig. 3a–c) are Gaussian, and thus we represent the CG bonded potentials by harmonic springs:
where x is the bond length, bending angle, or torsional angle. We obtain the parameters xo and ko from atomistic intramolecular distributions (Fig. 3a–c) using iterative Boltzmann inversion.
The intermolecular potential must describe van der Waals interactions and hydrogen bonding. Because the coarse-grained beads are neutral, electrostatic interactions are not considered. We represent the van der Waals interactions with a 6-12 Lennard-Jones potential:
where r represents the separation distance. To obtain the preferred separation distance σ and the potential well depth ε, we extract the data near the first peak of a published amorphous cellulose pair distribution function (Srinivas et al. 2011), apply Boltzmann Inversion, and fit inverted data points with Eq. (2). Based on the fitting, we assign σ as 5.4 Å and ε as 0.10 kcal/mol.
As with the atomistic simulation, the coarse-grained microfibril is solvated with water, which we also represent in CG form. He et al. (2010) reported a series of CG water models with different levels of coarse-graining, functional forms of the potential energy, and cut-off distances for the non-bonded interactions. We selected a CG water bead that represents two water molecules with a 12-6 Lennard-Jones potential (σ = 3.779 Å, and ε = 1.118 kcal/mol).
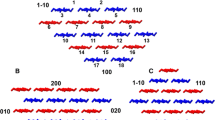
Our approach is to add specific intermolecular potentials sequentially, thus we performed an initial simulation of the cellulose microfibril with only the bonded and van der Waals potentials. As expected, the structure disorders from the original crystalline form, and water penetrates the microfibril and disturbs the packing (Fig. 4). This is reflected in the intermolecular pair distribution function (Fig. 5), which is far from the atomistic level target. We thus must add specific intermolecular potentials to represent preferential interchain interactions, including hydrogen bonds. To accomplish this, we first associate the peaks in the target pair distribution function with specific features in the cellulose microfibril structure. As shown in Fig. 6, the first four peaks correspond to adjacent bead preferred separation distances in the (11-0), (110), (010), and (100) planes respectively. We anticipate that the remaining peaks are secondary repeats of these distances, as they occur at roughly twice the distances of the first four. The atomistic and CG representation of the microfibril cross section are shown in Fig. 6e, in which the chains are extended into the paper. Miller indices were used to describe the planes and directions in the structure: the side surfaces of the diamond shape cross section are labeled as (11-0) and (110) planes; the layers of glucan units are aligned parallel to the unit cell axis b in (100) plane; the vertical stacking direction is the unit cell axis a, which is in the (010) plane. The equilibrium values of the directional intermolecular distance distributions (Fig. 6a–d) correspond to the pair distribution peak locations (Fig. 6f). The consistency of the distances suggests the origins of the structurally significant peaks. Because features in the pair distribution function peaks at separation distances above 9 Å most likely originate from the closer interactions, adding the four new potentials may be sufficient to reproduce long range features.

Snapshot of the position of cellulose CG beads after 15 ns. Only van der Waals potential is considered in the nonbonded potential term

Comparison of pair distribution functions obtained from CG simulation with van der Waals interaction only (dashed line) and the target from atomistic simulation trajectories (solid line)

The target pair distribution function decomposed into specific structural features. a Interlayer separation distance distribution in (110) planes; b interlayer separation distance distribution in (11-0) planes; c intralayer separation distance distribution in (100) planes; d interlayer separation distance distribution in (010) planes; e CG mapping of the cross section view of the microfibril, in which the structurally significant separation distances are shown; (in the atomistic cross-sectional view, a and b unit cell axes are highlighted in solid lines, and Miller indices of the crystal planes are highlighted in dashed lines), f target pair distribution function, in which the structurally significant peaks are labeled as their corresponding planes
Boltzmann Inversion can be used on the intermolecular distance distribution curves (Fig. 6a–d) to obtain a harmonic representation of these bonds, but such bonds are unbreakable and unrealistic. To represent the intermolecular constrains more realistically, we use Morse potentials, which weaken as distance between beads increases.
Here Do determines the depth of the energy well, ro is the distance of minimum energy, and the stiffness parameter α determines the curvature of the potential around ro. As the intermolecular bonds in (100) planes is a hydrogen bond, and the others are similar in strength, we assign Do in the Morse potential as 5.0 kcal/mol, which is representative of the O–H⋯O hydrogen bond energy strength (Steiner 2002). We determine α by requiring that the Morse potential retain the same curvature as the harmonic potential around ro. The difference between harmonic and Morse potentials should not be significant when simulating a 40-residue microfibril, but this choice may be crucial when simulating longer fibrils, in which intermolecular bonds may break and the formation of disordered regions may occur.
Result and discussion
Force field parameters and cellulose crystal structure
We add pseudo-bonds representing interchain interactions sequentially based on their physical meaning and significance. A glucose residue in the microfibril cross-section forms two intra-layer and six inter-layer bonds, where a layer [(100) plane, see Fig. 6e] contains parallel, hydrogen bonded glucose rings. It has been reported that the intralayer hydrogen bonds are the dominant intermolecular interaction in native cellulose microfibrils (Bu et al. 2009), and thus we add this interchain potential first. At higher temperatures (500 K), intralayer hydrogen bonds are weakened, and this enables formation of interlayer bonds (Matthews et al. 2011). We next add interlayer bonds between adjacent glucose residues in the (110) and (11-0) planes. As shown in Fig. 7, each addition brings the pair distribution function closer to the atomistic target, but the third peak at 7.7 Å is not captured. This is also an interlayer bond between glucose residues in the (010) plane. To model this, we add a potential between adjacent beads in this plane in order to fully reproduce the accurate crystal structure of cellulose microfibril.

Demonstration of potentials required to reproduce the pair distribution function peaks. The intermolecular interactions of simulation above are van der Waals interaction and a intralayer bonds in (100), b intralayer bonds in (100) and interlayer bonds in (110), c intralayer bonds in (100) and interlayer bonds in both (110) and (11-0), d intralayer bonds in (100) and interlayer bonds in (110), (11-0) and (010). Potentials are not yet tuned
Following addition of interchain bonds (morse potentials), the intrachain bonds (harmonic potentials) required refinement. We present the full force field in Table 1. As mentioned in the methodology section, the reason that we choose the ring center model is that it introduces softer torsion that minimizes the reduction in conformational freedom. This enables us to more accurately decompose the pair distribution function to individual type of intermolecular interactions. Based on the refined potential parameters (Table 1), the torsional potential is more than 30 times softer than the bending angle potential, which is consistent with our strategy of bead location selection. Additionally, the strength of interchain bonds can be evaluated by α, from which we note that the potential in the intralayer direction [i.e. (100)] is at least two times sharper than the interchain interactions. This agrees with the structure of cellulose microfibril, as the intralayer interaction is contributed mostly by hydrogen bonds, and the interlayer interactions are contributed mostly by packing and Iβ crystal structure. This force field maintains the target interchain and intrachain distributions (Fig. 8) when used in a 20 ns simulation of a 6 × 6×40 microfibril.

Comparison of CG simulation with atomistic targets: a bond length, b bending angle, c torsional angle distribution, and d pair distribution functions. Dashed curves distributions following simulation with the CG force field for 5 ns. Solid curves atomistic target distributions
Simulation with the CG force field does not disrupt the cellulose Iβ crystal structure: after 20 ns of CG simulation time, the structure is highly ordered with unit cell parameters within 2 % of X-ray diffraction results (Nishiyama et al. 2002). A snapshot of the final structure of the 6 × 6 × 40 simulation is shown in Fig. 9, and lattice parameters are presented in Table 2. Note that parameters a and b (Fig. 6e) are enforced directly by the Morse potentials. The 40-residue fibril twists with a helical angle of 1.6°/nm, similar to the atomistic simulation (1.4°/nm).

Cross sectional and longitudinal view of the coarse-grained cellulose microfibril (MD simulation snapshot)
Applying the CG force field to longer fibrils
Using the same force field, we simulated microfibrils between 100 and 400 glucose residues. The initial structures of the microfibrils are built based on experimentally determined cellulose Iβ lattice parameters. Each microfibril was placed in a 10-Å solvation shell, and the same minimization strategy was employed. We used 5 ns production runs. As shown in Fig. 10, microfibrils longer than 100 nm form kinks comprised of two bends. The kinked regions are 15–20 residues (7–10 nm) long, and the bending angles are 20°. Multiple kinks form in the 400-residue microfibril, with spacings of 50 and 100 nm. This is a unique observation, made possible by simulating microfibrils longer than 100 residues. We believe that kinks are a reproducible feature in fibrils longer than 100 nm, as this feature is observed in all the long fibril simulations we performed. It is interesting in light of reports of periodic disorder, both directly using SANS on ramie fibers (Nishiyama et al. 2003), and indirectly through acid hydrolysis (Araki et al. 1998; Bondeson et al. 2006; Nelson and Tripp 1953; Nickerson and Habrle 1947). The SANS study estimates that disordered regions are 2–3 nm, and the periodicity based on both methods is 50–150 nm. Thus, it is reasonable to conclude that the kinks are linked to the periodic disorder. Multiple simulations on long fibrils are required to generate adequate statistics on periodicity. Interestingly, the kinks appear to relieve twist. Compared to the 40-residue fibril simulation, the extent of twist in the longer fibrils is significantly weaker. The 40-residue fibril twists uniformly along the longitudinal direction, whereas the longer fibrils do not twist with the exception of one end of the 300 residue fibril. This suggests that kinks may occur to relive the twist which leads to the formation of periodic disorder.

CG simulation snapshot of microfibril 100 and 400 glucose units long
If kinks occur spontaneously, they must be energetically favorable. In Fig. 11, we compare the potential energy of 200-residue kinked and straight microfibrils. The straight configuration is obtained by fixing the positions of beads in the center interior chain, thus forcing the microfibril to remain straight and eliminating twist. As expected, the potential energy of the straight microfibril is higher than that of the kinked microfibril throughout the simulation. This strengthens the previous conclusion that kinks are a reproducible feature of long microfibrils. The difference per bead is 0.4 kcal/mol, about twice the energy of a trans-gauche rotation. As shown in Fig. 12, the energy difference is not distributed evenly among residues, but is concentrated at the kink location. In addition to the variation with residue position, we investigated the potentials that stabilize kinks. Energies for the eight different potentials are plotted in Fig. 12. It is observed that kinks cause energy penalty in the bonded potential, (010) potential, and (100) potential in the residues in the kinked region. (Fig. 12a, e, h) This suggests that the formation of kinks disturbs the crystal packing in that region, while the regions of the fibril away from kinks retain their unit cell structure. Because the formation of kinks disturbs crystal packing, intra-chain bond stretching/bending, and interchain interactions in the unit cell axis directions a and b (Fig. 6e) resists bending. In these potentials, the energetic cost is localized in the 10–20 residues directly involved in the kink. Interestingly, torsional and van der Waals potentials (Fig. 12c, d) provide the driving force for kink formation, as they decrease energy in kinked regions. Compared to the 10–20 residues affected in the unit cell potentials, torsional and van der Waals potentials are affected over a wider range: 40 (torsion) and 50 (van der Waals) residues. These potentials involve more force sites per bead (four for torsion and more than 20 for van der Waals) and thus it appears that these interactions initiate kinks, only later disrupting the unit cell structure.

Comparison of bended (lower black points) and non-bended (upper grey points) 200 residue microfibril potential energy

Various potentials against residue number for bended 200-residue microfibril. The type of potential is indicated in the y-axis, and residue number is shown in x-axis. a–c The intramolecular potential variation along the fibril. d–h The intermolecular potential variation. a, e, h contain a peak at kinked region, indicating those interactions are unfavor of the kink. Interestingly, these directions correspond to the principle axis of the cellulose microfibril crystal structure. This implies that the formation of the kink disturbs the local crystal structure of the microfibril. c, d form a well at kinked region, indicating that the long range interactions involving multiple beads are in favor of the kink formation
Conclusions
We have reported a solvated single-bead coarse-grained force field for the cellulose Iβ microfibril based on atomistic simulation. The force field is constructed such that chain configuration, intermolecular packing and hydrogen bonding of the CG system are consistent with that of the atomistic system, and then used to simulate long (100–400 residues in length) fibrils. The most important feature of these long fibril simulations is the appearance of kinks in the longitudinal direction. The kinks are spaced by 50–150 nm, and appear to relieve twist. Microfibril simulations with more than 200 residues do not twist with the exception of one end in the 300-residue case. The periodicity and kink size are on the same order of magnitude as reported for fibrils with periodic disorder (Nishiyama et al. 2003). Based on the above observations, we conclude that twist may be an end effect, which can be relieved by increasing the fibril length. Torsion and van der Waals interactions are favored in kink formation, and are responsible for initiating kinks. A primary cell wall model suggested recently that load-bearing structure in primary walls is dominated by biomechanical “hot spots” (<2 % of the total xyloglucan) in which xyloglucan and cellulose microfibrils are in close proximity (Park and Cosgrove 2012). It is not possible to have short stretches of microfibrils in close contact without bending. We suggest that kinks provide preferred locations to form “hot spots” while retaining the interfibril spacing. 13C NMR spectra show that both cellulose Iα (triclinic) and Iβ (monoclinic) exist in higher plants (Atalla and Vanderhart 1984; Newman et al. 1994). It has been shown that bending can cause the interconversion between the two allomorphs, and that the Iα:Iβ ratio is sensitive to the bending angle (Jarvis 2000). Our force field is designed to simulate cellulose Iβ, and the extent of bending is not sufficient to allow the microfibril to fully convert to Iα (which requires bending of 39°) (Jarvis 2000). It is likely that bends like those observed here initiate this interconversion. The coarse-grained force field is developed based on Iβ allomorph. Thus, its ability to study cellulose Iα structure and interconversion is limited. However, the natural occurrence of kinks implies that microfibrils can interconvert spontaneously.
References
Albersheim P, Darvill A, Roberts K, Sederoff R, Staehelin A (2010) Plant cell walls. Garland Science, New York
Araki J, Wada M, Kuga S, Okano T (1998) Flow properties of microcrystalline cellulose suspension prepared by acid treatment of native cellulose. Colloids Surf A 142:75–82
Atalla RH, Vanderhart DL (1984) Native cellulose: a composite of two distinct crystalline forms. Science 223:283–285
Ayton GS, Noid WG, Voth GA (2007) Systematic coarse graining of biomolecular and soft-matter systems. MRS Bull 32:929–934
Bahar I, Rader A (2005) Coarse-grained normal mode analysis in structural biology. Curr Opin Struct Biol 15:586–592
Bergenstrahle M, Berglund LA, Mazeau K (2007) Thermal response in crystalline Iβ cellulose: a molecular dynamics study. J Phys Chem B 111:9138–9145
Bergenstrahle M, Thormann E, Nordgren N, Berglund LA (2009) Force pulling of single cellulose chains at the crystalline cellulose–liquid interface: a molecular dynamics study. Langmuir 25:4635–4642
Bergenstråhle M, Wohlert J, Larsson PT, Mazeau K, Berglund LA (2008) Dynamics of cellulose–water interfaces: NMR spin-lattice relaxation times calculated from atomistic computer simulations. J Phys Chem B 112:2590–2595
Blaschek W, Koehler H, Semler U, Franz G (1982) Molecular weight distribution of cellulose in primary cell walls. Planta 154:550–555
Bond PJ, Holyoake J, Ivetac A, Khalid S, Sansom MSP (2007) Coarse-grained molecular dynamics simulations of membrane proteins and peptides. J Struct Biol 157:593–605
Bondeson D, Mathew A, Oksman K (2006) Optimization of the isolation of nanocrystals from microcrystalline cellulose by acid hydrolysis. Cellulose 13:171–180
Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M (1983) CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem 4:187–217
Bu L, Beckham GT, Crowley MF, Chang CH, Matthews JF, Bomble YJ, Adney WS, Himmel ME, Nimlos MR (2009) The energy landscape for the interaction of the family 1 carbohydrate-binding module and the cellulose surface is altered by hydrolyzed glycosidic bonds. J Phys Chem B 113:10994–11002
Damm W, Frontera A, Tirado-Rives J, Jorgensen W (1997) OPLS all-atom force field for carbohydrates. J Comput Chem 18:1955–1970
Darden T, York D, Pedersen L (1993) Particle mesh Ewald: an Nlog(N) method for Ewald sums in large systems. J Chem Phys 98:10088–10092
Depa P, Chen C, Maranas JK (2011) Why are coarse-grained force fields too fast? A look at dynamics of four coarse-grained polymers. J Chem Phys 134:014903
Doruker P, Jernigan RL, Bahar I (2002) Dynamics of large proteins through hierarchical levels of coarse-grained structure. J Comput Chem 23:119–127
Fernandes AN, Thomas LH, Altaner CM, Callow P, Forsyth VT, Apperley CD, Kennedy CJ, Jarvis MC (2011) Nanostructure of cellulose microfibrils in spruce wood. Proc Natl Acad Sci 108:1195–1203
Girard S, Muller-Plathe F (2004) Coarse-graining in polymer simulations. In: Karttunen M, Vattulainen I, Lukkarinen A (eds) Novel methods in soft matter simulations, vol 640., Lect Notes PhysSpringer, Berlin, pp 327–356
Glass DC, Moritsugu K, Cheng X, Smith JC (2012) REACH coarse-grained simulation of a cellulose fiber. Biomacromolecules 13:2634–2644
Guvench O, Greene SN, Kamath G, Brady JW, Vendable RM, Pastor RW, Mackerell AD (2008) Addictive empirical force field for hexopyranose monosaccharides. J Comput Chem 29:2543–2564
Guvench O, Hatcher E, Venable RM, Pastor RW, Mackerell AD (2009) CHARMM additive all-atom force field for glycosidic linkages between hexopyranoses. J Chem Theory Comput 5:2353–2370
Hanus J, Mazeau K (2006) The xyloglucan–cellulose assembly at the atomistic scale. Biopolymers 82:59–73
He X, Shinoda W, DeVane R, Klein ML (2010) Exploring the utility of coarse-grained water models for computational studies of interfacial systems. Mol Phys 108:2007–2020
Hynninen AP, Matthews JF, Backham GT, Crowley MF, Nimlos MR (2011) Coarse-grain model for glucose, cellobiose, and cellotetraose in water. J Chem Theory Comput 7:2137–2150
Izvekov S, Voth G (2005) A multiscale coarse-graining method for biomolecular systems. J Phys Chem B 109:2469–2473
Jarvis MC (2000) Interconversion of the Iα and Iβ crystalline forms of cellulose by bending. Carbohydr Res 325:150–154
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935
Kennedy CJ, Cameron GJ, Sturcova A, Apperley DC, Altaner C, Wess TJ, Jarvis MC (2007) Microfibril diameter in celery collenchyma cellulose: X-ray scattering and NMR evidence. Cellulose 14:235–246
Kirschner KN, Yongye AB, Tschampel SM, Gonzalez-Outeirino J, Daniels CR, Foley BL, Woods RJ (2008) GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J Chem Theory Comput 29:622–655
Kony D, Damm W, Stoll S, van Gunsteren W (2002) An improved OPLS–AA force field for carbohydrates. J Comput Chem 23:1416–1429
Koyama M, Sugiyama J, Itoh T (1997) Systematic survey on crystalline features of algal celluloses. Cellulose 4:147–160
Lins RD, Hunenberger PH (2005) A new GROMOS force field for hexopyranose-based carbohydrates. J Chem Theory Comput 26:1400–1412
Liu P, Izvekov S, Voth GA (2007) Multiscale coarse-graining of monosacharrides. J Phys Chem B 111:11566–11575
Lopez CA, Rzepiela AJ, de Vries AH, Dijkhuizen L, Hunenberger PH, Marrink SJ (2009) Martini coarse-grained force field: extension to carbohydrates. J Chem Theory Comput 5:3195–3210
Marrink SJ, de Vries AH, Mark AE (2004) Coarse grained model for semiquantitative lipid simulations. J Phys Chem B 108:750–760
Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, de Vries AH (2007) The MARTINI force field: coarse grained model for biomolecular simulations. J Phys Chem B 111:7812–7824
Matthews JF, Skopec CE, Mason PE, Zuccato P, Torget RW, Sugiyama J, Himmel ME, Crowley MF (2006) Computer simulation studies of microcrystalline cellulose Iβ. Carbohydr Res 341:138–152
Matthews JF, Bergenstrahle M, Beckham GT, Himmel ME, Nimlos MR, Brady JW, Crowley MF (2011) High-temperature behavior of cellulose I. J Phys Chem B 115:2155–2166
Matthews JF, Beckham GT, Bergenstrahle-Wohlert M, Brady JW, Himmel ME, Crowley MF (2012) Comparison of cellulose Iβ simulations with three carbohydrate force fields. J Chem Theory Comput 8:735–748
Mazeau K (2005) Structural micro-heterogeneities of crystalline Iβ-cellulose. Cellulose 12:339–349
Mazeau K, Heux L (2003) Molecular dynamics simulations of bulk native crystalline and amorphous structures of cellulose. J Phys Chem B 107:2394–2403
Mazeau K, Rivet A (2008) Wetting the (110) and (100) surfaces of Iβ cellulose studied by molecular dynamics. Biomacromolecules 9:1352–1354
McCann MC, Wells B, Roberts K (1990) Direct visualization of cross-links in the primary plant cell wall. J Cell Sci 96:323–334
Molinero V, Goddard WA (2004) M3B: a coarse grain force field for molecular simulations of malto-oligosaccharides and their water mixture. J Phys Chem B 108:1414–1427
Monticelli L, Kandasamy SK, Periole X, Larson RG, Tieleman DP, Marrink S-J (2008) The MARTINI coarse-grained force field: extension to proteins. J Chem Theory Comput 4:819–834
Moran JI, Alvarez VA, Cyras VP, Vazquez A (2008) Extraction of cellulose and preparation of nanocellulose from sisal fibers. Cellulose 15:149–159
Nelson ML, Tripp VW (1953) Determination of the leveling-off degree of polymerization of cotton and rayon. J Polym Sci 10:577–586
Newman RH (1999) Estimation of the lateral dimensions of cellulose crystallites using 13C NMR signal strengths. Solid State Nucl Magn Reson 15:21–29
Newman RH, Ha M-A, Melton LD (1994) Solid-state 13C NMR investigation of molecular ordering in the cellulose of apple cell walls. J Agric Food Chem 42:1402–1406
Nickerson RF, Habrle JA (1947) Cellulose intercrystalline structure: study by hydrolytic methods. Ind Eng Chem 39:1507–1512
Nishiyama Y, Langan P, Chanzy H (2002) Crystal structure and hydrogen-bonding system in cellulose Iβ from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc 124:9074–9082
Nishiyama Y, Kim U-J, Kim D-Y, Katsumata KS, May RP, Langan P (2003) Periodic disorder along ramie cellulose microfibrils. Biomacromolecules 4:1013–1017
O’Sullivan AC (1997) Cellulose: the structure slowly unravels. Cellulose 4:173–207
Paavilainen S, Rog T, Vattulainen I (2011) Analysis of twisting of cellulose nanofibrils in atomistic molecular dynamics simulations. J Phys Chem B 115:3747–3755
Park YB, Cosgrove DJ (2012) Change in cell wall biomechanical properties in the xyloglucan-difficient xxt1/xxt2 mutant of Arabidopsis. Plant Physiol 158:1933–1943
Plimpton S (1995) Fast parallel algorithms for short-range molecular dynamics. J Comput Phys 117:1–19
Queyroy S, Neyerts S, Brown D, Muller-Plathe F (2004) Preparing relaxed systems of amorphous polymers by multiscale simulation: application to cellulose. Macromolecules 37:7338–7350
Ragauskas AJ, Williams CK, Davison BH, Britovsek G, Cairney J, Eckert CA, Frederick WJ, Hallett JP, Leak DJ, Liotta CL, Mielenz JR, Murphy R, Templer R, Tschaplinski T (2006) The path forward for biofuels and biomaterials. Science 311:484–489
Reith D, Putz M, Muller-Plathe F (2003) Deriving effective mesoscale potentials from atomistic simulations. J Comput Chem 24:1624–1636
Ryckaert J-P, Ciccotti G, Berendsen HJC (1977) Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23:327–341
Shelley JC, Shelley MY, Reeder RC, Bandyopadhyay S, Moore PB, Klein ML (2001) Simulations of phospholipids using a coarse grain model. J Phys Chem B 105:9785–9792
Shih AY, Arkhipov A, Freddolino PL, Schulten K (2006) Coarse grained protein-lipid model with application to lipoprotein particles. J Phys Chem B 110:3674–3684
Srinivas G, Cheng X, Smith JC (2011) A solvent-free coarse-grain model for crystalline and amorphous cellulose microfibrils. J Chem Theory Comput 7:2539–2548
Steiner T (2002) The hydrogen bond in the solid state. Angew Chem Int Ed 41:48–76
Stevens M (2004) Coarse-grained simulations of lipid bilayers. J Chem Phys 121:11942–11948
Thomas LH, Forsyth VT, Šturcová A, Kennedy CJ, May RP, Altaner CM, Apperley DC, Timothy JW, Jarvis MC (2013) Structure of cellulose microfibrils in primary cell walls from collenchyma. Plant Physiol 161:465–476
Tozzini V (2005) Coarse-grained models for proteins. Curr Opin Struct Biol 15:144–150
Wohlert J, Berglund LA (2011) A coarse-grained model for molecular dynamics simulations of native cellulose. J Chem Theory Comput 7:753–760
Zhao Z, Shklyaev OE, Nili A, Mohamed MNA, Kubicki JD, Crespi VH, Zhong L (2013) Cellulose microfibril twist, mechanics, and implication for cellulose biosynthesis. J Phys Chem A 117:2580–2589
Acknowledgments
We thank Dr. Zhen Zhao and Dr. Linghao Zhong for providing the atomistic simulation coordinates of the solvated 6 × 6×40 microfibril. This work is supported as part of The Center for Lignocellulose Structure and Formation, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0001090. The simulations were performed on high performance computing systems supported and maintained by the Penn State Research Computing and Cyberinfrastructure (RCC).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Fan, B., Maranas, J.K. Coarse-grained simulation of cellulose Iβ with application to long fibrils. Cellulose 22, 31–44 (2015). https://doi.org/10.1007/s10570-014-0481-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-014-0481-2