
Abstract
CeO2 nanorods (CeO2-nrs), CeO2 nanocubes (CeO2-ncs) and CeO2 nanopolyhedrons (CeO2-nps) were prepared by hydrothermal method and then NiO crystallites were deposited on these supports by precipitation-deposition, respectively. The physic-chemical properties of Ni/CeO2 catalysts were characterized and performances for carbon dioxide methanation reaction were tested. The Ni/CeO2-nrs sample shows well metal dispersion and high concentration of oxygen vacancy, which leads to the high catalytic activity for CO2 methanation. Especially at 300 °C, the CO2 conversion could reach 60%. Further analysis reveals that the content of oxygen vacancy has a positive correlation with the surface area of catalyst. The largest surface area results in the most of oxygen vacancy on the Ni/CeO2-nrs catalyst, and then a large amount of CO2 could be activated at low temperatures. Meanwhile, large surface area facilitates the dispersion of active metals, and improves the degree of H2 activation. The combined effect results in the promotion of catalytic activity for CO2 methanation at low temperatures.
Graphic Abstract
CeO2 nanorods (CeO2-nrs), CeO2 nanocubes (CeO2-ncs) and CeO2 nanopolyhedrons (CeO2-nps) supported Ni catalysts were prepared. These catalysts ware tested for CO2 methanation and further characterized by BET, XPS, TEM, H2-TPR and TPD. The results showed that the content of oxygen vacancy has positive correlation with the specific surface area of catalyst.

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Carbon dioxide as an important component of greenhouse gas and C1 resource has been widely investigated for its capture, storage and utilization [1,2,3]. CO2 methanation (known as Sabatier reaction) which would reduce CO2 emissions and produce natural gas, is considered to be one of the most effective and practical technologies for CO2 recycling [4,5,6]. As an exothermic reaction, CO2 methanation is favored at low temperatures. However, inert CO2 shows a slow conversion rate at temperatures < 350 °C due to the kinetic limitation. Therefore, active catalyst is necessary to reduce the activation energy for CO2 methanation [7].
Noble metals such as Ru and Rh present high activity and selectivity for low-temperature CO2 methanation, but Ni-based catalysts are more commonly used due to their high activity and low cost [8]. Al2O3, ZrO2, SiO2, CeO2 and many other oxides have been exploited as supports for Ni catalysts in CO2 methanation. Sun et al. [9,10,11] prepared a nickel catalyst supported on nanosheet γ-Al2O3 and the results indicated that the well dispersed Ni crystallites with small diameter and moderate basic sites on support resulted in a good catalytic activity for CO2 methanation. In addition, this catalyst exhibited well stability at 325 °C during 40 h time-on-stream. Tada et al. [12] studied the CO2 methanation over the Ni-based catalysts with different supports (i.e., CeO2, α-Al2O3, TiO2, and MgO) and found that Ni/CeO2 catalyst showed high CO2 conversion, especially at temperatures lower than 400 °C.
The catalytic activity is highly correlated with the reaction mechanism (e.g., the reaction intermediate and route) and different catalysts may show different mechanisms [13, 14]. Pan et al. [15] compared the CO2 methanation over Ni/Al2O3 and Ni/Ce0.5Zr0.5O2 catalysts. The results revealed that CO2 adsorbed on medium basic sites in Ni/Ce0.5Zr0.5O2 catalyst and formed monodentate carbonates while CO2 adsorbed on strong basic sites in Ni/γ-Al2O3 will not participate in the reaction. Monodentate formate derived from monodentate carbonate on medium basic sites could be hydrogenated more quickly than bidentate formate derived from hydrogen carbonate which resulted in higher activity of Ni/Ce0.5Zr0.5O2 than Ni/γ-Al2O3. The oxygen vacancies also affect the formation of intermediate products and further influence the catalyst activity in the CO2 methanation. Jia et al. [16] found that the plasma decomposition caused the formation of Ni-ZrO2 interficial sites with more oxygen vacancies, which played crucial roles in CO2 activation and methanation. Wang et al. [17] compared the CO2 methanation mechanisms over Ru/CeO2 and Ru/α-Al2O3 catalysts, respectively. It was revealed that CO2 methanation underwent formate route over Ru/CeO2 catalyst, and the formate dissociation to methanol catalyzed by oxygen vacancy was the rate-determining step. In contrast, CO2 methanation underwent CO route over Ru surface in Ru/α-Al2O3 with the absence of oxygen vacancy, demonstrating active site dependent catalytic mechanism toward CO2 methanation. In addition, the catalytic activity evaluation and the oscillating reaction over Ru/CeO2 catalyst further proved that the oxygen vacancy catalyzed the rate-determining step with a much lower activation temperature compared with Ru surface in Ru/α-Al2O3 (125 versus 250 °C).
High oxygen storage capacity (OSC) coupled with a high oxygen transport capacity gives CeO2 a unique property for catalyst applications. Previous studies showed that the OSC of CeO2 was related to its exposed crystal planes. Oxygen storage takes place both at the surface and in the bulk as {110}/{100} planes exposed while the storage restricted at the surface as {111} planes exposed [18]. It has been reported that CeO2-supported metal catalysts (i.e., Ru, Ni et al.) were active for CO2 methanation, particularly at low temperatures. Sakpal [19] investigated the catalytic activity of Ru-CeO2 catalysts with different morphologies and found that the Ru/CeO2/r catalyst (CeO2/r = CeO2 rods) was easier to be reduced and contained more oxygen vacancies compared to Ru/CeO2/o and Ru/CeO2/c catalyst (CeO2/o = CeO2 octahedra, CeO2/c = CeO2 cubes). Characterization further revealed that the highest concentration of oxygen vacancies led to the most active catalyst and the adsorption of CO2 at an oxygen vacancy was the rate-determining step. A Ni catalyst supported on ceria nanorods was also compared with the Ni/CeO2 commercial products and state-of-the-art literature catalysts for CO2 methanation [20]. The high efficiency of the former catalyst was presumed to be the formation of highly active interfacial sites with abundance in Ce3+ for a low-temperature methanation process. Zhou et al. [21] prepared a Ni/CeO2 catalyst by the hard-template method and the catalytic properties were also investigated by CO2 methanation. In-situ FT-IR and in-situ XPS results indicated that CO2 molecules could be reduced by active metal Ni species and surface oxygen vacancies to generate active CO species and then promoted CO2 methanation. The CO2 conversion and CH4 selectivity reached 91.1% and 100% at 340 °C and atmospheric pressure.
To date, the majority of studies have shown the oxygen vacancy could promote CO2 methanation. However, few studies focus on the factors affecting the concentration of oxygen vacancy on the catalyst. Thus, this work prepared CeO2 with different morphologies, such as nanorods, nanocubes, and nanopolyhedrons (denoted as CeO2-nrs, CeO2-ncs, and CeO2-nps, respectively) as supports that dominantly expose {110}, {100}, and {111} planes, respectively. Subsequently, Ni-supported catalysts were prepared by precipitation–deposition, and applied in the CO2 methanation. The relationship between the physical and chemical structures of the catalysts and their catalytic performances are analyzed and then the main factors affecting the catalytic activities of Ni/CeO2 catalysts with different morphologies in the CO2 methanation are explored.
2 Experimental
2.1 Catalyst Preparation
The CeO2 supports with different morphologies were synthesized via a previously reported hydrothermal method [19]. NaOH (96.0 g) and Ce(NO3)3·6H2O (8.68 g) were dissolved in deionized water (350 and 50 mL) respectively. The two solutions were then mixed and stirred for 30 min, after which the resulting mixture was transferred to a 100 mL Teflon-lined stainless-steel autoclave, and hydrothermally treated at 180 °C for 24 h. The precipitate was then collected by filtration, and thoroughly washed with distilled water and ethanol. CeO2-ncs was obtained by drying at 60 °C for 10 h and subsequently calcined at 500 °C for 4 h. The synthesis of CeO2-nrs and CeO2-nps were carried out also with the above procedures, with the exception that a hydrothermal temperature of 100 °C was employed for CeO2-nrs, while for CeO2-nps, 0.16 g of NaOH was used.
The Ni/CeO2 catalysts were then prepared via a precipitation-deposition method. An aqueous solution of Ni(NO3)2·6H2O was mixed with the support at 60 °C over 2 h (the theoretical nickel loading is 20 wt.%). The pH was adjusted to 10 with a 1 mol·L−1 NaOH aqueous solution, and the suspension was further stirred and aged at 60 °C for 10 h. The precipitate was then collected by filtration, thoroughly washed with distilled water, dried at 60 °C for 12 h, and calcined at 500 °C for 4 h. Finally, the calcined catalyst was ground and screened to 180–425 μm prior to evaluation of the catalytic activity.
2.2 Catalyst Characterization and Analysis
N2 adsorption–desorption isotherms at − 196 °C were obtained on a Micrometrics ASAP 2020 HD88 analyzer. Before measurement, the samples were degassed under vacuum at 200 °C for 12 h. The crystal structure of the prepared catalysts was analyzed with X-ray power diffractometry (XRD, X’Pert MPD Pro, PANalytical) at its Cu Kα radiation of λ = 0.154 nm. The patterns were recorded with a scan angle range 10°–90° at a scanning speed of 8° min−1. The Ni loading on the supports was determined by X-ray fluorescence (XRF, AXIOX, PANalytical). Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) images were obtained on a JEOL JEM-2100 electron microscope operated at an acceleration voltage of 200 kV.
The temperature programmed reduction (TPR) and temperature programmed desorption (TPD) properties of the catalysts were determined using an Auto Chem II2920 instruments (Micrometrics) coupled with a mass spectrometer (MS, TILON, US). Prior to the measurements, the sample (~ 0.1 g) was heated from room temperature to 200 °C at a rate of 10 °C min−1, and then maintained for 1 h under a flow of He. After cooling the sample to 50 °C, the H2-TPR profile was recorded by increasing the temperature to 900 °C at a rate of 10 °C min−1 under a binary gas flow (10 vol.% H2/Ar). Prior to the CO2-TPD test, the catalyst (~ 0.1 g) was initially reduced in situ under a flow of H2/Ar at 600 °C for 2 h, and then cooled to 50 °C under an Ar atmosphere. The sample was then saturated with 10 vol.% CO2/Ar for 1 h and heated to 900 °C at a rate of 10 °C min−1 under a flow of He. The desorbed CO2 was detected simultaneously using a thermal conductivity detector (TCD) and by MS. For the H2-TPD measurements, the reduced sample (~ 0.1 g) was subjected to an atmosphere of 10 vol.% H2/He at 50 °C for 2 h prior to performing the measurements in a flow of He upon heating from 50 to 900 °C at a rate of 10 °C min−1. The hydrogen consumption was calculated based on the area of the hydrogen desorption peak. The Ni dispersion (D) and particle size (dNi) were calculated as follows:
where Y denotes the H2 adsorption capacity (mol g−1), M is the molecular weight of Ni (58.69 g mol−1), W is the Ni loading on the support (%), FSis the stoichiometric factor (H2/Ni = 1/2), σNi is the atomic cross-sectional area of Ni (0.0649 nm2), and ρNi is the density of Ni metal (8.902 g cm−3).
The X-ray photoelectron spectroscopy analysis was performed using an AXIS Ultra XPS spectrometer running at 15 kV and 15 mA with monochromomator Al Kα radiation (hν = 1486.7 eV). The binding energy (BE) values were related to the C 1s peak of contaminant carbon at 286.8 eV with an uncertainty of ± 0.2 eV. The obtained spectra were fitted using Gaussian–Lorentzian curves to more accurately extract the binding energies of the different element core levels.
2.3 Evaluation of the Catalytic Activity
The activity of the catalyst was evaluated at atmospheric pressure in a quartz fixed-bed reactor with an inner diameter of 16 mm. Before reaction, the catalyst (500 mg) was reduced under a stream of 10 vol.% H2/N2 (50 mL min−1) at 600 °C over 4 h. Then, the reactor was cooled down to reaction temperature under N2 flow (50 mL min−1), and a mixture gas of H2/N2/CO2 with volume ratio of 4/1/1 was introduced into the reactor and the gas hourly space velocity (GHSV) was 36,000 h−1. The reaction temperature was monitored by a thermocouple close to the bottom of the catalyst bed. The product composition was analyzed with a micro gas chromatograph (Micro3000, Agilent) equipped with TCD. The flow rates of H2, N2, and CO2 were controlled by mass flow meters, and N2 was used as an internal standard to calculate the volume flow of each component in the product. The CO2 conversion (XCO2) and CH4 selectivity (SCH4) were calculated with the following equations:
where the XCO2 and SCH4 is the CO2 conversion and CH4 selectivity; fin and fout is the molar feed rate of the import and export flow in the reactor; yCO2,in, yCO2,out, and yCH4,out is the volume fraction of the import and export of CO2 and CH4 in the reactor.
3 Results and Discussion
3.1 Characterization
Table 1 shows the main physic-chemical properties of the prepared Ni/CeO2 catalysts. The Ni/CeO2-nrs catalyst exhibited the highest surface area followed by the Ni/CeO2-nps and Ni/CeO2-ncs catalysts. As noted, a large surface area is conducive to the dispersion of active metals and the exposure of active sites [22].
TEM and EDX images of CeO2 supports with different morphologies or the corresponding Ni/CeO2 catalysts are presented in Fig. 1. Figure 1-A1 displays the CeO2 nanorods and the average length of these rods is about 100–200 nm while the diameter is approximate 15–20 nm. The interplanar spacing of 0.19 and 0.27 nm on the HRTEM images of an isolated nanosheet have lattice fringe directions attributed to {110} and {100}, as observed in Fig. 1-A2. In Fig. 1-B1, the CeO2-nps consists of nanoparticles with average diameter of about 9.5 nm. The corresponding HRTEM image (Fig. 1-B2) shows that the interplanar spacing of 0.31 nm has a lattice fringe direction attributed to {111} planes. For the CeO2 nanocubes (CeO2-ncs) shown in Fig. 1-C1, the particle size ranges from 15 to 100 nm, and the lattice structure is discerned with spacing of 0.27 nm (Fig. 1-C2) close to the lattice spacing measured for {100} planes. The TEM images of nickel-loading CeO2 catalysts shown in Fig. 1-A3, B3, and C3 indicate that there is no significant change in the morphology of CeO2 following the nickle loading. Furthermore, EDX-mapping in Fig. 1 (Fig. 1-A4, B4, and C4 for CeO2-nrs CeO2-nps and CeO2-ncs, respectively) could elucidate that most of Ni particles were incorporated in the CeO2 nanostructure in these three catalysts. The Ni/CeO2-nrs catalysts displayed the best Ni dispersion, and the elements Ni and Ce were uniformly distributed in the catalyst.

TEM and EDX images of CeO2 supports with different morphologies or the corresponding Ni/CeO2 catalysts
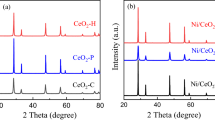
The XRD patterns of the CeO2 supports (Fig. 2a), and the calcined (Fig. 2b) and reduced (Fig. 2c) Ni/CeO2 catalysts are shown in Fig. 2. The peaks appeared at 28.5°, 33.1°, 47.5°, 56.3°, 59.1°, and 69.4° in Fig. 2a are attributed to the {111}, {200}, {220}, {311}, {222} and {400} planes of CeO2. No diffraction peaks of hexagonal crystal forms such as Ce(OH)3 and Ce(OH)CO3 are found, indicating the CeO2 supports exhibit good crystal purity [23]. The XRD patterns of the calcined Ni/CeO2 catalysts in Fig. 2b display additional diffraction peaks at 37.2°, 43.3° and 62.9°, corresponding to the {111}, {200}, and {220} planes of NiO, respectively. After reduced under a stream of 10 vol.% H2/N2 (50 mL min−1) at 600 °C for 4 h, the diffraction peaks of NiO in the catalysts disappear completely, while Ni peaks present at 44.6°, 51.9° and 76.5° corresponding to the {111}, {200} and {220} planes, respectively as shown in Fig. 2c. The crystal sizes of the metallic Ni calculated using the Scherrer-equation for the {200} plane at 51.9° are 8.2, 29.6, and 31.4 nm for Ni/CeO2-nrs, Ni/CeO2-nps and Ni/CeO2-ncs, respectively.

XRD profiles of CeO2 supports with different morphologies (a) and calcined (b) and reduced (c) Ni/CeO2 catalysts
The interaction between NiO and support in the catalysts was analyzed by H2-TPR (Fig. 3). The Ni/CeO2 catalysts mainly exhibit three reduction peaks. The peaks at 247–255 °C and 347–355 °C are attributed to the reduction of NiO. The former corresponds to the reduction of the bulk NiO that has no or weak interaction with CeO2, while the latter represents the reduction of the NiO that has strong interaction with support. In addition, the peak at 831–858 °C is attributed to the reduction of CeO2 [24]. The H2-TPR profiles show that the NiO particles in all Ni/CeO2 catalysts could be completely reduced to metallic nickel after reduced at 600 °C for 4 h in accordance with the result of XRD patterns in Fig. 2c.

H2-TPR profiles of Ni/CeO2 catalysts
The Ni/CeO2 catalysts were characterized by H2-TPD to explore their H2 adsorption capacity, and the results are shown in Fig. 4. The first peak before 400 °C belongs to the weakly active sites of the metal on the catalyst surface. The second peak after 400 °C is attributed to the active sites strongly chemisorbed on the surface. It can be found that the H2 desorption peaks of the three catalysts are mainly before 300 °C, and the H2 desorption area of Ni/CeO2-nrs is significantly higher than other two catalysts, which indicate that its H2 adsorption capacity is the strongest. The nickel dispersion and average nickel diameter calculated from the H2-TPD profiles are displayed in Table 1.The dispersion of active sites in the Ni-based catalysts follows an order of Ni/CeO2-nrs > Ni/CeO2-nps > Ni/CeO2-ncs.

H2-TPD profiles of Ni/CeO2 catalysts
The CO2 adsorption capacity plays an important role in maintaining the catalytic activity for CO2 methanation. Therefore, CO2-TPD of the CeO2 supports and Ni/CeO2 catalysts were carried out and the results are shown in Fig. 5. For CeO2 supports, the low-temperature desorption peak at 85–130 °C is attributed to the CO2 interacted with the weak basic sites on the surface (Fig. 5a). The broad adsorption peak between 260 and 294 °C is considered to be related to the moderate basic sites on the CeO2 [25,26,27]. The area of the peak attributed to the weak basic sites in CeO2-nrs is the largest indicating the CeO2 nanorods support is more conducive to CO2 adsorption at low temperatures. As shown in Fig. 5b, the desorption peaks corresponding to the weak and moderate basic sites overlaps between 80 and 300 °C after nickel loading. In addition, the Ni/CeO2-nrs catalyst exhibits the largest area of CO2 desorption peaks. The CO2 adsorptions are calculated to be 60, 25 and 19 μmol gcat−1 for Ni/CeO2-nrs, Ni/CeO2-nps, and Ni/CeO2-ncs, respectively.

CO2-TPD profiles of CeO2 supports with different morphologies (a) and Ni/CeO2 catalysts (b)
The electronic properties of the metal oxide and the adsorbed species on the catalysts also play an important role in the hydrogen and carbon dioxide activation. Figure 6 displays XPS spectra for the reduced Ni/CeO2 catalysts, revealing peaks assigned to O 1s, Ni 2p, and Ce 3d. Two obvious surface oxygen peaks are observed in the O 1s spectrum, where the binding energy at 529.1 ± 0.1 eV is assigned to lattice oxygen species (OL), and the peak at 530.9 ± 0.2 eV is characteristic of O2− in oxygen deficient regions (OV). The ratio of OV/OL can be used to represent the concentration of surface oxygen vacancies on CeO2 surface [19]. Table 2 shows that the ratio of OV/OL decreases in an order of Ni/CeO2-nrs > Ni/CeO2-nps > Ni/CeO2-ncs. It has been reported that CO2 molecules could be reduced by the oxygen deficient regions to form CO species to realize CO2 activation (CeO2−δ + CO2 → CeO2 + CO). 21.

X-ray photoelectron spetra of the reduced Ni/CeO2 catalysts
Peaks for different oxidation states of Ni in the reduced Ni/CeO2 catalysts were assigned based on Gaussian fitting and literatures [28, 29]. The split peak at 852.5 eV in the Ni 2p spectrum corresponds to metallic Ni, while the peak observed at 854.5, 856.1 and 857.8 eV corresponds to the binding energy of NiO which were strongly bonded with CeO2, and the accompanying peak appears at 861.1 eV. Table 2 shows that the ratio of Ni0/Ni2+ also follows an order of Ni/CeO2-nrs > Ni/CeO2-nps > Ni/CeO2-ncs. Kim et al. found that the content of Ni0 in the Ni/CeO2 catalyst correlates with the surface electron states of Ce, and it increased with the Ce3+ rising [30].
A complex peak pattern is shown and ten peaks are fitted for the Ce 3d spectrum. The v0 (879.6 eV), ν (881.8 eV), v′ (884.0 eV), v″ (888.6 eV), and v‴ (896.5 eV) labeled peaks in the figure refer to the spin–orbit coupling 3d5/2, while the μ0 (898.0 eV), μ (900.2 eV), μ′ (901.8 eV), μ″ (906.9 eV), and μ‴ (916.2 eV) peaks are associated with 3d3/2. The v‴, μ‴, v″, μ″, ν, and μ peaks are characteristics of Ce4+ corresponding to the Ce 3d94f0 O 2p6, Ce 3d94f1 O 2p5, and Ce 3d94f2 O 2p4 electrons, respectively. Meanwhile, the v′, μ′, v0 and μ0 peaks are characteristics of Ce3+ corresponding to the Ce 3d94f1 O 2p6 and Ce 3d94f2 O 2p5 electrons, respectively [31]. The ratio of the cumulative intensities of all peaks assigned to Ce3+ and the cumulative intensities of all peaks assigned to Ce4+ is a measure for the Ce3+/Ce4+ atomic ratio, as shown in Table 2. A higher ratio of Ce3+/Ce4+ indicates the presence of more oxygen vacancies, since the presence of oxygen vacancies maintains the electronic balance between Ce3+ and Ce4+ (Eq. 5) [27]. Some previous studies have shown that the presence of oxygen vacancies induces the production of O2− or OH groups on the surface due to adsorption of water and the CO2 adsorption capacity of a catalyst is related to the –OH and O2− species present on the support [32, 33]. Therefore, from Table 2 the Ni/CeO2-nrs catalyst contains a greater concentration of oxygen vacancies and Ni0, thereby rendering its CO2 adsorption capacity superior to those of the other two catalysts.
3.2 Catalytic Activity
The CO2 conversion and CH4 selectivity for CO2 methanation over the Ni/CeO2 catalysts at different temperatures are presented in Fig. 7, whereby the thermodynamic equilibrium values were calculated by minimizing the Gibbs free energy method. As shown in Fig. 7a, the CO2 conversion increases rapidly as raising the temperature from 250 to 400 °C, and then decreases gradually from 400 to 550 °C for all the three Ni/CeO2 catalysts. However, the CH4 selectivity decreases as increasing the temperature from 250 to 550 °C (Fig. 7b), because the water–gas shift reaction takes place at higher temperatures, which converts CO2 into CO instead of CH4 [34]. Meanwhile, as temperatures lower than 450 °C, the CO2 conversion follows an order of Ni/CeO2-nrs > Ni/CeO2-nps > Ni/CeO2-ncs. Especially, the CO2 conversion and CH4 selectivity at 300 °C are 60.1 and 96.9% for Ni/CeO2-nrs, respectively, while the CO2 conversions for the other two catalysts are both less than 10%. In addition, it is obvious that at lower temperatures (250–450 °C), the CO2 conversion is much lower than the equilibrium data which reveals that the CO2 methanation is subject to kinetic control.

CO2 conversion (a) and CH4 selectivity (b) for CO2 methanation over Ni/CeO2 catalysts
The promotion effect of oxygen vacancy on the catalytic activity of Ni-CeO2 catalyst in the CO2 methanation is also evaluated by the turnover frequency (TOF) values. To ignore the potential effect of mass and heat transfer, the TOF values was calculated at 250 °C (where CO2 conversion was less than 15%). The TOF of samples were calculated as moles of CH4 produced per mole of surface Ni atoms per second according to the methods reported [16, 35, 36]. From Fig. 8, it is observed that TOFs follow an order of Ni/CeO2-nrs (0.111 s−1) > Ni/CeO2-nps (0.078 s−1) > Ni/CeO2-ncs (0.066 s−1), confirming the CO2 conversion and CH4 selectivity in Fig. 7. The TOF values increase linearly with the concentration of oxygen vacancy in the catalyst, indicating the oxygen vacancy can improve the CO2 adsorption and activation, and then result in the improvement of the intrinsic activity of the catalyst.

The effect of surface oxygen vacancy (OV/OL) on turnover frequency (TOF) at 250 °C
3.3 Discussion
According to the results above, the Ni/CeO2-nrs catalyst exhibited well metal dispersion and CO2 adsorption capacity, thereby resulting in a significantly high CO2 conversion during CO2 methanation compared to the other two catalysts. Previous studies have shown [21] that the surface area of the support played an important role in the dispersion of active metals. However, the structure parameters affecting the CO2 adsorption capacity require further discussion.
The adsorption capacity of Ni-based catalysts toward CO2 is mainly related to their basic sites [37,38,39], whereby the type and concentration of basic sites are influenced by the oxygen vacancy present in the support [16]. Thus, the quantitative relationship between the oxygen vacancy and the specific surface area is investigated and the result is shown in Fig. 9. As indicated, the oxygen vacancy concentration and the Brauner–Emmett–Teller (BET) surface area are positively correlated, and the Ni/CeO2-nrs catalyst seems possessing the largest surface area and then the largest oxygen vacancy concentration.

The relationship between BET surface area and concentration of oxygen vacancy (OV/OL) in Ni/CeO2 catalysts
From Table 1, the surface area of the Ni/CeO2-nrs catalyst is approximately twice that of the Ni/CeO2-ncs catalyst. Therefore, an additional test was carried out to double the quality of Ni/CeO2-ncs for the CO2 methanation whereas with unchanged gas residence time with catalyst bed, and then the CO2 conversion and CH4 selectivity were compared with the performance of Ni/CeO2-nrs. As shown in Fig. 10, the CO2 conversion and CH4 selectivity over the Ni/CeO2-ncs catalyst are improved significantly and approach to that over Ni/CeO2-nrs in the temperature range of 250–550 °C. This result indicates that CO2 adsorption and activation capacity is rate-determining step for CO2 methanation, especially at low temperatures, because doubling the quantity of the Ni/CeO2-ncs catalyst could increase the total oxygen vacancy content for CO2 adsorption and activation, thereby further improve the CO2 conversion and CH4 selectivity.

CO2 conversion (a) and CH4 selectivity (b) for CO2 methanation over Ni/CeO2-ncs catalyst with different catalyst amount
4 Conclusions
Three Ni/CeO2 catalysts with different morphologies were prepared and the catalytic activity for CO2 methanation was evaluated. It was found that the Ni/CeO2 nanorods catalyst exhibited the highest CO2 conversion, especially at temperatures lower than 400 °C. At 300 °C, the CO2 conversion reached 60% compared to 10% achieved over Ni/CeO2 nanopolyhedrons and Ni/CeO2 nanocubes. However, catalytic activity of CO2 methanation over doubled quantity of Ni/CeO2 nanocubes catalyst approximately approached to that over Ni/CeO2 nanorods catalyst at the same gas residence time. Combining the characterizations, it could be concluded that the oxygen vacancy in the Ni/CeO2 catalyst, which is related with its morphology and surface area is conducive to the low-temperature adsorption of CO2, and then the catalytic activity for CO2 methanation.
References
Zhen W, Gao F, Tian B et al (2017) Enhancing activity for carbon dioxide methanation by encapsulating (111) facet Ni particle in metal–organic frameworks at low temperature. J Catal 348:200–211. https://doi.org/10.1016/j.jcat.2017.02.031
Ma Y, Liu J, Chu M et al (2020) Cooperation between active metal and basic support in Ni-based catalyst for low-temperature CO2 methanation. Catal Lett 150:1418–1426. https://doi.org/10.1007/s10562-019-03033-w
Hu L, Urakawa A (2018) Continuous CO2 capture and reduction in one process: CO2 methanation over unpromoted and promoted Ni/ZrO2. J CO2 Util 25:323–329. https://doi.org/10.1016/j.jcou.2018.03.013
Younas M, Loong KL, Bashir MJK et al (2016) Recent advancements, fundamental challenges, and opportunities in catalytic methanation of CO2. Energ Fuel 30:8815–8831. https://doi.org/10.1021/acs.energyfuels.6b01723
Li W, Zhang A, Jiang X et al (2017) Low temperature CO2 methanation: ZIF-67-derived Co-based porous carbon catalysts with controlled crystal morphology and size. Acs Sustain Chem Eng 5:7824–7831. https://doi.org/10.1021/acssuschemeng.7b01306
Danaci S, Protasova L, Lefevere J et al (2016) Efficient CO2 methanation over Ni/Al2O3 coated structured catalysts. Catal Today 273:234–243. https://doi.org/10.1016/j.cattod.2016.04.019
Muroyama H, Tsuda Y, Asakoshi T et al (2016) Carbon dioxide methanation over Ni catalysts supported on various metal oxides. J Catal 343:178–184. https://doi.org/10.1016/j.jcat.2016.07.018
Ma S, Tan Y, Han Y (2011) Methanation of syngas over coral reef-like Ni/Al2O3 catalysts. J Nat Gas Chem 20:435–440. https://doi.org/10.1016/S1003-9953(10)60192-2
Takenaka S, Shimizu T, Otsuka K (2004) Complete removal of carbon monoxide in hydrogen-rich gas stream through methanation over supported metal catalysts. Int J Hydrogen Energy 29:1065–1073. https://doi.org/10.1016/j.ijhydene.2003.10.009
Zyryanova MM, Snytnikov PV, Gulyaev RV et al (2014) Performance of Ni/CeO2 catalysts for selective CO methanation in hydrogen-rich gas. Chem Eng J 238:189–197. https://doi.org/10.1016/j.cej.2013.07.034
Sun J, Wang Y, Zou H et al (2019) Ni catalysts supported on nanosheet and nanoplate γ-Al2O3 for carbon dioxide methanation. J Energy Chem 29:3–7. https://doi.org/10.1016/j.jechem.2017.09.029
Tada S, Shimizu T, Kameyama H et al (2012) Ni/CeO2 catalysts with high CO2 methanation activity and high CH4 selectivity at low temperatures. Int J Hydrogen Energy 37:5527–5531. https://doi.org/10.1016/j.ijhydene.2011.12.122
Huang J, Li X, Wang X et al (2019) New insights into CO2 methanation mechanisms on Ni/MgO catalysts by DFT calculations: elucidating Ni and MgO roles and support effects. J CO2 Util 33:55–63. https://doi.org/10.1016/j.jcou.2019.04.022
Frontera P, Macario A, Ferraro M et al (2017) Supported catalysts for CO2 methanation: a review. Catalyst 7:59. https://doi.org/10.3390/catal7020059
Pan Q, Peng J, Sun T et al (2014) Insight into the reaction route of CO2 methanation: promotion effect of medium basic sites. Catal Commun 45:74–78. https://doi.org/10.1016/j.catcom.2013.10.034
Jia X, Zhang X, Rui N et al (2019) Structural effect of Ni/ZrO2 catalyst on CO2 methanation with enhanced activity. Appl Catal B Environ 244:159–169. https://doi.org/10.1016/j.apcatb.2018.11.024
Wang F, He S, Chen H et al (2016) Active site-dependent reaction mechanism over Ru/CeO2 catalyst toward CO2 methanation. J Am Chem Soc 138:6298–6305. https://doi.org/10.1021/jacs.6b02762
Mai H, Sun L, Zhang Y et al (2015) Shape-selective synthesis and oxygen storage behavior of ceria nanopolyhedra, nanorods, and nanocubes. J Phys Chem B 109:24380–24385. https://doi.org/10.1021/jp055584b
Sakpal T, Lefferts L (2018) Structure-dependent activity of CeO2 supported Ru catalysts for CO2 methanation. J Catal 367:171–180. https://doi.org/10.1016/j.jcat.2018.08.027
Varvoutis G, Lykaki M, Konsolakis M et al (2020) Remarkable efficiency of Ni supported on hydrothermally synthesized CeO2 nanorods for low-temperature CO2 hydrogenation to methane. Catal Commun 142:106036–106040. https://doi.org/10.1016/j.catcom.2020.106036
Zhou G, Liu H, Cui K et al (2016) Role of surface Ni and Ce species of Ni/CeO2 catalyst in CO2 methanation. Appl Surf Sci 383:248–252. https://doi.org/10.1016/j.apsusc.2016.04.180
Zhu P, Chen Q, Yoneyama Y (2014) Nanoparticle modified Ni-based bimodal pore catalysts for enhanced CO2 methanation. RSC Adv 4:64617–64624. https://doi.org/10.1039/C4RA12861C
Li L, Jiang B, Tang D et al (2018) Hydrogen production from chemical looping reforming of ethanol using Ni/CeO2 nanorod oxygen carrier. Catalysts 8:257. https://doi.org/10.3390/catal8070257
Damyanova S, Bueno JMC (2003) Effect of CeO2 loading on the surface and catalytic behaviors of CeO2-Al2O3-supported Pt catalysts. Appl Catal A Gen 253:135–150. https://doi.org/10.1016/S0926-860X(03)00500-3
Moghaddam SV, Rezaei M, Meshkani F et al (2018) Carbon dioxide methanation over Ni-M/Al2O3 (M: Fe, CO, Zr, La and Cu) catalysts synthesized using the one-pot sol–gel synthesis method. Int J Hydrogen Energy 43:16522–16533. https://doi.org/10.1016/j.ijhydene.2018.07.013
Yu Y, Chan YM, Bian Z et al (2018) Enhanced performance and selectivity of CO2 methanation over g-C3N4 assisted synthesis of Ni-CeO2 catalyst: kinetics and DRIFTS studies. Int J Hydrogen Energy 43:15191–15204. https://doi.org/10.1016/j.ijhydene.2018.06.090
Pino L, Vita A, Cipitì F et al (2011) Hydrogen production by methane tri-reforming process over Ni-ceria catalysts: effect of La-doping. Appl Catal B Environ 104:64–73. https://doi.org/10.1016/j.apcatb.2011.02.027
Li M, Veen AC (2018) Tuning the catalytic performance of Ni-catalysed dry reforming of methane and carbon deposition via Ni-CeO2-x interaction. Appl Catal B Environ 237:641–648. https://doi.org/10.1016/j.apcatb.2018.06.032
Chen X, Li M, Guan J et al (2012) Nickel-silicon intermetallics with enhanced selectivity in hydrogenation reactions of cinnamaldehyde and phenylacetylene. Ind Eng Chem Res 51:3604–3611. https://doi.org/10.1021/ie202227j
Kim MJ, Youn J, Kim HJ et al (2020) Effect of surface properties controlled by Ce addition on CO2 methanation over Ni/Ce/Al2O3 catalyst. Int J Hydrogen Energy 45:24595–24603. https://doi.org/10.1016/j.ijhydene.2020.06.144
Bêche E, Charvin P, Perarnau D et al (2008) Ce 3d XPS investigation of cerium oxides and mixed cerium oxide (CexTiyOz). Surf Interface Anal 40:264–267. https://doi.org/10.1002/sia.2686
Souza MMVM, Aranda DAG, Schmal M (2001) Reforming of methane with carbon dioxide over Pt/ZrO2/Al2O3 catalysts. J Catal 204:498–511. https://doi.org/10.1006/jcat.2001.3398
Drorouilly KJM, Averseng F et al (2013) Role of oxygen vacancies in the basicity of ZnO: from the model methylbutynol conversion to the ethanol transformation application. Appl Catal A Gen 453:121–129. https://doi.org/10.1016/j.apcata.2012.11.045
Stangeland K, Kalai DY, Li H et al (2018) Active and stable Ni based catalysts and processes for biogas upgrading: the effect of temperature and initial methane concentration on CO2 methanation. Appl Energy 227:206–212. https://doi.org/10.1016/j.apenergy.2017.08.080
Chen H, Mu Y, Shao Y et al (2019) Coupling non-thermal plasma with Ni catalysts supported on BETA zeolite for catalytic CO2 methanation. Catal Sci Technol 9:4135. https://doi.org/10.1039/C9CY00590K
Li LJ, Yi WJ, Liu TW et al (2017) Hydrogenation of 3-hydroxypropanal into 1,3-propanediol over bimetallic Ru–Ni catalyst. RSC Adv 7:32027–32037. https://doi.org/10.1039/C7RA01184A
Song F, Zhong Q, Yu Y et al (2017) Obtaining well-dispersed Ni/Al2O3 catalyst for CO2 methanation with a microwave-assisted method. Int J Hydrogen Energy 42:4174–4183. https://doi.org/10.1016/j.ijhydene.2016.10.141
Quindimil A, De-La-Torre U, Pereda-Ayo B et al (2018) Ni catalysts with La as promoter supported over Y- and BETA-zeolites for CO2 methanation. Appl Catal B Environ 238:393–403. https://doi.org/10.1016/j.apcatb.2018.07.034
Park JN, McFarland EW (2009) A highly dispersed Pd-Mg/SiO2 catalyst active for methanation of CO2. J Catal 266:92–97. https://doi.org/10.1016/j.jcat.2009.05.018
Acknowledgements
This work was supported by the Fund of International Science and Technology Cooperation Program of China (2019YFC1906802).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interest
There have no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ma, Y., Liu, J., Chu, M. et al. Enhanced Low-Temperature Activity of CO2 Methanation Over Ni/CeO2 Catalyst. Catal Lett 152, 872–882 (2022). https://doi.org/10.1007/s10562-021-03677-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-021-03677-7