
Abstract
LaBO3 (B = Fe, Mn, and FeMn) perovskite-type oxides were prepared by sol–gel method and then used as catalysts in CO hydrogenation for light olefins. The catalysts were characterized using XRD, H2-TPR, SEM, CO (CO2)-TPD, and XPS. The results showed that the lattice oxygen migration and oxygen vacancies promoted oxygen mobility by doping Mn2+ at the B site, Moreover, the presence of manganese as a promoter in the catalyst increased olefin selectivity compared with the olefin selectivity of the catalyst containing iron at the B-site and exhibited resistance to carbon deposition; while reducing the metal elements. In CO hydrogenation, potassium-promoted LaFeMnO3 catalysts afforded high catalytic activity and C2=–C4= selectivity. An O/P value of 5.0 and a C2=–C4= fraction of 54% were achieved for all hydrocarbons with low methane selectivity.
Graphic Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Direct conversion of syngas into light olefins via Fischer–Tropsch synthesis (FTS) has become popular as a non-oil route due to feedstock availability, simplicity, and high energy efficiency. However, the complexity and diversity of CO hydrogenation yield a wide range of products. C2–C4 hydrocarbons (less than 58%) ruled by the Anderson–Schulz–Flory (A–S–F) distribution and the high selectivity of CH4 have restricted industrial applications of CO hydrogenation [1]. Thus, new catalyst design strategies are needed to control the product distribution and improve the selectivity of light olefins.
Recently, perovskite-type oxides (PTOs) have been introduced to synthesize olefins from syngas due to their unique structure that allows defect formation and their adjustable valence states via elemental doping [2,3,4,5]. Moreover, ABO3-type perovskite oxides can be used as acid–base catalysts due to the presence of metal cations and oxygen vacancies. The change in the cation composition at A and B sites could provide controllable selectivity for target products [4]. K+ doping strongly affects the structure of B site elemental catalysts [5]. For Gd2−xSr1+xFe2O7 (x = 0, 0.1, 0.2, 0.3, and 0.4) [6], the substitution of Sr for Gd distorts the structure and affords heterovalent iron atoms (Fe3+ and Fe4+), thereby promoting syngas conversion into light olefins, especially ethylene and propylene. Furthermore, GdFeO3 and GdMnO3 exhibit good catalytic performance in CO hydrogenation [7], benefiting from the enhanced interaction between the species in the perovskite structure. Under the reaction conditions of 350–400 °C, 1 atm and H2/CO = 2, in the presence of GdFeO3 and GdMnO3, olefin selectivity of about 25% and 80%, respectively, was achieved. Differences in their catalytic activity can be attributed to different diffusion rates of weakly bound atomic hydrogen across the catalyst surface based on the surface polymerization reaction mechanism of the CHx radicals [2].
Although PTO catalysts used in CO hydrogenation have been investigated, the relationship between their structure (Fe and Mn in B sites) and catalytic performance was not discussed in detail. In this study, LaFeO3, LaMnO3, and LaFeMnO3 were prepared and then promoted by potassium. Combined with the systematic characterization of the structures and surface properties of the catalysts, the structure–catalytic activity relationships were studied.
2 Experimental Procedure
2.1 Perovskite Preparation
K/LaBO3 (B = Fe, Mn, and FeMn) was prepared by the sol–gel and impregnation methods. An appropriate amount of lanthanum nitrate, manganese nitrate or ferric nitrate, and citrate acid were simultaneously dissolved in deionized water to afford a mixed salt solution with a molar ratio of (La:Fe = 0.6; and La:Mn = 0.5). Thereafter, the prepared solution was reacted in a water bath at 80 °C for heating and stirring until the gel was formed. Then, the gel was dried in an oven at 105 °C for 12 h. Consequently, the dried sample was calcined at 400 °C for 1 h and 750 °C for 5 h with a ramp of 5 °C/min under static air. The obtained perovskites are denoted as LaBO3 (B = Fe, Mn, and FeMn). Finally, the perovskite samples were impregnated with potassium carbonate (2 wt% potassium), and the target catalysts were obtained.
2.2 Physicochemical Characterization
The morphologies of all fresh and used catalysts were characterized via scanning electron microscopy (SEM, KYKY-2008B). The catalysts were sprayed with gold at 25 kV.
X-ray diffraction (XRD) patterns were obtained using a Rigaku D/MAX2200PC spectrometer at 40 kV and 30 mA using monochromatic Cu Kα radiation. The samples were scanned from 5° to 80° with a scanning rate of 8°/min. The collected XRD patterns were analyzed using the JCPDS data base.
Temperature-programmed reduction (H2-TPR) was measured by a Micromeritics AutoChem II 2920 chemisorption analyzer equipped with a thermal conductivity detector (TCD). 50 mg of the sample was filled in a quartz tube. First, it was flushed under the flow of a He atmosphere (30 mL/min) at 350 °C for 1 h to remove the moisture and then cooled to 50 °C. Thereafter, the temperature was increased to 800 °C at a ramp rate of 10 °C/min under 10% H2/Ar flow. The H2 consumption was recorded by the TCD.
In situ temperature-programmed desorption of CO (CO-TPD) was performed on an AutoChem II chemisorption analyzer (Micromeritics). 50 mg sample was reduced using 10% H2/Ar for 4 h at 600 °C and purged by He (30 mL/min) for 1 h. Then, the sample was cooled in a He flow and then pulse-adsorbed by CO at 50 °C until saturation, followed by a purge with He for 1 h. The desorption signal was recorded as the temperature increased from 50 to 800 °C at a heating rate of 10 °C/min.
The in situ temperature-programmed-desorption of CO2 (CO2-TPD) was almost identical to CO-TPD, except that CO was replaced by CO2.
X-ray photoelectron spectroscopy (XPS) was performed using a Thermo Scientific ESCALAB 250 spectrometer for all samples with an Al Kα source. The base pressure of the chamber was less than 2 × 10−8 Pa. The binding energies (BEs) were calibrated relative to the adventitious carbon using the C1s peak at 284.8 eV.
2.3 Catalytic Reaction Testing
The catalytic performance of samples was evaluated using a stainless steel fixed-bed reactor (i.d. = 8 mm; length = 400 mm). 1 mL catalyst and about 2 mL quartz sand were packed in the middle of the tube, and a thermocouple was placed inside in contact with the catalyst to monitor the catalyst bed temperature. Before the catalytic tests, the perovskite was reduced at 600 °C for 4 h under 10%H2/N2 flow. The FTS reaction was performed at T = 320 °C, P = 2.0 MPa, GHSV = 1000 h−1, and H2/CO = 2(molar ratio). The gaseous products and reactants were detected on an online gas chromatograph (GC-9560) equipped with a TCD and flame ionization detector. The product selectivity was on a carbon basis. The CO conversion (XCO) and product selectivity (Si) were calculated by Eqs. (1) and (2), respectively:
where Fin and Fout are the molar flows of the feed gas and effluent (mol/h), respectively, YCO,in and YCO,out are the volume fractions of CO in the feed gas and effluent, respectively, and Yi,in and Yi,out are the volume fractions of CO2 in the feed gas and effluent, respectively.
3 Results and Discussion
3.1 Characterization of Perovskites
3.1.1 X-Ray Diffraction
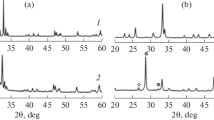
The XRD patterns of as-prepared perovskites exhibited the characteristic perovskite diffraction peaks, showing that cations entered the perovskite structure. From Fig. 1, all patterns exhibited strong reflexions corresponding to the presence of a well-defined perovskite structure with no diffraction peaks related to the Fe and Mn species, indicating that the orthorhombic LaBO3 phase was successfully prepared using the sol–gel method. This ensured that Fe and Mn ions were uniformly mixed at the atomic level and could interact due to proximity. The LaFeO3 intensity of the characteristic diffraction peak was significantly higher than that of the other samples. Moreover, the characteristic perovskite diffraction peak shifted to higher 2θ values with Mn substitution due to the difference in the size of Fe3+(0.064 nm) and Mn2+(0.08066 nm). Note that Mn2+ doping increases the lattice spacing of the perovskite, as confirmed by the XRD diffraction peak shifts [8]. In addition, as listed in Table 1, the crystalline size of LaBO3 calculated by the Scherrer equation varied with the B site, and the composite LaFeMnO3 had a smaller particle size (14.8 nm) than LaFeO3 (96.0 nm) and LaMnO3(68.6 nm). These results confirm that metal substitution at the B site influences the crystalline structure and affects the formation of metal phases on the catalyst surface due to reduction. Therefore, it influences the C2–C4 olefin selectivity [9]. Combined with the catalytic performance results, LaFeMnO3 appears to have a structure well suited for CO hydrogenation.

X-ray diffraction patterns of complex oxides
No noticeable difference was found between the calcined LaFeO3 and that reduced at 600 °C, as shown in Fig. 2, proving that iron in the perovskite structure cannot be easily reduced to metallic iron, which depends on the coupling of the coordinated metal cations with the abundant oxygen vacancies [10]. However, when the sample was reduced at 800°C, the Fe characteristic diffraction peak appeared, due to the partial reduction of Fe3+ to Fe0+. This indicates that this orthorhombic perovskite precursor can be used as a precursor of the FTS reaction [11]. The FTS reaction on iron-based catalysts undergoes some complex phase changes during the reduction carbonization and reaction process, according to H2-TPR and literature [12]. The main process is Fe2O3 → Fe3O4 → (FeO) → FeCX.

X-ray diffraction patterns of K/LaFeO3 after calcination and reduction
The crystal structure change of the modified perovskite LaFeO3 after FTS is shown in Fig. 3. According to the JCPDS database, fresh samples of LaFeO3 comprise a very small amount of starting metal oxide materials. In the XRD pattern of the catalyst after hydrogenation, the impurity peaks disappeared, providing that these partially reduced oxides were used as metallic active phases. The samples show strong characteristic diffraction peak intensities at 2θ = 32°, especially the used sample of LaFeMnO3. The characteristic diffraction peak intensity of the used LaFeMnO3 was substantially higher than that of the fresh samples. This indicates that the system crystallinity or crystalline domain of the perovskite-like structure increased. Moreover, it shows that the crystal structure of the perovskite-type catalyst was not been completely destroyed since the Fe-based perovskite itself had certain redox and oxygen release performances. The partially reduced perovskite phase strongly interacts with the modified perovskite phase. This prevents metal particle wear or excessive carbide formation [11]. Thus, good catalytic performance was retained after the syngas conversion to olefins.

X-ray diffraction patterns of complex oxides
3.1.2 BET Surface Area
Table 1 shows the B site significantly affected the specific surface area of the catalyst. The specific surface area of LaFeO3 was relatively low (11.52 m2/g). When the B site was occupied by Mn, the specific surface area of the catalyst was the highest. Upon Fe doping, the specific surface area of the modified perovskite LaFeMnO3 was relatively high (27 m2/g) since the radius of Mn2+ is larger than that of Fe3+. Thus, when the when the trivalent Fe3+ is partially replaced, the formed oxygen vacancies causes the shrinkage of the unit cell and reduces the porosity of the catalyst [7]. Therefore, the cell expansion after doping increased the comparative area.
As shown in Fig. 4, the N2 adsorption–desorption isotherms show that the catalyst has a typical type II isotherm with an H3 hysteresis ring structure, indicating that the catalyst had a mesoporous structure. The initial position of the retention ring was observed at P/P0 = 0.9, indicating that most of the formed pores were stacked pores, which may be due to the accumulation of catalyst particles for forming the porous structure during the decomposition of citrate by calcined samples [13]. For the LaFeO3 and LaMnO3 catalysts, the surface area and pore volume increased, and the initial hysteresis ring moved toward a lower relative pressure, and became larger. The LaFeMnO3 catalyst had a larger surface area than LaFeO3 (see Table 1). This denotes that the addition of Mn can increase the surface area of PTOs and promote the dispersion of iron nanoparticles.

N2 adsorption–desorption isotherms of the samples
3.1.3 H2-TPR Analysis
The reduction behavior of LaBO3 varies with the B site composition, and an orthorhombic perovskite structure makes metal reduction difficult. As shown in Fig. 5, no metal particles were detected in the temperature range corresponding to the hydrogen consumption measured by the TPR. The TPR profiles of the LaFeO3 sample showed a three-stage reduction, which can be ascribed to the reduction of Fe4+ to Fe3+ and a small number of Fe3+ to Fe2+, accompanied by the formation of oxygen vacancies on the surface of LaFeO3 [4]. The TPR profiles of the LaMnO3 sample can be divided into two steps. Due to the removal of a small quantity of surface-adsorbed oxygen species and the excess oxygen contained in the lattice as well as the reduction of Mn4+ to Mn3+, the reduction peak is nearly close [8]. When the B sites comprised both Fe and Mn, the reduction peaks moved toward a higher temperature. This indicates that the introduction of Fe could strengthen the Mn–O bonds near Fe, thereby restraining the mobility of the lattice oxygen. The second reduction peaks were distinctly shifted to a higher temperature, indicating that the addition of Fe inhibited the reduction of Mn species. Moreover, the electron transferring from iron ions to the adsorbed oxygen species could cause the transformation of some Fe3+ to Fe4+. Therefore, the hydrogen-consuming peaks of the LaFeMnO3 sample concentrated in the 400–450 °C temperature range and without the third hydrogen consumption peak, compared to that of LaFeO3, denote that the B site plays a decisive role in metal cation reduction as well as lattice oxygen migration and that the oxygen vacancies promote the oxygen mobility.

H2-TPR profiles of the samples
3.1.4 CO/CO2-TPD Analysis
To better understand the influence of K/LaBO3 on the adsorption of CO-TPD, a comparison with the K/Fe2O3 sample was performed, as shown in Fig. 6. PTOs have significantly different impacts on CO adsorption. For K/Fe2O3, only one desorption peak can be observed in the 350–520 °C temperature range. The desorption peaks in the 600–800 °C range for PTO catalysts were higher than those of iron oxide, indicating the stronger chemisorption of CO on K/LaBO3. Furthermore, as CO underwent both molecular adsorption to form linear and bridge species and dissociative adsorption [1,14], the desorption peak areas of K/LaFeMnO3 were larger than those of other pre-reduced samples, implying that the presence of both Fe and Mn at B sites enhanced CO adsorption. Interestingly, strong desorption peaks were also observed for the K/LaFeO3 samples after CO hydrogenation, which can be attributed to the different heats of adsorption of CO and H2 on the metal surface [2]. Overall, the K/LaFeMnO3 catalyst afforded excellent reaction performance, which is consistent with the catalytic activity test results.

CO-TPD profiles of the samples
The surface adsorption of CO2 plays an important role in CO hydrogenation. For all the reduced catalyst samples, three CO2 desorption peaks were observed, as shown in Fig. 7. The desorption temperatures below 150 °C, in the 200–400 °C range, and above 450 °C corresponded to weak, moderate, and strong basic sites, respectively. Compared with K/LaFeO3 and K/LaMnO3, K/LaFeMnO3 showed a larger desorption peak, and the CO2 desorption peak shifted to a higher temperature, suggesting that surface basicity was enhanced. In addition, the peak of the K/LaFeMnO3 sample indicates the increase in the number of alkaline sites. This kind of adsorption requires either one active metal site or one oxygen vacancy site on the surface of the perovskite to bond CO [7]. Since no reduction behavior from Fe3+ to Fe0 was detected by TPR and XPS results, the recorded TPD peak was assigned to the CO adsorbed on the oxygen vacancies. According to literature [5,15,16,17], weak basic sites offer active sites for CO insertion, and only the strongly adsorbed CO species in the strong basic sites can be dissociated and hydrogenated. Notably, the weak basic sites were attributed to the CO adsorbed on oxygen vacancies on the surface of K/LaBO3, which might also be the active sites for CO associative adsorption. In contrast, the strong basic sites were attributed to the dissociatively adsorbed CO. Thus, the addition of metal elements to the B sites increased the alkalinity of the catalyst surface.

CO2-TPD profiles of the samples
3.1.5 SEM and EDX Analysis
SEM images showed that the synthesized compounds had similar morphologies, small particle morphology, smooth particles, tight binding, porous structure, and no agglomeration. The particles were anisotropic, and the average diameter and length of the crystallites were 20–100 nm. Notably, the morphologies and particle size distribution of the particles prepared via the sol–gel method were uniform. Moreover, all the samples had porous surfaces. The morphology of the fresh LaFeO3 catalyst was a uniformly-sized sphere. This is due to the decomposition of the complexes and nitrates from the catalyst during the preparation process [15], in which yielded a loose and uniform surface morphology. After the FTS reaction, the edges of the spherical particles melted, as shown in Fig. 8, and the surface appeared to be slightly sintered. The spherical particles melted into agglomerates, which may be due to the oxidation of iron generated during the reaction [16]. Although the agglomeration of catalyst particles reduced the effective contact area with the perovskite catalyst, the catalytic activity remained stable.

SEM images of LaBO3 samples: a LaFeO3; b used LaFeO3; c LaMnO3; d used LaMnO3; e LaFeMnO3; f used LaFeMnO3
EDX shows the uniform distribution of La, Fe, and Mn on the surface of the used catalyst LaFeMnO3, and no carbon was detected as shown in Fig. 9. During the surface preparation process, Mn2+ successfully entered the crystal lattice [8]. The presence of oxygen vacancies on LaFeMnO3 combined with the modified effect of lanthanum made the catalyst highly resistant to carbon deposition.

EDX mapping of used K/LaFeMnO3
3.1.6 XPS Analysis
As shown in Fig. 10a, two main peaks and a satellite peak were observed in the Fe 2p profiles of all calcined catalyst precursors. The two main peaks located at 724.48 eV and 709.83 eV were assigned to Fe2+ and Fe3+, respectively [18]. As for the Mn 2p spectra of all calcined samples presented in Fig. 10a, two main peaks at 641.8 eV and 643.6 eV corresponded to Mn3+ and Mn4+, respectively [19]. Thus, the Mn species in the LaFeMnO3 perovskite structure were a mixture of Mn3+ and Mn4+. A similar result was also reported in other perovskites containing Mn. Figure 10b shows La 3d5/2 spectra characteristic of La3+ with the maximum at 833.48 eV and 837.68 eV. The fitting of the Fe 2p spectra in Fig. 10c showed that the peaks at 709.83 eV decreases, and an enhanced peak corresponding to the oxygen vacancy migration appeared at 710.08 eV. This result can be ascribed to the addition of Mn species. Furthermore, the relative peak intensity of Fe 2p1/2 and Fe 2p3/2 gradually decreased with the addition of Mn. The signal of bulk oxygen (O2−) shifted to small BE values, whereas the signal of surface oxygen (O1−) shifted to high values (Fig. 10d). This trend is probably due to the change in the effective doped Mn2+ with increasing lattice oxygen migration and oxygen vacancies [20,21]. The consumed surface oxygen species are replaced by the adsorption and dissociation of syngas, or by the migration of bulk oxygen species, promoting oxygen mobility and C2=–C4= selectivity when combined with the catalytic performance.

a Survey XPS spectrum of LaBO3 perovskite, and high-resolution XPS spectra of b La 3d, c Fe 2p, and d O 1s
3.2 Catalytic Performance
The catalytic results of several LaBO3-derived catalysts were evaluated (Fig. 11). Without K doping, the catalytic performances of LaFeO3 and LaMnO3 were weak. We found that the impregnated catalysts can significantly improve the selectivity of light olefins and CO conversion compared to LaFeO3 and K/LaFeO3 as well as LaFeMnO3 and K/LaFeMnO3. Potassium acts as an electronic assistant, adjusting the surface alkalinity of PTO samples and weakening the secondary hydrogenation of olefins. Thus, the adsorption of CO and the formation of the active phase of iron carbide were promoted [5]. However, the CO conversion of these catalysts was still below 20% under a typical FTS reaction pressure and space velocity, implying that the LaBO3 catalysts possessed inferior activity in CO hydrogenation compared to traditional iron-based catalyst. This is likely due to the unique perovskite structure and Fe oxides were not exposed. Consequently, the CO conversion was severely inhibited. LaMnO3 had a vigorous hydrogenation reaction, forming mainly carbon dioxide and methane due to the Mn site surface adsorption energies of CO and H2 being higher than that of the Fe site. The energy barrier to the reaction of adsorbed CO and H2 was increased with Fe incorporation into the B site [2,7]. Thus, in the presence of Fe and Mn, the CO conversion decreased, while the selectivity of light olefins increased due to the “seesaw” effect. The giving electron ability of Mn promoted the CO dissociation and weakened H2 adsorption [8]. Since Mn and Fe strongly interact to form homogeneous oxides, they coordinated with each other and promoted the distribution of products. Mn addition further promoted Fe dispersion and increased reduction from Fe2O3 to Fe3O4. The simultaneous doping of Fe and Mn ions can increase the oxygen vacancy content of the oxide, thus further improving the catalytic activity of the catalyst. Among all the derived catalysts, K/LaFeMnO3 had the best catalytic performance with an olefin/paraffin(O/P) value of 5.0 and C2=–C4= fraction of 54 wt%. Notably, the generation of C4+ hydrocarbon was significantly inhibited when the perovskite catalysts were used. The carbon chain seemed to be cut by scissors, and the production of C5+ products was below 8%. The ability of carbon chain growth reduced, leading to the lattice confinement and product distribution being deviated from the A–S–F distribution.

Catalytic performance of catalysts with time on stream. Reaction conditions: H2/CO = 2, 1000 h–1, 320 °C, and 2.0 MPa
4 Conclusions
LaBO3 (B = Fe, Mn, and FeMn) catalysts afforded excellent catalytic performance in the direct conversion of syngas to light olefins, high selectivity to higher olefins, and very good resistance to sintering and carbon deposition. K/LaFeMnO3 had the best catalytic performance with an O/P value of 5.0 and a C2=–C4= fraction of 54%. The lattice oxygen migration and oxygen vacancies promoted the oxygen mobility by doping Mn2+ at the B sites. Moreover, Mn addition further promoted Fe dispersion and increased its reduction from Fe2O3 to Fe3O4. Structure controlled LaBO3 (B = Fe, Mn, and FeMn) suppressed chain growth and improved the product distribution. The results demonstrate that the catalysts originated from LaFeMnO3 perovskites are stable and effective for the synthesis of olefins by CO hydrogenation.
References
Torres Galvis HM, De Jong KP (2013) Catalysts for production of lower olefins from synthesis gas: a review. ACS Catal 3(9):2130–2149
Sheshko TF, Markova EB, Sharaeva AA et al (2019) Carbon monoxide hydrogenation over Gd(Fe/Mn)O3 perovskite-type catalysts. Pet Chem 59(12):1307–1313
Sheshko TF, Serov YM, Dement’eva MV et al (2016) Catalytic hydrogenation of carbon monoxide over nanostructured perovskite-like gadolinium and strontium ferrites. Russ J Phys Chem A 90(5):926–931
Bedel L, Roger AC, Rehspringer JL et al (2005) La(1−y)Co0.4Fe0.6O3−d perovskite oxides as catalysts for Fischer–Tropsch synthesis. J Catal 235:279–294
Goldwasser MR, Dorantes VE, Perez ZMJ (2003) Modified iron perovskites as catalysts precursors for the conversion of syngas to low molecular weight alkenes. J Mol Catal A 193(1–2):227–236
Sheshko TF, Kryuchkova TA, Serov YM et al (2017) New mixed perovskite-type Gd2−x Sr1+x Fe2O7 catalysts for dry reforming of methane, and production of light olefins. Catal Ind 9(2):162–169
Sheshko TF, Sharaeva AA, Powell OK et al (2020) Carbon oxide hydrogenation over GdBO3 (B = Fe, Mn, Co) complex oxides: effect of carbon dioxide on product composition. Pet Chem 60(5):571–576
Gao S, Liu N, Liu J et al (2020) Synthesis of higher alcohols by CO hydrogenation over catalysts derived from LaCo1−xMnxO3 perovskites: effect of the partial substitution of Co by Mn. Fuel 261:116415
Torres Galvis HMT, Bitter JH, Davidian T et al (2012) Iron particle size effects for direct production of lower olefins from synthesis gas. J Am Chem Soc 134(39):16207–16215
Zhao K, Li L, Zheng A et al (2017) Synergistic improvements in stability and performance of the double perovskite-type oxides La2−xSrxFeCoO6 for chemical looping steam methane reforming. Appl Energy 197:393–404
Bedel L, Roger AC, Rehspringer JL et al (2004) Structure-controlled La-Co-Fe perovskite precursors for higher C2–C4 olefins selectivity in Fischer–Tropsch synthesis. Stud Surf Sci Catal 147:319–324
Chou W, Wu P, Luo M et al (2020) Effects of Al, Si, Ti, Zr promoters on catalytic performance of iron-based Fischer–Tropsch synthesis catalysts. Catal Lett 150(7):1993–2002
Jaroniec M, Solovyov LA (2006) Improvement of the Kruk–Jaroniec–Sayar method for pore size analysis of ordered silicas with cylindrical mesopores. Langmuir 22(16):6757–6760
Ao M, Pham GH, Sage V et al (2016) Structure and activity of strontium substituted LaCoO3 perovskite catalysts for syngas conversion. J Mol Catal A 416:96–104
Yang QL, Liu GL, Liu Y (2018) Perovskite-type oxides as the catalyst precursors for preparing supported metallic nanocatalysts: a review. Ind Eng Chem Res 57(1):1–17
Tien Thao N, Son LT (2016) Production of cobalt-copper from partial reduction of La(Co, Cu)O3 perovskites for CO hydrogenation. J Sci Adv Mater Devices 1(3):337–342
De Smit E, Weckhuysen BM (2008) The renaissance of iron-based Fischer–Tropsch synthesis: on the multifaceted catalyst deactivation behavior. Chem Soc Rev 37(12):2758–2781
Fang YZ, Liu Y, Zhang LH (2011) LaFeO3-supported nano Co-Cu catalysts for higher alcohol synthesis from syngas. Appl Catal A 397(1):183–191
Wang Y, Zheng Y, Wang Y et al (2019) Evaluation of Fe substitution in perovskite LaMnO3 for the production of high purity syngas and hydrogen. J Power Sources 449:227505
Xu Y, Dhainaut J, Dacquin JP et al (2021) La1−x(Sr, Na, K)xMnO3 perovskites for HCHO oxidation: the role of oxygen species on the catalytic mechanism. Appl Catal B 287:119955
Chang H, Bjørgum E, Mihai O et al (2020) Effects of oxygen mobility in La-Fe-based perovskites on the catalytic activity and selectivity of methane oxidation. ACS Catal 10(6):3707–3719
Acknowledgements
This work was supported by the National Natural Science Foundation of China (U20A20124).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ma, Lh., Gao, Xh., Ma, Jj. et al. K/LaFeMnO3 Perovskite-Type Oxide Catalyst for the Production of C2–C4 Olefins via CO Hydrogenation. Catal Lett 152, 1451–1460 (2022). https://doi.org/10.1007/s10562-021-03744-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-021-03744-z