
Abstract
Particulate matter (PM) is an environmental pollutant closely associated with human airway inflammation. However, the molecular mechanisms of PM-related airway inflammation remains to be fully elucidated. It is known that COX-2/PGE2 play key roles in the pathogenesis of airway inflammation. Filaggrin is a transmembrane protein contributing to tight junction barrier function. As such, Filaggrin prevents leakage of transported solutes and is therefore necessary for the maintenance of epithelial integrity. The objective of the present study was to investigate the regulatory mechanisms of COX-2/PGE2 and Filaggrin upon PM exposure both in vivo and in vitro. C57BL/6 mice received intratracheal instillation of PM for two consecutive days. In parallel, human bronchial epithelial cells (HBECs) were exposed to PM for 24 h. PM exposure resulted in airway inflammation together with upregulation of COX-2/PGE2 and downregulation of Filaggrin in mouse lungs. Corresponding dysregulation of COX-2/PGE2 and Filaggrin was also observed in HBECs subjected to PM. PM exposure led to the phosphorylation of ERK, JNK, and PI3K signaling pathways in a time-dependent manner, while blockade of PI3K with the specific molecular inhibitor LY294002 partially reversed the dysregulation of COX-2/PGE2 and Filaggrin. Moreover, pretreatment of HBECs with NS398, a specific molecular inhibitor of COX-2, and AH6809, a downstream PGE2 receptor inhibitor, reversed the downregulation of Filaggrin upon PM exposure. Taken together, these data demonstrated that the PI3K signaling pathway upregulated COX-2 as well as PGE2 and acted as a pivotal mediator in the downregulation of Filaggrin.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As industrialization has progressed worldwide, atmospheric pollution is recognized to be the leading contributor to global disease burden (Huang 2014). The major atmospheric pollutant, particulate matter (PM), is a mixture of solid particles and liquid droplets, which primarily deposits in the lungs through inhalation (Mukherjee and Agrawal 2018; Traboulsi et al. 2017). Substantial epidemiological investigations have revealed that PM exposure participates in the pathogenesis of airway diseases (Li et al. 2018b; Wang et al. 2018). Human bronchial epithelial cells (HBECs) act as the first line of defense against PM exposure, which ultimately leads to the malfunction of the cells (Chen et al. 2016; De Grove et al. 2018). The exposed HBECs secrete excessive cytokines, which in turn lead to irreversible pathological changes in HBECs (Wang et al. 2007). Considering the pivotal role of HBECs in the pathogenesis of airway diseases, research into the relevant mechanism affecting these cells might contribute to an understanding of PM-related airway disease.
Cyclooxygenase2 (COX-2) is an inducible rate-limiting enzyme, the expression of which is upregulated by mitogen and endotoxin (Rumzhum and Ammit 2016). COX-2 is a well-known inflammatory mediator, which mainly converts arachidonic acid into prostaglandins including prostaglandin E2 (PGE2) (Yagami et al. 2016). Previous research studies have demonstrated that PM exposure upregulates the expression of COX-2 in multiple systems including cardiovascular and neuron systems, and upregulated COX-2 subsequently exerts a variety of pathogenic effects. Jie et al. reported that COX-2 mediates PM-induced apoptosis of vascular endothelial cells in cardiovascular injury (Yin et al. 2017). Additionally, Ben et al. reported that PM exposure stimulated COX-2-mediated excitatory synaptic transmission of neurons in neurodegenerative disease (Li et al. 2018a). However, the pathogenic role of COX-2 in the malfunction of HBECs remains to be elucidated. Filaggrin belongs to the keratin gene family and is involved in the differentiation of epithelial cells (Zhong et al. 2005). The loss-of-function in mutations of FLG in humans is correlated with airway hyperresponsiveness, a characteristic of PM-related airway disease (Berg et al. 2012; Cubero et al. 2016; Palmer et al. 2007). A systemic review of the literature concerning COX-2 and Filaggrin pointed out that PM downregulates Filaggrin via COX-2 expression/PGE2 production, leading to skin barrier dysfunction (Lee et al. 2016). Given the functional similarity between the bronchial epithelium and the epidermis in maintaining epithelial barrier integrity, it is meaningful to investigate the expression of COX-2 and Filaggrin upon PM exposure and explore the potential relevant molecular mechanisms.
Therefore, an in vivo mouse model of PM exposure was adopted to investigate the corresponding lung morphological changes in addition to the expression pattern of COX-2 and Filaggrin. Moreover, a HBEC in vitro model of PM exposure was employed to explore the potential relevant mechanism. We report that PM exposure upregulates COX-2 and downregulates Filaggrin via the PI3K/AKT signaling pathway. We propose that COX-2, as well as downstream PGE2, leads to Filaggrin downregulation.
Materials and methods
Reagents and antibodies
The standard reference airborne PM (standard reference material 1649b), mainly composed of polycyclic aromatic hydrocarbons, polychlorinated biphenyl congeners, pesticides, and doxins, was purchased from the National Institute of Standards and Technology (Gaithersburg, MD, USA). The specific molecular inhibitors SP600125, U0126, LY294002, AH6809, and NS398 were purchased from Selleck (Houston, TX, USA). Antibodies against COX-2, phospho-ERK, ERK, phospho-JNK, JNK, phospho-AKT, AKT, and EpCAM were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-Filaggrin antibody was obtained from GeneTex (San Antonio, TX, USA) and anti-GAPDH antibody was purchased from Beyotime (Shanghai, China). mRNA primers were synthesized by Sangon Biotech (Shanghai, China). The reagents for real-time qPCR were purchased from TaKaRa Bio (Shiga, Japan). The reagents used for western blotting were obtained from Solarbio Life Science (Beijing, China).
Cells and animals
HBECs were purchased from the Chinese Academy of Sciences (Shanghai, China) and cultured at 37 °C in a 5% CO2 incubator in RPMI-1640 medium (Gibco, Waltham, MA, USA) supplemented with 10% fetal bovine serum (Gibco, Waltham, MA, USA) and 50 U/mL penicillin and streptomycin (Gibco, Waltham, MA, USA). Male C57BL/6 mice (specific pathogen-free), with body weights ranging from 20 to 22 g, were purchased from Beijing Vital River Laboratory Animal Technology Company (Beijing, China). All protocols for animal experiments were approved by the Animal Experiment Center Ethics Committee, Wenzhou Medical University.
In vitro and in vivo PM treatments
For in vitro experiments, PM was resuspended in PBS at a stock concentration of 4 mg/cm3 and HBECs were exposed to PM at 0, 25, 50, 100, 200, and 400 μg/cm3 for 24 h. Additionally, HBECs were pretreated with a specific molecular inhibitor 60 min prior to PM exposure. For in vivo experiments, PM was resuspended in PBS at 100 μg (in 25 μL PBS) and mice were exposed for two consecutive days via daily intratracheal instillation. All mice were sacrificed 24 h after the last intratracheal instillation of PM.
Real-time qPCR
Total RNA was isolated through a guanidinium isothiocyanate/chloroform-based technique (TRIzol™ Invitrogen, Carlsbad, USA). RNA was subsequently reverse transcribed to cDNA using a SuperScript First-strand Synthesis System (Invitrogen, USA). SYBR Green PCR master kit was used with appropriate concentrations (10 nM) of forward and reverse primers in a total volume of 20 μL. Optimization was conducted for each gene-specific primer prior to the experiment to confirm that 10 nmol/L primer concentrations did not produce nonspecific primer-dimer amplification signals in no-template control wells. Quantitative RT-PCR was performed using an ABI 7000 PCR instrument (Eppendorf, Hamburg, Germany) with the two-stage program parameters provided by the manufacturer, as follows: 1 min at 95 °C and then 40 cycles of 5 s at 95 °C and 30 s at 60 °C. Sequences of the primer sets used for this analysis are as follows: COX-2: 5′-TGAGTGTGGGATTTGACCAG-3′ (forward [F]) and 5′-TGTGTTTGGAGTGGGTTTCA-3′ (reverse [R]); FLG: 5′-TGATGCAGTCTCCCTCTGTG-3′ (F) and 5′-TGTTTCTCTTGGGCTCTTGG-3′ (R); and for human glyceraldehyde-3-phosphate dehydrogenase (GAPDH): 5′-CCACCCATGGCAAATTCCATGGCA-3′ (F) and 5′-TCTACACGGCAGGTCAGGTCCACC-3′ (R). Specificity of the produced amplification product was confirmed by examination of dissociation reaction plots. Each sample was tested in triplicate with quantitative RT-PCR. Each group had six wells (Table 1).
Western blotting
Total protein from lung tissues and HBECs were lysed in RIPA buffer containing phenylmethanesulfonyl fluoride (PMSF, Beyotime) and phosphatase inhibitor (Beyotime). The concentration was measured using a bicinchoninic acid (BCA) protein assay kit (Thermo Scientific, Waltham, MA, USA). An equal quantity of lysate (45 μg) was loaded onto a 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel for electrophoresis after which the proteins were transferred to a PVDF membrane. The PVDF membrane was first incubated overnight at 4 °C with different dilutions of a primary antibody and then incubated with an appropriate secondary antibody for 1 h at room temperature. Immunoreactive bands were detected with a Bio-Rad Laboratories system using ECL reagents (Thermo Scientific, Waltham, MA, USA) and the relevant signals measured using Image Lab software.
Histological analysis
The lungs were embedded in paraffin and 5-μm sections were generated and stained with hematoxylin and eosin (H&E) according to the manufacturer’s instructions. Histological analysis was based on a method previously described (Lee et al. 2006) using an Olympus BX53 inverted microscope (Olympus, Melville, NY, USA). Three independent blinded investigators graded the score. The degree of peribronchial and perivascular inflammation was evaluated on a subjective scale of 1 to 4. A value of 1 was adjudged when no inflammation was observed, a value of 2 for occasional cuffing with inflammatory cells, a value of 3 when most bronchi or vessels were surrounded by a thin layer (1 to 5) of inflammatory cells, and a value of 4 when most bronchi or vessels were surrounded by a thick layer (> 5) of inflammatory cells.
Immunofluorescence staining
Mice exposed to PM for two consecutive days were sacrificed (24 h after the second PM treatment) and the lungs fixed in 4% paraformaldehyde, infiltrated with 30% sucrose/PBS under a pressure of 20 cmH2O, embedded in Tissue-Tek OCT compound (Sakura Finetek, Torrance, CA, USA), and then fresh-frozen in liquid nitrogen. Five- and 50-μm-thick sections were stored at − 80 °C and then fixed in ice-acetone for 10 min. The lung tissues were then incubated overnight with mouse monoclonal antibodies against EpCAM, COX-2, or Filaggrin, respectively, and then incubated with the corresponding secondary antibodies. Counterstaining was performed with DAPI (4,6′-diamidino-2-phenylindole). Confocal microscopic images were collected using a Leica TCS SL laser scanning confocal microscope (Leica Microsystems, Mannheim, Germany).
Statistical analysis
All numerical results were presented as mean ± SD. At least three independent biological repeats of experiments were carried out for statistical analyses. A one-way ANOVA with Bonferroni post hoc test was used to evaluate statistical significance between data sets. The level of statistical significance was set at P < 0.05.
Results
In vivo PM exposure led to lung inflammation mainly confined to the conducting airway in C57BL/6 mice
A mouse model of PM exposure was adopted with intratracheal instillation of 100 μg/day/mouse of PM suspension for two consecutive days (Fig. 1a). Vehicle (PBS only)-treated mice were used as controls (n = 8). Lung histological changes were evaluated through H&E staining between vehicle and PM-treated experimental mice (n = 8) (Fig. 1b). Conducting airway inflammation was observed in PM-treated mice. Supporting these histological changes, the proinflammatory cytokines IL-6 and IL-8 measured by ELISA were higher in the PM group compared to those in the vehicle group (Fig. 1d, e). These results indicate that PM exposure induces conducting airway inflammation.

PM exposure in C57BL/6 mice leads to lung inflammation mainly confined to the conducting airways. a Protocol of PM exposure. b Representative images of lung sections stained with H&E. c Semiquantitative inflammatory score of lung sections stained with H&E. d Level of IL-6 in BALF measured by ELISA. e Level of IL-8 in BALF measured by ELISA. Values represent means ± SD, *P < 0.05 or **P < 0.01, compared with the vehicle group; n = 3
Dysregulation of COX-2/PGE2 and Filaggrin in mouse lungs upon PM exposure in vivo
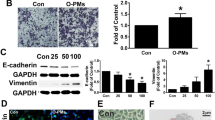
Next, we explored the potential dysregulation of COX-2/PGE2 and Filaggrin in lung tissues of mice upon PM exposure. Immunohistochemical staining revealed slightly elevated COX-2 expression in the bronchial epithelium of lung tissues (Fig. 2a). In addition, immunohistochemical staining showed slightly downregulated Filaggrin around the bronchial epithelium (Fig. 2b). This result was also confirmed by western blot analysis (Fig. 2c–e). In summary, PM exposure of the conducting airway of the mice led to the dysregulation of COX-2 and Filaggrin.

Dysregulation of COX-2/PGE2 and Filaggrin in mouse lungs upon PM exposure. Immunofluorescence staining of lung sections from mice challenged with PM for two consecutive days: EpCAM (green), COX-2/Filaggrin (red), and DAPI (blue), respectively. a Representative image of COX-2 (red) in lung tissues. b Representative image of Filaggrin (red) in lung tissues. c Western blot analysis of the expression of COX-2 and Filaggrin in whole lung tissue. The optical densities of protein bands are illustrated in d and e. Values represent means ± SD, *P < 0.05 or **P < 0.01, compared with the vehicle group; n = 3
PM exposure induced the dysregulation of COX-2/PGE2 and Filaggrin in HBECs in vitro
To validate the dysregulation of COX-2/PGE2 and Filaggrin in vitro, human bronchial epithelial cells (HBECs, also termed BEAS-2B, an epithelial virus-transformed cell line) were stimulated with increasing doses of PM. Our results indicate that PM induced COX-2 mRNA and protein upregulation in HBECs in a dose-dependent manner. Upregulation of PGE2 at the protein level was also observed (Fig. 3a, c, d, e). Conversely, PM induced the downregulation of Filaggrin mRNA and protein in HBECs in a dose-dependent manner (Fig. 3b, d, f). These data demonstrated that the effect of in vivo PM exposure on COX-2/PGE2 and Filaggrin expression can be reproduced in vitro using HBECs, thereby opening the way to more mechanistic insights into this regulation. Some of these mechanisms are explored in the following set of experiments.

PM exposure induced the dysregulation of COX-2/PGE2 and Filaggrin in HBECs. a, b HBECs were challenged with PM in a dose-dependent (0, 25, 50, 100, 200, and 400 μg/cm3) for 24 h. The mRNA of COX-2 and Filaggrin were measured by real-time PCR. c The level of PGE2 in supernatant was measured by ELISA. d The protein expression of COX-2 and Filaggrin were measured by western blot analysis. The optical densities of protein bands are illustrated in e and f. Values represent means ± SD, *P < 0.05 or **P < 0.01, compared with the vehicle group; n = 3
PM induced the dysregulation of COX-2/PGE2 and Filaggrin via activation of the PI3K pathway
To investigate the signaling pathway involved in the process of PM exposure, HEBCs were stimulated with PM for different times. The results of western blot analysis demonstrated that PM led to activation of ERK, JNK, and PI3K signaling in a time-dependent manner, respectively. The phosphorylation level of p-ERK and p-JNK reached at a peak 15 min after exposure to PM, while p-AKT reached a peak after 5 min (Fig. 4a–d). Pretreatment of HBECs with a specific PI3K inhibitor 1 h prior to PM exposure blocked the PM-induced dysregulation of COX-2/PGE2 and Filaggrin in a dose-dependent manner (Fig. 5a–c), while specific ERK and JNK inhibitors did not work (Fig. 5d–j). These data illustrated that PM exposure triggered changes in COX-2/PGE2 and Filaggrin expression via activation of the PI3K signaling pathway.

PM induced phosphorylation of MAPK and PI3K pathways in a time-dependent manner. a HBECs were stimulated with 200 μg/cm3 PM in a time-dependent manner (0, 5, 15, 30, 60, and 120 min). The phosphorylation of ERK, JNK, and PI3K were measured by western blotting. The optical densities of p-AKT/AKT, p-ERK/ERK, and p-JNK/JNK bands are illustrated in b, c, and d. Values represent means ± SD, *P < 0.05 or **P < 0.01, compared with the vehicle group; #P < 0.05 or ##P < 0.01, compared with the PM group; n = 3

PM induced the dysregulation of COX-2/PGE2 and Filaggrin via the PI3K pathway. a, d, g HBECs were pretreated with specific molecular inhibitors of PI3K, ERK, and JNK and then stimulated with PM. The protein expression of COX-2 and Filaggrin were measured by western blot analysis. The optical densities of protein bands are illustrated in b, c, e, f, h, and i. Values represent means ± SD, *P < 0.05 or **P < 0.01, compared with the vehicle group; #P < 0.05 or ##P < 0.01, compared with the PM group; n = 3
Targeting COX-2/PGE2 partially reversed PM-induced downregulation of Filaggrin
To elucidate the potential effect of COX-2/PGE2 in the downregulation of Filaggrin expression upon PM exposure, HBECs were pretreated with specific COX-2/PGE2 receptor inhibitors before PM exposure. Both COX-2 inhibitor and PGE2 receptor inhibitor prevented PM-induced downregulation of Filaggrin in a dose-dependent manner (Fig. 6a–c). Our results, therefore, indicate that downregulation of Filaggrin upon PM exposure is mediated via the upregulation of COX-2/PGE2.

Targeting COX-2/PGE2 partially reversed PM-induced downregulation of Filaggrin. a HBECs were pretreated with specific molecular inhibitors of COX-2 and PGE2 receptor then stimulated with PM. The protein expression of Filaggrin was measured by western blotting. The optical densities of protein bands are illustrated in b and c. Values represent means ± SD, *P < 0.05 or **P < 0.01, compared with the vehicle group; #P < 0.05 or ##P < 0.01, compared with the PM group; n = 3
Discussion
A growing body of epidemiological evidence suggests that exposure to PM is closely correlated with the mortality and morbidity of respiratory diseases (Kim et al. 2017). However, the pathophysiological processes triggered by PM exposure are complex, involving mucus hyperproduction, bronchial epithelial injury, and impaired airway antibacterial defense leading to loss of epithelial integrity (Aloui et al. 2016; Chen et al. 2018; Xu et al. 2018a; b). COX-2 is a well-known critical mediator of the inflammatory process responsible for the synthesis of downstream PGE2. Numerous researchers have previously reported ROS-dependent upregulation of COX-2 in HBECs upon PM exposure (Fernando et al. 2019; Wang et al. 2017), while the effect of COX-2 in the differentiation of epithelial cells was scarcely analyzed. In comparison with the diverse effects of COX-2, the bronchial epithelial expression of Filaggrin is scarcely reported. For example, Ying et al. (2006) demonstrated a lack of Filaggrin expression in normal bronchial mucosa, while GeneCards (http://www.genecards.org) indicated that Filaggrin was expressed in the bronchial epithelium. The published literature focusing on Filaggrin is mainly restricted to a skin condition known as dermatosis (Batista et al. 2015; Orfali et al. 2017; Proksch et al. 2016; Scharschmidt et al. 2009). Loss of epidermal Filaggrin expression correlates with allergic sensitization through enhanced percutaneous exposure to environmental substances, which, in turn, provokes an adaptive immune response (Jones 2007; Saunders et al. 2016). As hyperresponsiveness is a characteristic of PM-related airway disease (Yang et al. 2019), whether this is mediated via decreased bronchial epithelial Filaggrin expression remains yet to be clarified.
Our work indicated that short-term PM exposure primarily leads to bronchitis, not alveolitis. Considering the difficulty in primary culture of bronchial epithelial cells from the mouse, immortalized HBECs were adopted for in vitro experiments, which might be of greater translational potential. The present research further indicated that COX-2 is primarily localized in the bronchial epithelium. Associated with such expression, we have reported for the first time Filaggrin downregulation in the bronchial epithelium upon PM exposure. Given the pivotal role of Filaggrin in the differentiation of epithelial cells, downregulated Filaggrin expression might lead to impaired epithelial integrity. As such, Filaggrin is a promising therapeutic target and future research into the function of Filaggrin is required. Using loss of function approaches in vitro for FLG will allow a determination of whether a decrease in FLG is sufficient to mimic the effect of PM exposure in the bronchial epithelium. In addition, the maintenance of an epithelial barrier using air–liquid interface in vitro culture systems could be established. Furthermore, in vivo experiments allowing FLG overexpression in the bronchial epithelium in the context of PM exposure (corresponding to a rescue experiment) will allow us to establish whether decreased FLG in pathological circumstances is required to elicit a deleterious epithelial phenotype in addition to impacting inflammation. Finally, it will be important to validate the results obtained so far in vitro and in vivo with PM in humans. Of particular interest will be the analysis of the BALF from humans exposed to different levels of PMs. Such populations are unfortunately not available in China. A quite recent development allowing gaps between basic and translational research to be filled has been the use of human lungs either from end-stage patients or from patients who died from nonrespiratory diseases. Precision cut lung slides allow the testing in vitro of the response of human lungs to different stimuli. The readouts include analysis by immunofluorescence, qPCR, and flow cytometry for different specific cell populations in addition to isolation of well-defined epithelial stem cells by FACS and evaluation of their self-renewal and differentiation capabilities using organoid assays (Gkatzis et al. 2018).
It has been previously reported that oxidative stress is primordial to explain the toxic effects generated by PM exposure. Important studies have demonstrated that PM exposure leads to ROS-dependent activation of signaling pathways mediating the pathological processes, including the MAPK signaling pathway (Dou et al. 2018; Kim et al. 2019; Liu et al. 2019; Tsai et al. 2017). Previous research reported ERK, JNK, and PI3K/AKT activation in the lung following lipopolysaccharide stimulation (Chen et al. 2011; Shi et al. 2018). In our PM exposure model, we also demonstrated the corresponding phosphorylation of ERK, JNK, and PI3K/AKT. The specific molecular inhibitors of ERK, JNK, and PI3K were used to investigate the role of these signaling pathways in PM-exposed HBECs. We demonstrated that the PI3K signaling pathway is dominant in mediating the dysregulation of COX-2 and Filaggrin, illustrated by the results of upregulation of COX-2/PGE2 and downregulation of Filaggrin being reversed by a specific molecular inhibitor of PI3K. The PM/PI3K/AKT axis appears to be sufficient to explain the upregulation of COX-2/PGE2 and downregulation of Filaggrin. In addition, COX-2 and PGE2 are themselves key mediators of Filaggrin downregulation.
Further research will be required to investigate the pathogenic role associated with Filaggrin gene dysregulation in PM-related airway disease not only under short-term conditions but also long-term PM exposure. Both in vivo and in vitro approaches will be useful to delineate the molecular mechanisms involved. For example, using the HBECs, it will be important to delineate the dynamic transcriptional changes occurring after PM exposure. This will likely allow the determination of the identity of important functional targets downstream of PMs. In addition, the cell line used for this study, HBECs, likely represents a heterogeneous cell population, which remains to be further defined. As the bronchial epithelium is composed of basal cells, club cells, variants of club cells, ciliated cells, goblet cells, and neuroendocrine cells, the identity of the cells which have been transformed and their proliferative and differentiation capabilities are unknown. Using single cell transcriptomics, the response to PM exposure of the different subpopulations present in HBECs could be determined. In particular, this will allow identification of the trajectory of differentiation (or de-differentiation if the cells affected are indeed already differentiated) in the context of PM exposure (Fig. 7).

Mechanism of PM-induced airway epithelial injury (schematic). PM enters the airway by respiratory movement of the human body and deposits on the airway epithelial cells. PM activates the PI3K/AKT pathway in the airway epithelium and upregulates the expression of inflammatory factors such as COX-2, PGE2, IL-6, and IL-8, resulting in airway epithelial inflammation. Subsequently, airway epithelial inflammation causes a downregulation of Filaggrin expression via the COX-2/PGE2 pathway
In summary, the present research illustrated a critical role of the PI3K signaling pathway in PM-induced dysregulation of COX-2/PGE2 and Filaggrin, both in vitro and in vivo. Upregulated COX-2 and production of PGE2 leads to the downregulation of Filaggrin in the bronchial epithelium, which we propose is causative for the disruption of epithelial integrity. Future development of novel therapies targeting the PM/PI3K/AKT/COX-2/PGE2/Filaggrin axis is promising in protecting against PM-related destruction of epithelial integrity in humans.
References
Aloui R, Magne F, Devouassoux G, Deverchere J, Ritter P, Bentaher A, et al. Effects of fine particulate matter from on bronchial epithelial cells. Rev Mal Respir. 2016;33(9):767–74. https://doi.org/10.1016/j.rmr.2016.02.010.
Batista DI, Perez L, Orfali RL, Zaniboni MC, Samorano LP, Pereira NV, et al. Profile of skin barrier proteins (filaggrin, claudins 1 and 4) and Th1/Th2/Th17 cytokines in adults with atopic dermatitis. J Eur Acad Dermatol Venereol. 2015;29(6):1091–5. https://doi.org/10.1111/jdv.12753.
Berg ND, Husemoen LL, Thuesen BH, Hersoug LG, Elberling J, Thyssen JP, et al. Interaction between filaggrin null mutations and tobacco smoking in relation to asthma. J Allergy Clin Immunol. 2012;129(2):374–80, 380 e371-372. https://doi.org/10.1016/j.jaci.2011.08.045.
Chen C, Fang X, Wang Y, Li Y, Wang D, Zhao X, et al. Preventive and therapeutic effects of phosphoinositide 3-kinase inhibitors on acute lung injury. Chest. 2011;140(2):391–400. https://doi.org/10.1378/chest.10-3060.
Chen ZH, Wu YF, Wang PL, Wu YP, Li ZY, Zhao Y, et al. Autophagy is essential for ultrafine particle-induced inflammation and mucus hyperproduction in airway epithelium. Autophagy. 2016;12(2):297–311. https://doi.org/10.1080/15548627.2015.1124224.
Chen X, Liu J, Zhou J, Wang J, Chen C, Song Y, et al. Urban particulate matter (PM) suppresses airway antibacterial defence. Respir Res. 2018;19(1):5. https://doi.org/10.1186/s12931-017-0700-0.
Cubero JL, Isidoro-Garcia M, Segura N, Benito Pescador D, Sanz C, Lorente F, et al. Filaggrin gene mutations and new SNPs in asthmatic patients: a cross-sectional study in a Spanish population. Allergy Asthma Clin Immunol. 2016;12:31–10. https://doi.org/10.1186/s13223-016-0137-x.
De Grove KC, Provoost S, Brusselle GG, Joos GF, Maes T. Insights in particulate matter-induced allergic airway inflammation: focus on the epithelium. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology. 2018;48(7):773–86. https://doi.org/10.1111/cea.13178.
Dou C, Zhang J, Qi C. Cooking oil fume-derived PM2.5 induces apoptosis in A549 cells and MAPK/NF-small ka, CyrillicB/STAT1 pathway activation. Environ Sci Pollut Res Int. 2018;25(10):9940–8. https://doi.org/10.1007/s11356-018-1262-5.
Fernando IPS, Jayawardena TU, Kim HS, Lee WW, Vaas A, De Silva HIC, et al. Beijing urban particulate matter-induced injury and inflammation in human lung epithelial cells and the protective effects of fucosterol from Sargassum binderi (Sonder ex J. Agardh). Environ Res. 2019;172:150–8. https://doi.org/10.1016/j.envres.2019.02.016.
Gkatzis K, Taghizadeh S, Huh D, Stainier DYR, Bellusci S. Use of three-dimensional organoids and lung-on-a-chip methods to study lung development, regeneration and disease. Eur Respir J. 2018;52(5). https://doi.org/10.1183/13993003.00876-2018.
Huang YC. Outdoor air pollution: a global perspective. J Occup Environ Med. 2014;56(Suppl 10):S3–7. https://doi.org/10.1097/jom.0000000000000240.
Jones G. Susceptibility to asthma and eczema from mucosal and epidermal expression of distinctive genes. Curr Allergy Asthma Rep. 2007;7(1):11–7.
Kim, H.J., Choi, M.G., Park, M.K., and Seo, Y.R. 2017. Predictive and prognostic biomarkers of respiratory diseases due to particulate matter exposure. Journal of Cancer Prevention 22(1): 6-15. Doi: https://doi.org/10.15430/jcp.2017.22.1.6.
Kim M, Kim JH, Jeong GJ, Park KY, Lee MK, Seo SJ. Particulate matter induces proinflammatory cytokines via phosphorylation of p38 MAPK possibly leading to dermal Inflammaging. Exp Dermatol. 2019. https://doi.org/10.1111/exd.13943.
Lee KS, Lee HK, Hayflick JS, Lee YC, Puri KD. Inhibition of phosphoinositide 3-kinase delta attenuates allergic airway inflammation and hyperresponsiveness in murine asthma model. FASEB Journal : official publication of the Federation of American Societies for Experimental Biology. 2006;20(3):455–65. https://doi.org/10.1096/fj.05-5045com.
Lee CW, Lin ZC, Hu SC, Chiang YC, Hsu LF, Lin YC, et al. Urban particulate matter down-regulates filaggrin via COX2 expression/PGE2 production leading to skin barrier dysfunction. Sci Rep. 2016;6:27995. https://doi.org/10.1038/srep27995.
Li B, Guo L, Ku T, Chen M, Li G, Sang N. PM2.5 exposure stimulates COX-2-mediated excitatory synaptic transmission via ROS-NF-kappaB pathway. Chemosphere. 2018a;190:124–34. https://doi.org/10.1016/j.chemosphere.2017.09.098.
Li R, Zhou R, Zhang J. Function of PM2.5 in the pathogenesis of lung cancer and chronic airway inflammatory diseases. Oncol Lett. 2018b;15(5):7506–14. https://doi.org/10.3892/ol.2018.8355.
Liu B, Wu SD, Shen LJ, Zhao TX, Wei Y, Tang XL, et al. Spermatogenesis dysfunction induced by PM2.5 from automobile exhaust via the ROS-mediated MAPK signaling pathway. Ecotoxicol Environ Saf. 2019;167:161–8. https://doi.org/10.1016/j.ecoenv.2018.09.118.
Mukherjee A, Agrawal M. A global perspective of fine particulate matter pollution and its health effects. Rev Environ Contam Toxicol. 2018;244:5–51. https://doi.org/10.1007/398_2017_3.
Orfali, R.L., Zaniboni, M.C., and Aoki, V. 2017. Profile of skin barrier proteins and cytokines in adults with atopic dermatitis. Giornale italiano di dermatologia e venereologia : organo ufficiale, Societa italiana di dermatologia e sifilografia 152(2): 140-147. Doi: https://doi.org/10.23736/s0392-0488.16.05533-4
Palmer CN, Ismail T, Lee SP, Terron-Kwiatkowski A, Zhao Y, Liao H, et al. Filaggrin null mutations are associated with increased asthma severity in children and young adults. J Allergy Clin Immunol. 2007;120(1):64–8. https://doi.org/10.1016/j.jaci.2007.04.001.
Proksch E, Dahnhardt D, Dahnhardt-Pfeiffer S, Folster-Holst R. Epidermal barrier disorders in dermatoses. Der Hautarzt; Zeitschrift fur Dermatologie, Venerologie, und verwandte Gebiete. 2016;67(11):907–21. https://doi.org/10.1007/s00105-016-3883-2.
Rumzhum NN, Ammit AJ. Cyclooxygenase 2: its regulation, role and impact in airway inflammation. Clinical and Experimental Allergy: journal of the British Society for Allergy and Clinical Immunology. 2016;46(3):397–410. https://doi.org/10.1111/cea.12697.
Saunders SP, Moran T, Floudas A, Wurlod F, Kaszlikowska A, Salimi M, et al. Spontaneous atopic dermatitis is mediated by innate immunity, with the secondary lung inflammation of the atopic march requiring adaptive immunity. J Allergy Clin Immunol. 2016;137(2):482–91. https://doi.org/10.1016/j.jaci.2015.06.045.
Scharschmidt TC, Man MQ, Hatano Y, Crumrine D, Gunathilake R, Sundberg JP, et al. Filaggrin deficiency confers a paracellular barrier abnormality that reduces inflammatory thresholds to irritants and haptens. The Journal of Allergy and Clinical Immunology. 2009;124(3):496–506, 506.e491-496. https://doi.org/10.1016/j.jaci.2009.06.046.
Shi L, Dong N, Ji D, Huang X, Ying Z, Wang X, et al. Lipopolysaccharide-induced CCN1 production enhances interleukin-6 secretion in bronchial epithelial cells. Cell Biol Toxicol. 2018;34(1):39–49. https://doi.org/10.1007/s10565-017-9401-1.
Traboulsi H, Guerrina N, Iu M, Maysinger D, Ariya P, Baglole CJ. Inhaled pollutants: the molecular scene behind respiratory and systemic diseases associated with ultrafine particulate matter. Int J Mol Sci. 2017;18(2). https://doi.org/10.3390/ijms18020243.
Tsai MH, Hsu LF, Lee CW, Chiang YC, Lee MH, How JM, et al. Resveratrol inhibits urban particulate matter-induced COX-2/PGE2 release in human fibroblast-like synoviocytes via the inhibition of activation of NADPH oxidase/ROS/NF-kappaB. Int J Biochem Cell Biol. 2017;88:113–23. https://doi.org/10.1016/j.biocel.2017.05.015.
Wang X, Adler KB, Erjefalt J, Bai C. Airway epithelial dysfunction in the development of acute lung injury and acute respiratory distress syndrome. Expert Review of Respiratory Medicine. 2007;1(1):149–55. https://doi.org/10.1586/17476348.1.1.149.
Wang J, Huang J, Wang L, Chen C, Yang D, Jin M, et al. Urban particulate matter triggers lung inflammation via the ROS-MAPK-NF-kappaB signaling pathway. Journal of Thoracic Disease. 2017;9(11):4398–412. https://doi.org/10.21037/jtd.2017.09.135.
Wang C, Xu J, Yang L, Xu Y, Zhang X, Bai C, et al. Prevalence and risk factors of chronic obstructive pulmonary disease in China (the China Pulmonary Health [CPH] study): a national cross-sectional study. Lancet (London, England). 2018;391(10131):1706–17. https://doi.org/10.1016/s0140-6736(18)30841-9.
Xu F, Cao J, Luo M, Che L, Li W, Ying S, et al. Early growth response gene 1 is essential for urban particulate matter-induced inflammation and mucus hyperproduction in airway epithelium. Toxicol Lett. 2018a;294:145–55. https://doi.org/10.1016/j.toxlet.2018.05.003.
Xu F, Luo M, He L, Cao Y, Li W, Ying S, et al. Necroptosis contributes to urban particulate matter-induced airway epithelial injury. Cellular Physiology and Biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2018b;46(2):699–712. https://doi.org/10.1159/000488726.
Yagami T, Koma H, Yamamoto Y. Pathophysiological roles of cyclooxygenases and prostaglandins in the central nervous system. Mol Neurobiol. 2016;53(7):4754–71. https://doi.org/10.1007/s12035-015-9355-3.
Yang SI, Lee SY, Kim HB, Kim HC, Leem JH, Yang HJ, et al. Prenatal particulate matter affects new asthma via airway hyperresponsiveness in schoolchildren. Allergy. 2019;74(4):675–84. https://doi.org/10.1111/all.13649.
Yin J, Xia W, Li Y, Guo C, Zhang Y, Huang S, et al. COX-2 mediates PM2.5-induced apoptosis and inflammation in vascular endothelial cells. Am J Transl Res. 2017;9(9):3967–76.
Ying S, Meng Q, Corrigan CJ, Lee TH. Lack of filaggrin expression in the human bronchial mucosa. J Allergy Clin Immunol. 2006;118(6):1386–8. https://doi.org/10.1016/j.jaci.2006.08.030.
Zhong CY, Zhou YM, Douglas GC, Witschi H, Pinkerton KE. MAPK/AP-1 signal pathway in tobacco smoke-induced cell proliferation and squamous metaplasia in the lungs of rats. Carcinogenesis. 2005;26(12):2187–95. https://doi.org/10.1093/carcin/bgi189.
Funding
The work was supported by Zhejiang Provincial Science Technology Department Foundation (2020361718), Wenzhou Technology Foundation (Y20180125), Zhejiang Provincial Natural Science Foundation (LZ15H010001, LQ20H010002), The National Nature Science Foundation of China (81570075, 81770074), and The National Key Research and Development Program of China (2016YFC1304000). Saverio Bellusci acknowledges grants from the Deutsche Forschungsgemeinschaft (DFG; BE4443/1-1, BE4443/4-1, BE4443/6-1, KFO309 P7, and SFB1213-projects A02 and A04).
Author information
Authors and Affiliations
Contributions
ND and SB drafted the manuscript and were responsible for the interpretation of the data. CJS, LJL, YRH, JJC, and BBW collected the primary data and generated the figures, and CCS and ND contributed to the literature review. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
All protocols for animal experiments were approved by the Animal Experiment Center Ethics Committee, The First Affiliated Hospital of Wenzhou Medical University.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Figure S1
PM induced the dysregulation of PGE2 via the PI3K pathway. HBECs were pretreated with specific molecular inhibitors of ERK/JNK/PI3K then stimulated with PM. The protein expression of PGE2 was measured by ELISA. Values represent means ± SD, *: P < 0.05 or **: P < 0.01, compared with the Vehicle group; #: P < 0.05 or ##: P < 0.01, compared with the PM group; n = 3 (PNG 75 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Song, C., Liu, L., Chen, J. et al. Evidence for the critical role of the PI3K signaling pathway in particulate matter-induced dysregulation of the inflammatory mediators COX-2/PGE2 and the associated epithelial barrier protein Filaggrin in the bronchial epithelium. Cell Biol Toxicol 36, 301–313 (2020). https://doi.org/10.1007/s10565-019-09508-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10565-019-09508-1