
Abstract
Hemicelluloses such as xylans play an increasing role as renewable raw materials for technological applications. The complex and variable composition of hemicelluloses requires powerful analytical techniques in order to assess their composition. In the present study, the neutral fraction of hydrothermally isolated xylan from beech wood was characterized by capillary electrophoresis with laser-induced fluorescence detection (CE-LIF) upon derivatization with 8-aminopyrene-1,3,6-trisulfonic acid. Reproducible separation of the xylo-oligosaccharides was achieved using a polyvinyl alcohol coated capillary and a 25 mM sodium acetate buffer, pH 4.75, as background electrolyte at an applied voltage of −30 kV. Intermediate precision expressed as relative standard deviation was below 2.0 % for migration times and below 10 % for relative peak areas except for the oligomers present at very low concentrations only. At the same time, derivatization conditions proved to be robust as well. Samples obtained by fractionation of the xylan were subsequently characterized by CE-LIF. In addition, capillary electrophoresis with mass spectrometry detection indicated the presence of small amounts of xylo-oligosaccharides containing additional sugar moieties such as 4-O-methylglucuronic acid. Moreover, minor components containing acetyl groups could be detected. The presence of these impurities was confirmed by nuclear magnetic resonance analysis of the fractions. In conclusion, although none of the techniques applied here gave a complete picture of the composition of the investigated xylan or its fractions, the combination provided insight into the complexity of the sample.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Xylan-type polysaccharides are the main hemicellulose components of dicotyledons and monocotyledons with the exact composition depending on the taxonomic family (Ebringerová et al. 2005). In hardwood, the most common hemicelluloses are O-acetyl-4-O-methylglucuronoxylans, which possess a backbone of β-(1 → 4)-linked d-xylopyranose units containing 4-O-methyl-α-d-glucopyranosyl uronic acid attached to position 2 of d-xylopyranose. The glucuronic acid moieties can also be non-methylated. The ratio of xylose to glucuronic acid varies between 4:1 and 16:1. Some xylans containing 4-O-methyl-α-d-glucopyranosyl uronic acid and/or α-l-arabinofuranose attached to positions 3 and other sugar moieties such as arabinose, rhamnose, galactose, or glucose may be present as well (Ebringerová et al. 2005; Ebringerová and Heinze 2000). In higher plants, xylans are also acetylated. However, the acetyl groups are hydrolyzed under alkaline conditions during the typical extraction procedures while they can be partially preserved when the extraction is performed with hot water (Ebringerová and Heinze 2000). At present, xylans have been playing an increasing role as renewable raw materials for technological applications including the food, cosmetics, and pharmaceutical industry (Ebringerová and Heinze 2000; Ebringerová and Hromádková 1999; Ebringerová et al. 2005; Deutschmann and Dekker 2012).
The characterization of oligosaccharides from natural sources is a challenging task due to the complex composition. Spectroscopic techniques including nuclear magnetic resonance (NMR) spectroscopy (Teleman et al. 2002; Daus et al. 2011; Hoffman et al. 2005) or mass spectrometry (MS) such as matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) (Kabel et al. 2002b; Daus et al. 2011; Chong et al. 2011) have been applied to xylan analysis. With regard to separation techniques, high performance anion exchange chromatography combined with pulsed amperometric detection (HPAEC-PAD) has often been used for xylan characterization (Hilz et al. 2006; Kabel et al. 2002a, b). Other chromatographic techniques such as porous-graphitized carbon liquid chromatography with evaporative light scattering (PGC-ELSD) or mass spectrometric detection (Westphal et al. 2010) or reversed phase high performance liquid chromatography in combination with mass spectrometric detection (RP-HPLC-MS) (Vismeh et al. 2013) have also been employed. However, chromatographic techniques are often not able to separate all linear and branched oligosaccharides present in a mixture.
As a high resolution analytical technique, capillary electrophoresis (CE) is well suited for carbohydrate analysis (El Rassi 1999). In CE, analyte separation is based on differences in the electrophoretic mobility of charged solutes in a conductive medium under the influence of an electric field due to their different charge density (charge to mass ratio). As most oligosaccharides lack ionizable functional groups, derivatization prior to analytical separation is often required. Oligosaccharides also lack chromophores so that derivatization is used to introduce a suitable chromophore for UV or laser-induced fluorescence (LIF) detection as well. For both purposes several reagents exist for CE as well as HPLC which are attached at the carbonyl groups of saccharides via reductive amination as summarized in (Harvey 2011; Ruhaak et al. 2010). For CE, 8-aminonaphthalene-1,3,6-trisulfonic acid (ANTS) (Guttman et al. 1994) and 8-aminopyrene-1,3,6-trisulfonic acid (APTS) (Guttman et al. 1996; O’Shea et al. 1998) are often employed for labeling because they provide a unique charge to mass ratio for all uncharged oligosaccharides in addition to sensitive LIF detection. A drawback of these labels is their relatively low ionization yield in electrospray ionization mass spectrometry (ESI-MS) (Pabst et al. 2009; Bunz et al. 2013a).
CE-LIF and CE-MS have been applied to the analysis of xylan-derived oligosaccharides. For example, APTS-labeled xylo-oligosaccharides from Eucalyptus wood have been analyzed by CE-LIF and CE-ESI-MS (Kabel et al. 2006). Efficient separation of APTS-labeled oligosaccharides composed of up to 18 xylose units could be achieved within 15 min including the separation of oligosaccharides composed of different sugar residues but with identical degrees of polymerization. Furthermore, O-acetylated xylo-oligosaccharides could be identified in the presence of their non-acetylated counterparts. Xyloglucans from black currants were analyzed by HPLC and CE (Hilz et al. 2006). Compared to HPAEC or RP-HPLC higher resolution was observed for the CE upon APTS-labeling of the oligosaccharides. Moreover, CE separated diacetylated oligomers from monoacetylated or non-acetylated oligomers. HPLC and CE were also compared for the determination of xylo-oligosaccharides in enzymatically hydrolyzed pulp (Metsämuuronen et al. 2013). In this case, the oligosaccharides were not labeled but a high pH background electrolyte (pH 12.6) was employed that rendered a negative charge to the saccharides due to dissociation of the hydroxyl groups. CE showed better performance than HPLC in the separation of low molecular xylo-oligomers. Furthermore, CE-LIF and CE-MS has also been used for the characterization of other hemicelluloses, for example, for arabino-oligosaccharides (Westphal et al. 2010) or galactoglucomannans (Doliška et al. 2009). The use of CE for monosaccharide and oligosaccharide analysis in the pulp and paper industry has been reviewed (Hiltunen and Sirén 2013).
The aim of the work was to characterize the neutral fraction of hydrothermally isolated xylan from beech wood and fractions obtained by preparative size exclusion chromatography by CE-LIF after the labeling with APTS. CE-MS was employed in order to obtain further information on the composition of the oligosaccharides. Furthermore, NMR measurements of unlabeled oligosaccharide samples were performed.
Experimental
Chemicals and materials
d-Xylose (purity ≥99 %), ε-aminocaproic acid, D2O and maltose were obtained from Sigma-Aldrich (Taufkirchen, Germany). Xylotriose (purity >95 %) and xylohexaose (purity 95 %) were purchased from Megazyme International Ireland (Bray, Ireland). The glucose ladder and APTS were obtained from Beckman Coulter (Krefeld, Germany). All other chemicals and reagents were of the highest purity commercially available and used as obtained. Milli-Q water (Milli-Q Direct 8 system, Millipore, Schwalbach, Germany) was used for buffer and sample preparation. Background electrolytes were filtered through 0.20 µm CHROMAFIL PET filters (Macherey-Nagel, Düren, Germany).
Isolation of xylan from beech wood
Low molar mass xylan was obtained by hydrothermal treatment as described earlier (Garrote and Parajó 2002; Puls and Dietrichs 1991). Briefly, wood chips from beech were treated with saturated steam of 170–200 °C under pressure (10–20 bar) and fiberized in hot stage by expansion to atmospheric pressure (DeLong 1978). The water-soluble xylans present in the hydrolysates were separated by precipitation in methanol. Subsequently, the low molar mass xylan (HTX) was treated with the anion exchange resin IRA96 (chloride form) in order to remove glucuronic acid substituted oligosaccharides. The neutral fraction (HTX-n) was obtained by freeze-drying (Daus et al. 2011).
The water soluble xylan contains 60 % of HTX-n. After the hydrolysis of HTX-n with 0.07 M perchloric acid, the monosaccharide composition was determined by HPLC. In this communication examined HTX-n contained 87.2 % of xylose and 1.7 % of 4-O-methylglucuronic acid (Daus et al. 2011).
Fractionation of HTX-n by preparative size-exclusion chromatography
A portion of 10 g of HTX-n was intensively shaken with 100 mL of ultrapure water to receive a saturated solution. The undissolved residue (HTX-n-residual) was centrifuged at 5,000 U/min for 20 min (centrifuge Janetzki T23) and 10 mL portions of saturated HTX-n solution were fractionated by preparative size-exclusion chromatography (SEC) on the Sephadex LH-20 column (2 cm × 3 m) purchased from GE Healthcare Life Science. The preparative SEC-device was self-configured from two pumps Pharmacia P5000 (GE Life Science), auto-sampler Labomatic LS-3000 (Labomatic Instruments AG, Alschwill, Switzerland), refractometric detector Schambeck RI-2000A (Schambeck SFD GmbH, Bad Honnef, Germany) and fraction collector Labcontrol-2000 (Labomatic Instruments AG, Alschwill, Switzerland). The device was controlled by Prepcon 5 software from SCPA (Weyhe-Leeste, Germany). Ultrapure water was used as the mobile phase at the flow rate of 2 mL/min. The eluate was collected into 11 fractions (HTX-n-1–HTX-n-11) followed by freeze-drying.
APTS derivatization
HTX-n and its fractions (0.2–0.3 mg) were dissolved in 5 µL of aqueous APTS solution (0.2 M) and maltose (3 µL, 0.6 mg/mL in water) was added as internal standard. To this mixture 1.75 µL of acetic acid (30 %, v/v), and 5 µL of aqueous NaCNBH3 solution (100 mg/mL in water) were added and the solution was allowed to react for 4 h at 60 °C. A standard mixture was prepared as described for HTX-n using 77 μg of each standard (xylose, xylotriose and xylohexaose). For the comparison of the migration behavior, 0.1 mg of glucose ladder and 0.177 mg of HTX-n were used for labeling. The mixed sample of HTX-n with glucose ladder contained 45 µg of each analyte. For CE-LIF analysis, the standard mixture was diluted 100,000–1,000,000 times with water while HTX-n and its fractions were diluted 10,000–100,000 times.
CE-LIF
CE experiments were conducted on a BioRad CE-LIF2 System (Bio-Rad Laboratories, Hercules, CA, USA). The excitation wavelength of the argon-ion laser was 488 nm, the emitted light was measured at 520 nm. The CE instrument was controlled by the BioFocus RunControl software (version 6.0), electropherogram integration was performed by the BioFocus System Integration software. The separation was performed in a 50/45.5 cm, 50 µm id polyvinyl alcohol (PVA)-coated capillary (Beckman Coulter, Krefeld, Germany). A new capillary was rinsed with water and subsequently with the background electrolyte for 5 min each at a pressure of 100 psi followed by the application of a voltage of −30 kV for 2 min. Between runs, the capillary was rinsed with water and the background electrolyte for 3 min each. Overnight the capillary was filled with water. The samples were injected hydrodynamically at a pressure of 3 psi*sec. The background electrolyte (BGE) consisted of 25 mM acetic acid adjusted to pH 4.75 by addition of 1 M NaOH. Analyte separation was carried out at a temperature of 20 °C at an applied voltage of −30 kV (detection at the anode). The generated currents were in the range of 13 µA.
CE-MS
CE-MS experiments were conducted on an Agilent HP3D CE system (Agilent Technologies, Waldbronn, Germany) coupled to a quadrupole-time-of-flight mass spectrometer (QqTOF MS, micrOTOFQ, Bruker Daltonik, Bremen, Germany) via a commercial CE-ESI-MS interface (Agilent Technologies, Waldbronn, Germany). A 60 cm, 50 µm id PVA-coated capillary was used. The BGE consisted of 40 mM ε-aminocaproic acid (EACA) and 131 mM acetic acid (Bunz et al. 2013b). Samples were injected at 50 mbar for 14–20 s. In addition to the separation voltage of −30 kV a pressure of 10 mbar was applied. ESI was performed in the negative mode at a constant voltage of 4,000 V. The sheath-liquid consisted of 50 % aqueous isopropyl alcohol containing 0.2 % triethylamine and was supplied by a syringe pump (Cole-Parmer, Illinois, USA) equipped with a 5 mL syringe (5MDF-LL-GT, SGE Analytical Science Pty Ltd, Melbourne, Australia) at a flow rate of 4 µL/min. The pressure of the nebulizer gas (nitrogen) was set to 0.2 bar, the drying gas (nitrogen) flow rate to 4 L/min, and the drying temperature to 170 °C. The ion energy in the quadrupole was 5.0 eV, the energy in collision cell was 7.0 eV. Argon was used as collision gas. Calibration was performed with sodium formate cluster (5 mM NaOH, 0.2 % formic acid in isopropyl alcohol:water 1:1) prior to each sequence. Single point correction was performed using the internal standard (G2). The mixture of standards was diluted 100 times while the HTX-n fractions were diluted 50–200 times prior to CE-MS analysis. Data processing in the m/z range 100–2,500 Da was achieved with the DataAnalysis 4.0 software (Bruker Daltonik, Bremen, Germany).
NMR
13C NMR spectra were acquired on a Bruker Avance 400 MHz instrument (Bruker Biospin, Rheinstetten, Germany) with 200,000 scans (70 °C). The sample concentration was 100 mg/mL in D2O.
Results and discussion
CE-LIF analysis of HTX-n
The neutral fraction of the xylo-oligosaccharides (HTX-n) was obtained by hydrothermal treatment of beech wood and subsequent removal of the acidic oligosaccharides. The resulting oligosaccharides, which consist primarily of linear β-(1 → 4)-linked d-xylopyranose units, were derivatized with APTS by reductive amination and subsequently analyzed in a PVA-coated capillary using 25 mM sodium acetate buffer, pH 4.75, as background electrolyte. Under these conditions the electroosmotic flow is suppressed so that the analytes migrate to the detector at the anode based on their charge density (charge-to-hydrated radius ratio). A typical electropherogram of HTX-n is shown in Fig. 1a illustrating the complexity of the sample. Besides major peaks, which can be attributed to xylose oligomers, further small peaks could be detected. For peak identification, the commercially available standards xylose (X1), xylotriose (X3) and xylohexaose (X6) were analyzed separately as well as spiked into a HTX-n sample (data not shown). The tentative assignment of the unknown major xylo-oligosaccharide peaks was based on the regular increase of the electrophoretic mobility of the individual oligosaccharides within a homo-oligosaccharide ladder (Mittermayr and Guttman 2012).

Electropherograms of a HTX-n, b a glucose ladder and c a mixed sample of the glucose ladder and HTX-n analyzed by CE-LIF in a PVA-coated capillary (50/45.5 cm; 50 µm) using 25 mM sodium acetate buffer, pH 4.75, as background electrolyte at an applied voltage of −30 kV. d Electropherogram of HTX-n using 40 mM ε-aminocaproic acid and 131 mM acetic acid as background electrolyte with CE-LIF. X stands for xylo-oligomer, G stands for gluco-oligomer; the numbers indicate the degree of polymerization. R1 and R2 correspond to reagent peaks
It has been shown that the electrophoretic mobility, µ e , within a series of homo-oligosaccharides can be correlated to the molecular mass, M, according to
where q represents the charge of the molecules and the exponent x a value between 1/2 and 1 depending on the shape of the oligosaccharides. An exponent of 2/3 suggests a random coil structure of the analytes while a linear conformation is suggested for an exponent of 1 (Mittermayr and Guttman 2012). A glucose ladder composed of α-(1 → 4)-linked gluco-oligosaccharides was compared to the HTX-n fraction. The electropherogram of these standards is shown in Fig. 1b. The relationship between the electrophoretic mobility and the molecular mass of the xylo- and gluco-oligosaccharides expressed as their log values is summarized in Fig. 2. Close inspection of the graph representing the α-(1 → 4)-linked gluco-oligosaccharides revealed linear fittings in 2 separated sections of DP below 9 and above 9, which has also been observed before with a slope of −0.62 for DP 1–9 and −0.68 for DP 9–24 indicating a relaxed helical structure of the oligosaccharides with some minor change in the overall geometry around DP 9 (Mittermayr and Guttman 2012). In the present study, a slope of −0.68 (r2 = 0.9997) was found for the range from 1 to 9 units of the glucose ladder while a slope of −0.75 (r2 = 0.9998) resulted for the saccharides with a DP above 9. The latter value is higher than the previously reported data (Mittermayr and Guttman 2012) but still close to a value of 2/3 so that a helical structure can also be assumed for the gluco-oligosaccharides in the present study. The β-(1 → 4)-linked xylo-oligosacchrides of HTX-n also displayed 2 separate relationships between electrophoretic mobility and molecular mass (Fig. 2). The first comprised the low polymerization range with DP up to 3 while the other included longer oligomers with DP 4–13 with slopes of −0.58 (r2 = 0.9955) for X1–X3 and −1.06 (r2 = 0.9998) in the case of X4–X13. A similar behavior, i.e. deviation for X1-X3 from the line of oligomers with a higher DP has been observed for oligosaccharides composed of β-(1 → 4)-linked cello-oligosaccharides and N-acetylchito-oligosaccharides (Mittermayr and Guttman 2012). This indicates a change in the geometry of the oligosaccharides at a DP around 3 that may be explained by the stronger effect of the APTS label on the overall geometry of the saccharides with low DP. The slope of about −1, found in the present study for the oligosaccharides with DP > 3, indicates an extended, rod-like structure for xylo-oligosaccharides. Slopes of −0.51 and −0.81 have been reported for β-(1 → 4)-linked saccharides with DP up to 3 and above 3, respectively, (Mittermayr and Guttman 2012) which are in reasonable agreement with our data.

Dependence of the electrophoretic mobility on the molecular weight for β-(1 → 4)-xylo-oligosaccharides (filled diamond) and α-(1 → 4)-malto-oligosaccharides (filled square) expressed as their log values. The separations were conducted in 25 mM sodium acetate buffer, pH 4.75, in a PVA coated capillary (50/45.5 cm; 50 µm id) at −30 kV
Regarding the migration order, the β-(1 → 4)-linked xylo-oligomers migrated faster than the α-(1 → 4)-linked glucose oligomers up to a DP of 6. G7 and X7 displayed similar mobilities while in the range of DP ≥ 8 the α-(1 → 4)-malto-oligosaccharides migrated faster than the corresponding β-(1 → 4)-xylo-oligomers. This migration behavior can be explained by the different geometry of the oligosaccharides. α-(1 → 4)-linked malto-oligosaccharides have a helical structure, while the β-(1 → 4)-xylo-oligosaccharides presumably possess a linear, rod-like shape as shown for β-(1 → 4)-cello-oligomers (Mittermayr and Guttman 2012).
The plot of the electrophoretic mobilities predicted the comigration between different species, which was subsequently confirmed by analysis of a mixture of HTX-n and the glucose ladder as shown in Fig. 1c. Comigration of X4 and G3, X9 and G10 as well as X11 and G13 occurred as assumed. Furthermore, only partial resolution between X3 and G2, X5 and G4, and X7 and G7 was observed due to similar mobilities. Comigration is also predicted for X12 and G15, and X13 and G16 but was not experimentally verified due to the low concentration of the oligosaccharides in the mixture.
Assay repeatability and precision
In order to estimate the precision of the CE-LIF assay, repeatability and intermediate precision was determined. Repeatability was derived from the analysis of 3 APTS-labeled samples of an HTX-n batch injecting each sample 3 times. A freshly prepared background electrolyte was used for each sample. Under these conditions, the electrophoretic mobilities of the xylo-oligosaccharides were highly reproducible with RSD values between 0.36 and 0.51 %. Intermediate precision was derived by injecting one sample 17 times over a period of time of 3 months using several separately prepared background electrolytes. RSD values of the electrophoretic mobilities of the oligosaccharides varied between 1.08 and 1.98 %.
Analyte quantitation was performed using the normalization procedure of the corrected peak areas (area divided by the migration time) assuming comparable response factors in order to determine the xylo-oligomer distribution pattern. Only the major peaks assigned to the xylo-oligosaccharides were taken into consideration. Repeatability was estimated from a triplicate injection of an APTS-derivatized sample. None of the RSD values exceeded 3.3 % (data not shown) with the highest value observed for X18 which can be explained by the low overall content of this oligosaccharide. Intermediate precision was estimated from the analysis of 3 independently derivatized samples which were analyzed a total of 16 times over a period of time of about 3 months using different freshly prepared background electrolytes. The distribution of the HTX-n xylo-oligomers from these experiments is shown in Fig. 3. RSD values below 9.8 % were observed for the oligomers X2 to X16. The RSDs of X1, X17 and X18 were 17.2, 11.2 and 14.8 %, respectively. The latter two values are due to the low overall concentration of the oligomers in the 0.6–0.4 % range. The relatively high RSD value of X1 may be caused by comigration with arabinose which is not baseline separated in the present system (See “Characterization of HTX-n fractions”).

Intermediate precision of the distribution pattern of HTX-n. One batch of HTX-n was derivatized with APTS in three independent experiments and each sample was assayed by CE-LIF within a period of time of 3 months. A total of 16 injections was performed. Only the linear xylo-oligomers with a DP of 1–18 were evaluated
Complete calibration could not be performed due to the lack of standards for the DP range of the xylo-oligosaccharides. However, the repeatability and intermediate precision data indicate that the assay allows the comparison of the composition of different batches of HTX-n.
Effect of derivatization conditions on distribution pattern
For HTX-n, the effect of the conditions of the derivatization of HTX-n with APTS on the distribution pattern of the samples was investigated by variation of the derivatization time, solvent, and amount of APTS reagent (data not shown). HTX-n and their fractions were generally derivatized at 60 °C. Variation of the reaction time between 2 and 8 h did not result in significant changes of the statistical variation in the oligosaccharide distribution pattern obtained by CE-LIF analysis stated above. Thus, further derivatization reactions were carried out for 4 h. Although some changes in the distribution pattern were observed especially for the oligomers with DP 1–4 upon variation of the APTS concentration in the range of 0.1–0.4 M, these changes still were within the general variation found for the distribution pattern under the standard APTS concentration of 0.2 M as discussed above (“Assay repeatability and precision”). Therefore, it was concluded that the APTS concentration in the investigated range does not significantly affect the oligosaccharide distribution pattern.
Due to the polar nature of HTX-n oligosaccharides, the solvents water, DMSO/water (1:1, v/v), and DMSO were tested. Solvents usually employed for APTS derivatization such as THF (Guttman et al. 1996) were not investigated due to the limited solubility of the xylo-oligosaccharides in these solvents. Comparable oligosaccharide distribution patterns were observed with somewhat lower relative abundance of X1–X4 and a higher relative abundance of the X7–X15 oligomers after derivatization in DMSO/water and DMSO as compared to water. Moreover, the derivatization yield was significantly decreased in DMSO. Therefore, water was used as solvent for further derivatizations. Overall, the results suggest that the derivatization procedure is not sensitive to minor changes of the experimental conditions and yield reproducible distribution patterns for the HTX-n xylo-oligosaccharide mixture.
Effect of separation medium on distribution pattern
It is a known phenomenon in CE that buffer concentration can affect peak resolution. Because of the complex composition of the sample with many minor peaks migrating close to the main peaks of the linear xylo-oligomers, the effect of the concentration of the background electrolyte on the distribution pattern was investigated by analyzing one sample using background electrolytes with increasing buffer concentrations from 25 to 100 mM. With the exception of the xylose trimer (X3), the tetramer (X4), the decamer (X10) and the hexadecamer (X16), essentially identical distribution patterns were observed for the sample independent of the buffer concentration (Online Resource 1). When expressing the variability over all tested buffer concentrations, RSD values of 8.63, 7.24, 9.86, and 8.93 % were determined for X3, X4, X10, and X16, respectively. This can be explained by comigration or lower resolution of minor peaks from the major peaks of the xylo-oligosaccharides. All other RSD values were between 2.43 and 6.58 %. These data are within the range determined for assay precision, i.e. within RSD of 9.8 %. Nonetheless, the data indicate that the background electrolyte should be prepared carefully in order to achieve reproducible data for distribution pattern of complex oligosaccharide mixtures.
Characterization of HTX-n fractions
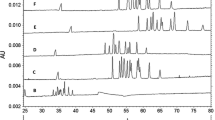
In addition, HTX-n was fractionated into 12 fractions that were characterized by CE-LIF. As examples, fractions HTX-n-3, HTX-n-5 and HTX-n-7 will be discussed in detail (for complete data see Online Resource 2). The CE-LIF electropherograms of the fractions are displayed in Fig. 4. Peak identification of the linear xylo-oligomers was performed by comparison of the migration times obtained with the unfractionated sample as well as by CE-MS measurements (see below). Oligosaccharides with low DP in the X2–X4 range were found in all fractions while higher DP oligomers were found in the first fractions only. Thus, fraction HTX-n-3 contains a relatively high amount of X8, X9 and X10. Interestingly, a considerable amount of X3 was also present. Fraction HTX-n-5 contained mainly X5, X6 and X7 while fraction HTX-n-7 is dominated by X4. In addition to the linear xylo-oligomers, further peaks were detected which could not be identified. Therefore, CE-MS measurements of the fractions were performed in order to obtain further information on their composition.

CE-LIF electropherograms of fractions HTX-n-7 (a), HTX-n-5 (b) and HTX-n-3 (c). The separations were conducted in 25 mM acetate buffer, pH 4.75, in a PVA coated capillary (50/45.5 cm; 50 µm id) at −30 kV. X stands for xylo-oligomer, G stands for gluco-oligomer; the numbers indicate the degree of polymerization, “X′” corresponds to satellite peak. R1 and R2 correspond to reagent peaks. U stands for unknown peak
For CE-MS, a volatile background electrolyte has to be employed. Thus, an ε-aminocaproic acid/acetic acid buffer was evaluated (Bunz et al. 2013b). The CE-LIF separation of HTX-n using the ε-aminocaproic acid/acetic acid buffer is shown in Fig. 1d. Good peak resolution was also observed for this buffer although direct comparison of the ε-aminocaproic acid/acetic acid background electrolyte and the sodium acetate buffer, pH 4.75, is difficult to assess due to the different composition which may result in a slightly altered migration behavior of the analytes. Moreover, the electric field strength in both systems is different. Upon hyphenation with an MS instrument the resolution will be reduced due to the additional influence of the sheath liquid. For an evaluation of the system and the ionization behavior of the APTS-labeled oligosaccharides a standard mixture containing X1, X3 and X6 was analyzed first. As described in the literature for APTS-labeled analytes (Pabst et al. 2009) a low ionization yield in ESI-MS was observed in the present study resulting in low molecular ion intensity. Both, [M-H]− and [M-2H]2− ions of the APTS-labeled oligosaccharides were detected (data not shown). Except for X1, a higher intensity was found for the [M-2H]2− ion so that these ions were selected in the analysis of the fractions. Peak assignment was performed according to the accurate masses only. Fragmentation experiments were not undertaken due to the low intensity of the ions caused by the low ionization efficiency of the APTS labeled oligosaccharides. Interestingly, using argon as collision gas fragmentation of X3 to X2 and of X6 to X4 was noted. Replacement of argon by nitrogen as collision gas resulted in decrease of fragmentation but at the expense of a significant lower sensitivity. Therefore, argon was used for the investigation of the fractions despite some fragmentation of the xylo-oligosaccharides.
The m/z data of the [M-2H]2− ions compared to the theoretical masses along with the tentative identification of oligosaccharides within the fractions are summarized in Table 1. In fraction HTX-n-7, the xylo-oligomers X3, X4 and X5 were identified by the respective [M-2H]2− ions in CE-MS. Apart from the internal standard, G2, the masses corresponding to X1 and X2 as well as gluco-oligomers G3 and G4 were found but only at very low intensity indicating a small concentration of the compounds. Furthermore, a peak with m/z 492.5663 (labeled X4′ in Fig. 4a) was detected which corresponds to a compound with the same mass as X4. It may be speculated that the compound could be a branched oligosaccharide composed of 4 xylose units or an oligosaccharide containing arabinose. Arabinose and xylose possess isobaric masses and can, therefore, not be distinguished based on their molecular mass.
As indicated by CE-LIF, the xylose oligomers X1, X5, X6, and X7 could be confirmed by CE-MS in fraction HTX-n-5 (Table 1). In contrast, X3 and X8, which were assigned in CE-LIF by comparison of migration times could not be detected by CE-MS. This can be explained by the fact that peak assignment in CE-LIF was done by migration time comparison only. Thus, other components migrating at the same time as a xylo-oligomer cannot be distinguished in CE-LIF. This is especially true in the case of incompletely separated peaks as occurring, for example, at about 4.1 min or 4.6 min in the electropherogram of fraction HTX-n-5 in Fig. 4b.
The peaks with identical masses as X6 and X7 (labeled X6′ and X7′ in Fig. 4b) were tentatively assigned either branched xylo-oligomers or oligomers containing also arabinose as discussed above. Compounds comigrating with these satellite peaks X6′ and X7′ were found at m/z 631.6154 and 697.6356, respectively. Based on the relative mass differences of about 7 of the [M-2H]2− ions as compared to the masses of X6 and X7 these compounds could be tentatively assigned as xylo-oligosaccharides containing one sugar unit with a methoxy substituent (X5 + MeOX; X6 + MeOX) or, alternatively, as xylo-oligomers branched with one rhamnose unit (X5 + Rha; X6 + Rha). These isobaric oligosaccharides cannot be distinguished based on their masses only. Furthermore, a peak with a mass trace at m/z 587.5887 was detected which migrated before X5. This m/z value can be interpreted as the [M-2H]2− ion of a xylo-oligosaccharide tetramer containing a 4-O-methylglucuronic acid moiety (X4 + MGA). A xylan with an acidic saccharide migrates faster than the corresponding neutral xylan due to the additional negative charge. Thus, the unknown compounds migrating between X3 and X5 in the CE-LIF electropherograms can be tentatively assigned as such acidic oligosaccharides. The analogous X5 + MGA and X6 + MGA oligomers with m/z 653.6116 and 719.6294 could also be detected as well as compounds with m/z 674.6156 and 740.6330, which would correspond to a monoacetylated X5 + MGA and X6 + MGA oligomers. Finally the mass trace of G6 was also observed for a small peak. In CE-LIF a split peak was observed at a migration time of about 3.4 min (Fig. 4b) which was not resolved in CE-MS. Based on mass trace of m/z 294.5010 corresponding to the [M-2H]2− ion, the peak pair (X1-U1; see Fig. 4b) can be assigned to the isobaric sugars xylose and arabinose. Only partial separation between these compounds was also observed when analyzing standards by CE-LIF with xylose migrating faster than arabinose (data not shown).
Fraction HTX-n-3 represented the most complex sample of the fractions. Besides the xylo-oligomers X2-X12 further mass traces corresponding to xylo-oligomers containing an additional 4-O-methylglucuronic moiety as well as acetyl residues could be identified (Table 1) albeit at low levels. As stated above, the lower resolution observed in CE-MS as compared to CE-LIF and the use of different background electrolytes hampers a direct comparison of both modes and makes peak assignment in CE-LIF based on the CE-MS experiments ambiguous, although mobilities of the xylose ladder obtained by CE-LIF and CE-MS show a perfect linear relation (r2 = 0.9989; data not shown) (Bunz et al. 2013b). In general, CE-MS confirmed the identity of the major components of the fractions and indicated the presence of related oligosaccharides as minor impurities such as gluco-oligomers or 4-O-methylglucuronic acid containing xylans.
NMR measurements
It has been shown previously, that NMR spectroscopy is a powerful tool for the characterization of xylans and their derivatives (Daus et al. 2011). Figure 5 shows the 13C-NMR spectra of the fractions HTX-n-3, HTX-n-5 and HTX-n-7 in D2O. Comparing the intensity of the signals of the anomeric C1 atoms of the reducing end [C1(Xredβ) at 96.5 ppm and C1(Xredα) at 92.0 ppm] and the non-reducing end [C1(Xnr) at 101.9 ppm] of the oligosaccharide with the signals of the internal xylose units, it can be concluded that the average chain length of the xylo-oligosaccharides decrease in the order HTX-n-3 > HTX-n-5 > HTX-n-7. This is in accordance with the CE-LIF and CE-MS analyses of the fractions as discussed above.

13C-NMR spectra of fractions HTX-n-3, HTX-n-5 and HTX-n-7
Besides the intense 13C-NMR signals that could be attributed to neutral xylans, very small signals at 21.0 ppm, 59.9 ppm (marked with a triangle in Fig. 5) and 173.8 ppm could be detected in fractions HTX-n-3 and HTX-n-5. These signals can be attributed to the acetyl CH3 group (21.0 ppm) as well as the 4-O-methoxy group (triangle) and the carbonyl signal (173.8 ppm) of the acetyl group. These data support the presence of small amounts of acidic 4-O-methylglucuronoxylans as well as acetyl xylo-oligosaccharide derivatives in these fractions which has also been indicated by the CE-MS measurements. Furthermore, the small signal at 61.0 ppm (C 6; marked with the asterisk in Fig. 5) confirms the presence of traces of hexose-oligomers in the samples, e.g. gluco-oligomers, which has also been indicated by the CE-MS data. However, it is not possible to assign any signal found in the NMR spectra to a structural feature, in particular the signals of very low intensity cannot be assigned unambiguously.
Conclusions
HTX-n and their fractions obtained by SEC were characterized by CE-LIF following derivatization with APTS. The assay employing a PVA-coated capillary and a 25 mM sodium acetate buffer, pH 4.75, under reversed polarity of the applied voltage resulted in reproducible conditions, allowing the determination of the composition with regard to the xylo-oligomers. Thus, CE-LIF appears to be a powerful tool for the analysis of HTX-n. The assay proved to be robust so that minor changes in the derivatization procedure or the separation conditions such as buffer concentration did not affect the overall performance with regard to the xylo-oligomer distribution pattern. CE-MS and NMR experiments were conducted in order to obtain further information on the composition of HTX-n fractions indicating the presence of minor amounts of xylo-oligosaccharides containing, for example, 4-O-methylglucuronic acid, acetyl-, or methyl substituents. Furthermore, small amounts of gluco-oligomers appear to be present in HTX-n. Overall, the combination of methods used proved to be powerful for the characterization of the components providing insight into the complexity of the HTX-n samples.
Abbreviations
- APTS:
-
8-Aminopyrene-1,3,6-trisulfonic acid
- BGE:
-
Background electrolyte
- CE:
-
Capillary electrophoresis
- DMSO:
-
Dimethyl sulfoxide
- DP:
-
Degree of polymerization
- EACA:
-
ε-Aminocaproic acid
- ESI-MS:
-
Electrospray ionization-mass spectrometry
- G:
-
Gluco-oligomer (numbers indicate degree of polymerization)
- HPAEC-PAD:
-
High performance anion-exchange chromatography with pulsed amperometric detection
- HTX:
-
Hydrothermally treated beech wood xylan
- HTX-n:
-
Neutral fraction of hydrothermally treated beech wood xylan
- LIF:
-
Laser-induced fluorescence
- MALDI-MS:
-
Matrix assisted laser desorption/ionization mass spectrometry
- MGA:
-
4-O-methylglucuronic acid
- NMR:
-
Nuclear magnetic resonance
- PGC-ELSD:
-
Porous graphitized carbon liquid chromatography with evaporative light scattering detection
- PVA:
-
Polyvinyl alcohol
- RP-HPLC:
-
Reversed phase high performance liquid chromatography
- RSD:
-
Relative standard deviation
- SEC:
-
Size-exclusion chromatography
- THF:
-
Tetrahydrofuran
- X:
-
Xylo-oligomer (numbers indicate degree of polymerization)
References
Bunz S-C, Cutillo F, Neusüß C (2013a) Analysis of native and APTS-labeled N-glycans by capillary electrophoresis/time-of-flight mass spectrometry. Anal Bioanal Chem 405:8277–8284
Bunz S-C, Rapp E, Neusüss C (2013b) Capillary electrophoresis/mass spectrometry of APTS-labeled glycans for the identification of unknown glycan species in capillary electrophoresis/laser-induced fluorescence systems. Anal Chem 85:10218–10224
Chong SL, Nissilä T, Ketola RA, Koutaniemi S, Derba-Maceluch M, Mellerowicz EJ, Tenkanen M, Tuomainen P (2011) Feasibility of using atmospheric pressure matrix-assisted laser desorption/ionization with ion trap mass spectrometry in the analysis of acetylated xylooligosaccharides derived from hardwoods and Arabidopsis thaliana. Anal Bioanal Chem 401:2995–3009
Daus S, Petzold-Welcke K, Kötteritzsch M, Baumgaertel A, Schubert US, Heinze T (2011) Homogeneous sulfation of xylan from different sources. Macromol Mater Eng 296:551–561
DeLong EA (1978) Verfahren zur Behandlung von Lignocellulosematerial und dadurch gewonnenes Lignocelluloseprodukt in Teilchenform. DE Patent 2830476C2 (in German)
Deutschmann R, Dekker RFH (2012) From plant biomass to bio-based chemicals: latest developments in xylan research. Biotechnol Adv 30:1627–1640
Doliška A, Strnad S, Ribitsch V, Kleinschek KS, Willför S, Saake B (2009) Analysis of galactoglucomannans from spruce wood by capillary electrophoresis. Cellulose 16:1089–1097
Ebringerová A, Heinze T (2000) Xylan and xylan derivatives—biopolymers with valuable properties, 1. Macromol Rapid Commun 21:542–556
Ebringerová A, Hromádková Z (1999) Xylans of industrial and biomedical importance. Biotechnol Genet Eng 16:325–346
Ebringerová A, Hromádková Z, Heinze T (2005) Hemicellulose. Adv Polym Sci 186:1–67
El Rassi Z (1999) Recent developments in capillary electrophoresis and capillary electrochromatography of carbohydrate species. Electrophoresis 20:3134–3144
Garrote G, Parajó JC (2002) Non-isothermal autohydrolysis of Eucalyptus wood. Wood Sci Technol 36:111–123
Guttman A, Cooke N, Starr CM (1994) Capillary electrophoresis separation of oligosaccharides: I. Effect of operational variables. Electrophoresis 15:1518–1522
Guttman A, Chen FTA, Evangelista RA, Cooke N (1996) High-resolution capillary gel electrophoresis of reducing oligosaccharides labeled with 1-aminopyrene-3,6,8-trisulfonate. Anal Biochem 233:234–242
Harvey DJ (2011) Derivatization of carbohydrates for analysis by chromatography, electrophoresis and mass spectrometry. J Chromatogr B 879:1196–1225
Hiltunen S, Sirén H (2013) Analysis of monosaccharides and oligosaccharides in the pulp and paper industry by use of capillary zone electrophoresis: a review. Anal Bioanal Chem 405:5773–5784
Hilz H, de Jong LE, Kabel MA, Schols HA, Voragen AGJ (2006) A comparison of liquid chromatography, capillary electrophoresis, and mass spectrometry methods to determine xyloglucan structures in black currants. J Chromatogr A 1133:275–286
Hoffman M, Jia Z, Pena MJ, Cash M, Harper A, Blackburn AR, Darvill A, York WS (2005) Structural analysis of xyloglucans in the primary cell walls of plants in the subclass Asteridae. Carbohydr Res 340:1826–1840
Kabel MA, Carvalheiro F, Garrote G, Avgerinos E, Koukios E, Parajo JC, Girio FM, Schols HA, Voragen AGJ (2002a) Hydrothermally treated xylan rich by-products yield different classes of xylo-oligosaccharids. Carbohydr Polym 50:47–56
Kabel MA, Schols HA, Voragen AGJ (2002b) Complex xylo-oligosaccharides identified from hydrothermally treated Eucalyptus wood and brewery´s spent grain. Carbohydr Polym 50:191–200
Kabel MA, Heijnis WH, Bakx EJ, Kuijpers R, Voragen AGJ, Schols HA (2006) Capillary electrophoresis fingerprinting, quantification and mass-identification of various 9-aminopyrene-1,4,6-trisulfonate-derivatized oligomers derived from plant polysaccharides. J Chromatogr A 1137:119–126
Metsämuuronen S, Lyytikäinen K, Backfolk K, Sirén H (2013) Determination of xylo-oligosaccharides in enzymatically hydrolysed pulp by liquid chromatography and capillary electrophoresis. Cellulose 20:1121–1133
Mittermayr S, Guttman A (2012) Influence of molecular configuration and conformation on the electromigration of oligosaccharides in narrow bore capillaries. Electrophoresis 33:1000–1007
O’Shea MG, Samuel MS, Konik CM, Morell MK (1998) Fluorophore-assisted carbohydrate electrophoresis (FACE) of oligosaccharides: efficiency of labeling and high-resolution separation. Carbohydr Res 307:1–12
Pabst M, Kolarich D, Pöltl G, Dalik T, Lubec G, Hofinger A, Altmann F (2009) Comparison of fluorescent labels for oligosaccharides and introduction of a new postlabeling purification method. Anal Biochem 384:263–273
Puls J, Dietrichs HH (1991) Separation of lignocelluloses into highly accessible fibre materials and hemicellulose fraction by the steaming-extraction process. Communication for European communities, EUR7091, energy biomass conference, pp 348–353
Ruhaak LR, Zauner G, Huhn C, Bruggink C, Deelder AM, Wuhrer M (2010) Glycan labeling strategies and their use in identification and quantification. Anal Bioanal Chem 397:3457–3481
Teleman A, Tenkanen M, Jacobs A, Dahlman O (2002) Characterization of O-acetyl-(4-O-methylglucuruno)xylan isolated from birch and beech. Carbohydr Res 337:373–377
Vismeh R, Humpula JF, Chundawat SPS, Balan V, Dale BE, Jones AD (2013) Profiling of soluble neutral oligosaccharides from treated biomass using solid phase extraction and LC-TOF MS. Carbohydr Polym 94:791–799
Westphal Y, Kühnel S, Schols HA, Voragen AGJ, Gruppen H (2010) LC/CE-MS tools for the analysis of complex arabino-oligosaccharides. Carbohydr Res 345:2239–2251
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Jáč, P., Elschner, T., Reiter, C. et al. Characterization of hydrothermally isolated xylan from beech wood by capillary electrophoresis with laser-induced fluorescence and mass spectrometry detection. Cellulose 21, 3993–4007 (2014). https://doi.org/10.1007/s10570-014-0456-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-014-0456-3