
Abstract
The Pd-catalyzed direct arylation of pyrroles is an important research field for organic synthesis and catalysis chemistry. However, imidazolidin-2-ylidene based Pd-NHC complexes (NHC=N-heterocyclic carbene) have not yet been employed as catalysts for the direct C5 mono-arylation of C2-substituted N-methylpyrrole derivatives with aryl halides. Therefore, we now report the synthesis and characterization of new 1,3-bis(substituted benzyl) imidazolinium salts as carbene precursors, and their corresponding Pd-PEPPSI-NHC type complexes (PEPPSI=Pyridine Enhanced Precatalyst Preparation Stabilization and Initiation). The catalytic properties of these complexes have been evaluated in the direct C5 mono-arylation of N-methylpyrrole-2-carboxaldehyde with a wide variety of (hetero)aryl halides. This environmentally attractive procedure has also been found to be tolerant to a wide variety of functional groups on the aryl halides such as formyl, acetyl, nitrile, fluoro or trifluoromethyl, and good yields have been obtained in presence of 1 mol% catalyst loading at 120 °C.
Graphic Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Arylpyrroles are ubiquitous structural units widely found in a variety of pharmaceuticals, natural products, and functional materials [1,2,3,4,5,6]. Several synthetic arylpyrrole derivatives have been shown to possess interesting biological and biomedical properties [7,8,9]. For example, as shown in Fig. 1, Atorvastatin (A) is a member of the drug class known as statins, used for lowering blood cholesterol [10, 11], Tanaproget (B) is a progesterone-receptor agonist [12]. The 2-aryl-1H-pyrrole derivative C has been reported to act as lipoxygenase inhibitor [13]. The 2,5-diaryl-1H-pyrrole derivative D has been identified as p38 mitogen-activated protein (MAP) kinases inhibitor [14]. The 2,3-diaryl-1H-pyrrole derivative E has been reported as cyclooxygenase-2 (COX-2) selective inhibitor [15]. The pyridyl substituted 2,3,5-triaryl-1H-pyrrole derivative F has been reported to be a glucagon receptor agonist, which is able to block glucose production [16].

Selected examples of biologically active arylpyrrole framework
Due to the widespread biological applications of arylpyrroles, the development of new and more convenient synthetic procedures is a topic of ongoing interest in modern organic synthesis [17,18,19,20,21,22,23,24,25,26]. In this context, the classical Pd-catalyzed cross-couplings such as Suzuki–Miyaura, Stille or Negishi reactions play an important role in synthesis of pyrrole derivatives, and they allow the formation of a wide variety of arylpyrroles [27, 28]. These classical cross-couplings make possible either the coupling of aryl halides with organometallic derivatives of pyrroles or the coupling of halopyrroles with aryl-metal derivatives (that have a relatively high price, toxicity, and sensitivity to air and moisture). Nevertheless, these procedures require the preliminary preparation of an organometallic derivative of the pyrrole or of the aryl compound, which can be tricky, and produce stoichiometric amounts of metallic salts as by-products [29].
In recent years, Pd-catalyzed direct arylation of pyrroles by via C–H bond activation has become a versatile tool for the construction of arylpyrroles. This process has gained considerable recent momentum as a significantly environmentally and economically attractive alternative to classical Pd-catalyzed cross-coupling reactions [30, 31]. In 1985, Ohta et al. reported one of the first examples of the direct arylation of several heteroaromatics, including pyrroles with aryl halides [32, 33]. Since these results, the Pd-catalyzed intermolecular direct arylation of substituted [34,35,36,37,38,39,40,41], and non-substituted [42,43,44,45,46,47,48,49,50,51,52,53,54], pyrroles with aryl halides has proved to be a very powerful method for the synthesis of a wide variety of arylpyrroles. Despite the fact that Pd-catalyzed direct arylation of heteroaromatics have become invaluable for catalysis chemistry, and significant advances have been reported, N-heterocyclic carbene-based PEPPSI-type Pd-complexes (Pd-PEPPSI-NHCs) have been weakly applied as the catalysts in the direct arylation of heteroaromatics to date [55,56,57,58,59,60,61,62].
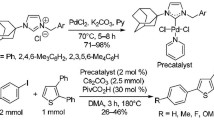
The first use of Pd-NHC complexes as a catalyst in the direct arylation of pyrroles was reported by Sames et al. in 2006 [43]. In this study, Sames described a mixed NHC/phosphine, [Pd(NHC)(PPh3)I2], complex (Pd-1) (NHC = imidazol-2-ylidene) as the catalyst (Fig. 2a), for the direct arylation of pyrroles with bromobenzene and aryl iodides. At about the same time, Sanford et al. reported [Pd(IMes)(OAc)2] complex (Pd-2), (Fig. 2b), featuring carboxylate ligands as the catalyst for the direct arylation of pyrroles with aryl iodonium salts [35]. These studies were performed with aryl iodides or bromides as the coupling partners. In 2012, Lee et al. reported only two examples of the direct arylation of pyrroles with aryl chlorides catalyzed by a highly electron-rich mixed NHC/phosphine, [Pd(NHC)(PCy3)Cl2], complex (Pd-3) bearing a functionalized NHC and a tricyclohexylphosphine (Fig. 2c) [49]. However, in 2013, our research group reported the first comprehensive study on the direct arylation of pyrroles with aryl chlorides catalyzed by [Pd(NHC)2X2] type complexes (Pd-4), (Fig. 2d) [50]. We found that Pd-(bis-NHC) complexes derived from the benzimidazol-2-ylidene scaffold were highly effective catalysts in the direct regioselective C2- or C5-arylation of a range of pyrrole derivatives using electron-deficient aryl chlorides. In 2014, Huynh et al. reported the synthesis and application of [Pd(NHC)2Br2] type complexes (Pd-5) (NHC = benzimidazol-2-ylidene), (Fig. 2e), with an alkyl thioether side chain in the direct arylation of N-methylpyrrole with aryl bromides [41]. In 2017, Yiğit et al. investigated [Pd(NHC)2Cl2] type complexes (Pd-6) (NHC = perhydrobenzimidazol-2-ylidene), (Fig. 2f), as the catalysts in the direct C2- or C5-arylation of pyrroles [51]. These complexes showed high catalytic activity using electronically activated aryl chlorides as the coupling partners.

Different-type of Pd-NHC precatalysts used for the direct arylation of pyrroles
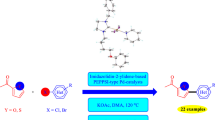
Up to now, in a limited number of studies, the direct arylation of heteroaromatics catalyzed by (benz)imidazol-2-ylidene based Pd-NHC complexes have been reported [35, 43, 49,50,51,52,53,54,55,56,57,58,59,60,61,62]. But, to the best of our knowledge, no reports are available on the direct arylation of pyrroles with aryl halides catalyzed by Pd-PEPPSI-NHC complexes bearing imidazolidin-2-ylidene ligand. In this regards, herein, we now report the synthesis and characterization of four 1,3-bis(para-substituted benzyl) imidazolinium salts (3a-3d) as carbene precursors, and their corresponding four [Pd(NHC)(Py)Br2] type complexes (4a-4d) as the catalysts. The catalytic properties of all Pd-PEPPSI-NHC complexes have been evaluated in the direct C5-arylation of the N-methylpyrrole-2-carboxaldehyde with aryl bromides and some unreactive aryl chlorides in presence of 1 mol% catalyst loading (Scheme 1). The C5-arylated pyrroles have been regioselectively obtained in moderate to high yields. To the best of our knowledge, this work is the first report of the direct C5-arylation of N-methylpyrrole-2-carboxaldehyde with aryl halides catalyzed by imidazolidin-2-ylidene based Pd-PEPPSI-NHC complexes.

Pd-PEPPSI-NHC catalyzed direct C5-arylation of N-methylpyrrole-2-carboxaldehyde with (hetero)aryl halides
2 Results and Discussion
2.1 General Synthesis
A convenient synthetic approach to imidazolinium salts 3a-3d and their palladium complexes 4a-4d was devised starting from synthesis of N-(2-(2-ethoxyphenoxy)ethyl)ethylenediamine 1. The general synthesis pathway of the compounds 1–4 are shown in the Scheme 2.

The synthetic route of imidazolinium salts (3a-3d), and their Pd-PEPPSI-NHC complexes (4a-4d)
2.2 Preparation and Characterization of N-(2-(2-Ethoxyphenoxy)ethyl)Ethylenediamine
It can be seen in Scheme 2, N-alkylation of the ethylenediamine with 2-(2-ethoxyphenoxy)ethyl bromide gave the N-(2-(2-ethoxyphenoxy)ethyl)ethylenediamine (1) as colourless viscous liquid in high yield (85%). The compound 1 was characterized by 1H NMR and 13C NMR and FT-IR spectroscopy, and elemental analysis studies. In the 1H NMR spectra of 1, the signal of the N-H protons of ethylenediamine were detected as splayed singlet at δ = 1.40 ppm with 3H intensity. In 13C NMR spectra, aliphatic carbon resonances of ethylenediamine were appeared at δ = 41.85 and 48.64 ppm as single signal. FT-IR data clearly indicated that N-(2-(2-ethoxyphenoxy)ethyl)ethylenediamine 1 exhibit a characteristic ν(C-N) and ν(N–H) band. In the FT-IR spectra, C–N bond vibration of compound 1 was observed as a sharp band at 1038 and 1248 cm−1. N–H bond vibration was also appeared as a broadband at 3296 cm−1. Elemental analysis data of the 1 was also consistent with the expected structure.
2.3 Preparation and Characterization of 1-(2-(2-ethoxyphenoxy)ethyl)imidazoline
The 1-(2-(2-ethoxyphenoxy)ethyl)imidazoline 2 was obtained by cyclization of the N-(2-(2-ethoxyphenoxy)ethyl)ethylenediamine 1 with N,N-dimethylformamide dimethyl acetal, as yellowish viscous liquid in high yield (96%). The formation of compound 2 was confirmed by the characteristic signals in 1H NMR and 13C NMR spectra. In 1H NMR spectra, C(2)-H proton downfield resonance of 2 was observed as sharp singlet at δ = 6.89 ppm. In 13C NMR spectra, C(2) carbon resonance of imidazoline ring was appeared at δ = 157.85 ppm, while C(4) and C(5) carbon resonances were observed at δ = 46.95 and 49.22 ppm. The FT-IR spectrum of compound 2 displays the characteristic ν(C=N) vibration band typically at 1603 cm−1. These data suggests the formation of imidazoline ring. Elemental analysis data was also consistent with the expected structure.
2.4 Preparation and Characterization of Imidazolinium Salts
The 1,3-bis(para-substituted benzyl) imidazolinium salts 3a-3d were synthesized as carbene precursors by interaction of the 1-(2-(2-ethoxyphenoxy)ethyl)imidazoline with para-substituted benzyl halides (Scheme 2). The reactions were carried out in anhydrous dimethylformamide (DMF) at 80 °C for 16 h, and the target salts were obtained as white solids in moderate to high yields. The imidazolinium salts were fully characterized by the combination of 1H NMR, 13C NMR, and IR spectroscopic techniques and elemental analyses. In the 1H NMR spectra, the signal of the acidic C(2)-H proton down-field resonance of imidazolinium ring for 3a-3d salts were observed as sharp singlets at δ = 10.03, 10.00, 10.23 and 9.95 ppm, respectively. Aliphatic CH2 protons of para-substituted benzyl substituents for imidazolinium salts 3a-3d were detected as singlet at δ = 4.79, 4.88, 4.72 and 4.77 ppm, respectively. In the 13C NMR spectra, C(2)-carbon resonances of the 3a-3d salts appeared at δ = 158.85, 159.14, 159.24 and 158.70 ppm, respectively as singlet. These downfield signals indicates the formation of imidazolinium salts. Also, aliphatic carbon resonances of para-substituted benzyl substituents for the 3a-3d salts were detected at δ = 50.31, 50.31, 50.15 and 50.26 ppm, respectively. The IR data clearly indicated that the imidazolinium salts 3a-3d exhibit a characteristic vC(2)-N vibration band typically between 1644-1645 cm−1. Elemental analysis data were also consistent with the expected structures.
2.5 Preparation and Characterization of Pd-PEPPSI-NHC Complexes
The Pd-PEPPSI-NHC complexes 4a-4d were prepared by metallation of the corresponding 1,3-bis(substituted benzyl) imidazolinium salts (3a-3d) with PdCl2. The reactions were carried out in presence of pyridine as N-donor ligand in acetonitrile (MeCN) at 80 °C for 16 h, and the target complexes were obtained as air-stable, yellowish solids between 39 and 50% yields. Pd-PEPPSI-NHC complexes 4a-4d are soluble in most organic solvents, such as CH2Cl2, CHCl3, EtOAc and DMSO, with the exception of non-polar ones, such as pentane, hexane and Et2O. Formation of Pd-NHC complexes is supported by NMR, IR spectroscopies and elemental analysis techniques. In the 1H NMR spectra of complexes 4a-4d, the characteristic down-field signals for the acidic C(2)-H protons of the imidazolinium salts 3a-3d disappeared in the 1H NMR spectra of the Pd-PEPPSI-NHC complexes. In addition, the down-field signals of pyridine ligand between δ = 7.24–9.00 ppm in the 1H NMR spectra indicates the formation of pyridine coordinated Pd-PEPPSI-NHC complexes. 13C NMR chemical shifts provide a useful diagnostic tool for Pd-carbene complexes. In the 13C NMR spectra of complexes 4a-4d, characteristic signal of C(2)-carbon of imidazolinium salts 3a-3d between δ = 158.70–159.24 ppm were completely disappeared, and the characteristic Pd-C(carbene) bond signals of the complexes 4a-4d were observed as singlet. In the 13C NMR spectra, the carbene signals of the Pd-PEPPSI-NHC complexes 4a-4d were observed at δ = 180.89, 181.51, 180.88 and 180.86 ppm, respectively. Also, characteristic down-field signals of the aromatic carbons of the pyridine ligand supports the formation of pyridine coordinated Pd-PEPPSI-NHC complexes. The IR data clearly indicated that, complexes 4a-4d exhibit a characteristic v(CN) stretching frequency peaks typically between 1591 and 1594 cm−1. Due to the electrons donation from the imidazolidin-2-ylidene ligand to the palladium centre, the C-N bond is weakened, and as a result, a decreasing in the v(CN) stretching frequency is expected. Also, the microanalysis data of the Pd-PEPPSI-NHC complexes agrees closely with the theoretical requirements of their structures.
2.6 Optimization of the Reaction Conditions for the Direct Arylation of N-Methylpyrrole-2-Carboxaldehyde
To test the applicability of Pd-PEPPSI-NHC complexes on the direct arylation of pyrroles, as can be seen in the Eq. 1, the reaction of N-methylpyrrole-2-carboxaldehyde with 3-bromoquinoline was examined as a model reaction. Then, we directed our efforts towards the Pd-PEPPSI-NHC catalyzed direct C5-arylation of N-methylpyrrole-2-carboxaldehyde. Last two decades, the Pd-catalyzed direct arylation of pyrroles was successfully performed using N,N-dimethylacetamide (DMA) and potassium acetate (KOAc) combination [40, 46, 48, 50,51,52,53,54,55,56,57,58,59,60,61,62]. Based on previously reported conditions for the Pd-catalyzed direct arylation of pyrroles, we employed KOAc as the base and DMA as the solvent in this study. The effect of the temperature, reaction time and catalyst loading were examined on the reaction. Yields were calculated with respect to 3-bromoquinoline from the GC results. Selected results from our preliminary studies are summarized in Table 1.

As can be seen in the Eq. 1, such arylations are known to occur preferentially at the α-positions to the nitrogen atom following the typical reactivity profiles of the pyrrole ring. Thus, under the direct arylation conditions C2-substituted pyrroles such as N-methylpyrrole-2-carboxaldehyde react at C5-position. To prove the arylation at the C5-position, we made also chemical characterizations of some well-known products in the literature [49, 51, 65] by NMR (see ESI file, pages S21-S25). In all case, we observed regioselective mono-arylation on only C5-position of N-methylpyrrole-2-carboxaldehyde in our preliminary studies, because the hydrogens at the C5-position of the N-methylpyrrole-2-carboxaldehyde is more reactive than C3- and C4-positions [63].
The arylation of N-methylpyrrole-2-carboxaldehyde with 3-bromoquinoline was carried out at 150 °C for 4 h without the addition of any Pd-catalyst in order to examine the effect of the catalyst on the reaction. However, under this conditions, no formation of the 5-(quinolin-3-yl)-1-methyl-2-formylpyrrole, 5i was obtained (Table 1, entry 1). In order to identify the most active catalyst among the Pd-PEPPSI-NHC complexes 4a-4d, reactions were carried out at 150 °C in precence of 1 mol% catalyst loading. In the presence of 4a-4d catalysts, 84%, 80%, 87% and 94% yields were observed, respectively (Table 1, entries 2–5). As a result of these preliminary studies, it was observed that the most active catalyst was complex 4d with 94% yield under same conditions (Table 1, entry 5). It was observed that the catalytic activity of complexes 4a-4d was enhanced by having sterically hindered and bulky groups on the NHC ligand. Then, well-known palladium complexes such as [Pd(PPh3)4] and PdCl2 were used as catalysts for comparison with the 4a-4d complexes. When [Pd(PPh3)4] complex was used under the same conditions, 80% yield was observed (Table 1, entry 6). However, when PdCl2 complex without any phosphine or NHC ligands was used, 56% yield was observed (Table 1, entry 7). When in situ generated palladium/NHC catalytic system with 2 mol% of NHC precursor 3d and 1 mol% of PdCl2 was used at 150 °C for 4 h, only 77% yield was observed (Table 1, entry 8). Thus, it was understood that isolated Pd-PEPPSI-NHC complex 4d was more active than in situ generated palladium/NHC catalytic system. Later, the effect of temperature on the yield was examined. When the temperature is reduced from 150 to 120 °C in presence of 1 mol% catalyst 4d, the yield decreased to 94% after 4 h (Table 1, entry 9). However, this decrease on the yield is within acceptable limits. When the reaction temperature was decreased from 120 to 90 °C, it was observed that the yield decreased up to 65% after 4 h (Table 1, entry 10). At this temperature, the reaction did not take place in satisfactory yield. Therefore, it was decided that the optimum temperature for the model reaction was 120 °C. Next, the effect of the reaction time on the yield was examined. When the reaction time was regularly reduced from 4 to 1 h, no significant difference was observed on the yield (Table 1, entries 11–13). But, when the reaction time was reduced from 1 to 30 min., the yield dropped to 50% (Table 1, entry 14). Therefore, the optimum reaction time for the model reaction was decided to be 1 h. Finally, the effect of catalyst-loading on the yield was also investigated. When the catalyst-loading was decreased from 1 to 0.5 mol% at 120 °C, only 64% yield was achieved after 1 h (Table 1, entry 15).
After these preliminary studies summarized in Table 1, we tried, to evaluate the scope and limitations of the synthesized Pd-PEPPSI-NHC complexes 4a-4d as the catalysts for the direct C5-arylation of N-methylpyrrole-2-carboxaldehyde with (hetero)aryl bromides bearing electron-withdrawing groups at the para- or ortho-position (Eq. 2), and even some unreactive aryl chlorides (Eq. 3). A wide range of functional groups on the (hetero)aryl halides such as aldehyde, acetyl, nitrile, fluoro and trifluoromethyl were well tolerated in presence of 1 mol% catalyst loading at 120 °C. The results of 4a-4d catalyzed direct C5-arylation of N-methylpyrrole-2-carboxaldehyde with (hetero)aryl bromides are summarized in Table 2.

Initially, under the optimal condition, the reaction of N-methylpyrrole-2-carboxaldehyde with a neutral aryl bromide such as bromobenzene was examined. In presence of Pd-complexes 4c and 4d, we observed good yields of the target product, 5-phenyl-1-methyl-2-formylpyrrole, 5a [64], (Table 2, entries 3 and 4). The reaction of N-methylpyrrole-2-carboxaldehyde with an electron-rich aryl bromide such as p-bromotoluene generated the 5-(4-methylphenyl)-1-methyl-2-formylpyrrole, 5b [65], in 90% yield in the presence of 4d catalyst after 2 h. In the presence of para-substituted electron-deficient aryl bromide such as 4-bromobenzaldehyde, the expected compound, 5-(4-formylphenyl)-1-methyl-2-formylpyrrole, 5c [51, 65], was obtained in moderate to high yields using only 1 mol% catalyst after 1 h (Table 2, entries 9–12). This compound was obtained in 70% isolated yields using 4d catalyst (Table 2, entry 12). The coupling of 1-methylpyrrole-2-carboxaldehyde with electron-poor aryl bromide such as 4-bromoacetophenone proceeds nicely. 4-Bromoacetophenone gave the 5-(4-acetylphenyl)-1-methyl-2-formylpyrrole, 5d [49], with moderate to high yields (Table 2, entries 13–16). In the presence of 4d catalyst, 78% isolated yield was obtained (Table 2, entry 16). The poorly activated 4-fluorobromobenzene was also a good substrate to afford the desired products 5-(4-fluorophenyl)-1-methyl-2-formylpyrrole, 5e [65], at between 52 and 83% yields (Table 2, entries 17–20). The reaction of 1-methylpyrrole-2-carboxaldehyde with 4-bromobenzotrifluoride gave the 5-(4-trifluoromethylphenyl)-1-methyl-2-formylpyrrole, 5f [49, 65], in 65% isolated yield in the presence of 4d catalyst (Table 2, entry 24). When sterically hindered electron-donating 2-bromotoluene was used as aryl halide, 70% yield of the 5-(2-methylphenyl)-1-methyl-2-formylpyrrole, 5 g [49], was obtained in the presence of 4d catalyst after 4 h (Table 2, entry 28). 2-Bromobenzonitrile gave the 2-(5-formyl-1-methylpyrrol-2-yl)-benzonitrile, 5 h [51, 65], with moderate to high yields after 4 h (Table 2, entries 29–32). This product was obtained in 68% isolated yield in the presence of 4d catalyst (Table 2, entry 32). Then, we examined the reactivities of electron-deficient heterocycles such as 3-bromoquinoline and 2-bromothiophene as heteroaryl bromides. A selective reaction was observed using 3-bromoquinoline. With this substrate, the target product 5-(quinolin-3-yl)-1-methyl-2-formylpyrrole, 5i [65], was obtained in 80% isolated yield in presence of 4d catayst after 1 h (Table 2, entry 36). When 2-bromothiophene was used as the heteroaromatic coupling partner, 72% yield of 5-(thiophene-2-yl)-1-methyl-2-formylpyrrole, 5j [66], was obtained in the presence of 4d catalyst after 2 h (Table 2, entry 40).
The direct arylation of heteroaromatics with aryl chlorides, and especially with pyrroles, is still a very challenging reaction. However, we observed that the reaction of N-methylpyrrole-2-carboxaldehyde with five aryl chloride derivatives in presence of catalyst 4d gave expected products in good yields (Eq. 3). But, we had to increase the reaction time to 15 h. The results of 4d catalyzed direct C5-arylation of N-methylpyrrole-2-carboxaldehyde with aryl chlorides are summarized in Table 3.

When chlorobenzene was used in the presence of 4d catalyst, which is the most active catalyst, 5a was obtained in 75% yield after 15 h (Table 3, entry 1). However, when p-chlorotoluene was used, 5b was obtained in 63% yield (Table 3, entry 2). When 4-chlorobenzaldehyde was used as the coupling partner, 5c was obtained in 87% yield after 15 h (Table 3, entry 3). High yields of expected C5-arylated product 5d was obtained for the coupling with 4-chloroacetophenone by using catalysts 4d (Table 3, entry 4). The coupling of the electron-deficient aryl chloride, 4-chlorobenzotrifluoride, with N-methylpyrrole-2-carboxaldehyde also proceeded to give 5f. However, when 4-chlorobenzotrifluoride was used in the presence of 4d catalyst, high yields could not be obtained despite the 15 h reaction (Table 3, entry 5).
As a results, we investigated the catalytic activities of Pd-PEPPSI-NHC complexes 4a-4d in the direct C5-arylation of N-methylpyrrole-2-carboxaldehyde with (hetero)aryl halides. The Pd-catalyzed direct C5-arylation of C2-substituted pyrroles with a wide variety of aryl halides has been previously reported by many groups [34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51]. But, to the best of our knowledge, this work is the first report of the direct C5-arylation of N-methylpyrrole-2-carboxaldehyde with aryl halides catalyzed by Pd-PEPPSI complexes based on saturated imidazolidin-2-ylidene ligand. In some cases, palladium complexes based on saturated imidazolidin-2-ylidene ligand for the direct C-H arylation of heteroaromatics are known to exhibit higher efficiency than their analogue palladium complexes based on unsaturated imidazol-2-ylidene ligand [56, 67]. Also, in the previous works, palladium complexes containing unsaturated ring NHC ligands such as benzimidazol-2-ylidene or imidazol-2-ylidene have been used as the catalyst, but palladium complexes using saturated ring NHC ligands such as imidazolidin-2-ylidene are highly limited. For example, Sames’s imidazol-2-ylidene based [Pd(NHC)(PPh3)I2] complex (Pd-1) was used for C(sp2)-H arylation of SEM-protected pyrrole-2-carbonitrile (SEM = trimethylsilylethoxymethyl) with aryl iodides in DMA at 125 °C (Scheme 3, a) [43]. Lee’s imidazol-2-ylidene based [(NHC)Pd(PCy3)Cl2] complex (Pd-3) was used for the direct C5−H arylation of N-methylpyrrole-2-carboxaldehyde with aryl chlorides in DMA at 110 °C (Scheme 3, b) [49]. Huynh’s benzimidazol-2-ylidene based [Pd(NHC)2Br2] complexes with an alkyl thioether side chain (Pd-5) were used in the direct C(sp2)−H arylation of N-methylpyrrole (Scheme 3, c) [41]. Our benzimidazol-2-ylidene based [Pd(NHC)2X2]-type complexes (Pd-4) were used for the direct C5-arylation of N-methylpyrrole-2-carboxaldehyde with aryl chlorides in DMA at 150 °C (Scheme 3, d) [50]. Also, in the most of the reported works, similar substrates have been employed with high catalyst loading [44, 45, 47, 48], and higher reaction time have been chosen for both aryl bromides and aryl chlorides [42, 44, 45, 47, 48]. But, in the present work the catalyst loading was reduced to 1 mol%, and the reaction time was shortened to 1–4 h for (hetero)aryl bromides, and 15 h for aryl chlorides (Scheme 3, e). Moreover, in the present study N-methylpyrrole-2-carboxaldehyde can be regioselectively arylated at the C5-position, and satisfactory results were obtained.

Comparison of the previous works with this work for the direct arylation of N-fused-pyrroles
The abundance of para-substituents on the N-benzyl moieties on the imidazolin-2-ylidene ligands were found to play a key role in governing the efficiencies of catalysts. Small differences on the reactivities of the catalysts 4a-4d were observed due to similar nature of the NHC moieties. However, it can be said that the complex 4d bearing NHC ligands with 2-(2-ethoxyphenoxy)ethyl and 4-tert-butylbenzyl substituents exhibited better catalytic activity than the others. We attributed these performance differences to well-accordance electronic and steric properties of the NHC ligand. It is known that oxidative additions of electron-withdrawing substrates to electron rich Pd-complexes and reductive elimination of the product from large, sterically hindered Pd-complexes proceed more readily. Therefore, the presence of an NHC ligand bearing a different second donating group such as ether side chains on the metal may radically increase the catalytic performance of the catalyst. The chelating nature of these ligands promotes production of highly stable complexes. The hemilabile part of such ligands is capable of reversible dissociation to produce vacant coordination sites, allowing complexation of substrates during the catalytic cycle. At the same time the strong-donor carbene moiety remains connected to the metal centre. In this arylation, we believe that the bulky and electron-donor NHC ligands bearing 2-(2-ethoxyphenoxy)ethyl substituents in complexes 4a-4d provide the synergetic steric and electronic effects to confer the metal center the appropriate properties to make optimum for the key steps of the catalytic cycles.
3 Conclusion
In summary, we prepared new 1,3-bis(para-substituted benzyl) imidazolinium salts as carbene precursors, and their Pd-PEPPSI-NHC type complexes in this work. These Pd-PEPPSI-NHC complexes were tested as catalysts for the direct C5-arylation of N-methylpyrrole-2-carboxaldehyde with a wide variety of (hetero)aryl bromides and some aryl chlorides. Under the tested conditions, the direct arylation efficiently proceeded in moderate to high yields in the presence of 1 mol% catalyst loading. Furthermore, this catalytic system exhibits a high functional group tolerance and a broad substrate scope. Also, it revealed that the NHC ligands with sterically bulky backbone on N-benzyl moieties, played a crucial role in the catalytic performance. Finally, the Pd-catalyzed direct arylation represents an effective way to carry out the arylation of pyrroles, a task that otherwise requires several synthetic steps or harsh reaction conditions. Therefore, further studies focused on the sythesis of new imidazolidin-2-ylidene ligands and their different type Pd-complexes, and their catalytic application in the direct arylation of heteroarenes as catalysts are currently underway by our research group.
4 Experimental
4.1 General Remarks
All manipulations were performed in Schlenk-type flasks under argon atmosphere. The melting point measurements were determined in open capillary tubes with an Electrothermal-9200 melting points apparatus. The C, H and N elemental analysis measurements were determined by LECO CHNS-932 elemental analyser. The IR spectra were recorded on GladiATR unit (Attenuated Total Reflection) in the range of 450–4000 cm−1 with a Perkin Elmer Spectrum 100 fourier-transform infrared spectrometer. Routine 1H NMR and 13C NMR spectra were recorded with a Bruker Ascend™ 400 Avance III HD NMR spectrometer with sample solutions prepared in CDCl3. The chemical shifts (δ) were reported in parts per million (ppm) relative to tetramethylsilane (TMS) as internal standard. Coupling constants (J values) were given in hertz (Hz). NMR multiplicities were abbreviated as follows: s = singlet, d = doublet, t = triplet, p = pentet, dd = doublet of doublets, tt = triplet of triplets, ddd = doublet of doublet of doublets, m = multiplet. 1H NMR spectra were referenced to residual protiated solvents (δ = 7.28 ppm for CDCl3), 13C NMR chemical shifts were reported relative to deuterated solvents (δ = 77.16 ppm for CDCl3). The catalytic solutions were analyzed with a Shimadzu GC 2025 equipped with GC-FID sensor and RX-5 ms column of 30 m length, 0.25 mm diameter and 0.25 μm film thickness.
4.2 Preparation of N-(2-(2-Ethoxyphenoxy)Ethyl)Ethylenediamine (1)
The N-(2-(2-ethoxyphenoxy)ethyl)ethylenediamine (1) was prepared by the N-alkylation of the ethylenediamine with 2-(2-ethoxyphenoxy)ethyl bromide. Lithium (0.287 g; 40.8 mmol) was in small pieces added in freshly distilled and dried ethylenediamine (30 mL) under argon atmosphere at 110 °C. The solution, which was stirred for 1 h, was then cooled to room temperature and 2-(2-ethoxyphenoxy)ethyl bromide (10 g; 40.8 mmol) was added to this solution. Then, anhydrous toluene (30 mL) was added to solution. After 1 min, lithium bromide precipitate started to form. The mixture was stirred for a further 1 h at 110 °C, and it was then cooled to room temperature. The precipitated lithium bromide was removed by filtered off, and was washed with 20 mL of toluene. Then, all volatiles were removed under vacuum. The crude product was distilled under reduced vacuum. The N-(2-(2-ethoxyphenoxy)ethyl)ethylenediamine (1) was isolated as colourless gel in 85% yield.
Yield 85%, 7.760 g (colourless gel); bp: 80–85 °C (under ~ 50 Torr pressure); FT-IR (vC-N): 1038 and 1248 cm−1; (vN-H): 3296 cm−1. 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 1.34 (t, J = 7.0 Hz, 3H, NCH2CH2OC6H4(OCH2CH3)-2); 1.40 (splayed singlet, 3H, N-Hs of ethylenediamine); 2.64–2.68 and 2.72–2.75 (m, 4H, NCH2CH2N); 2.93 (t, J = 5.3 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 3.98 (q, J = 7.0 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 4.02 (t, J = 5.3 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 6.77–6.85 (m, 4H, arom. Hs of NCH2CH2OC6H4(OCH2CH3)-2). 13C NMR (101 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 14.93 (NCH2CH2OC6H4(OCH2CH3)-2); 41.85 and 48.64 (NCH2CH2N); 52.41 (NCH2CH2OC6H4(OCH2CH3)-2); 64.40 (NCH2CH2OC6H4(OCH2CH3)-2); 69.13 (NCH2CH2OC6H4(OCH2CH3)-2); 113.66, 114.86, 120.98, 121.64, 148.69 and 149.20 (arom. Cs of NCH2CH2OC6H4(OCH2CH3)-2). Elemental analysis calcd. (%) for C12H20N2O2: C 64.26, H 8.99, N 12.49; found (%): C 64.94, H 8.62, N 12.75. (For the 1H NMR, 13C NMR and FT-IR spectrum of the N-(2-(2-ethoxyphenoxy)ethyl)ethylenediamine (1), see ESI file, pages S1-S2).
4.3 Preparation of 1-(2-(2-Ethoxyphenoxy)Ethyl)Imidazoline (2)
The 1-(2-(2-ethoxyphenoxy)ethyl)imidazoline (2) was prepared by the cyclization of the N-(2-(2-ethoxyphenoxy)ethyl)ethylenediamine (1) with N,N-dimethylformamide dimethyl acetal. For the preparation of compound 2, the N-(2-(2-ethoxyphenoxy)ethyl)ethylenediamine (1) (7.76 g; 34.6 mmol) was reacted with N,N-dimethylformamide dimethyl acetal (3.35 g; 38.0 mmol) at 90–110 °C for 3 h. End of the reaction, unreacted excess acetal were removed under vacuum. Then, the crude product was distilled under reduced vacuum. The 1-(2-(2-ethoxyphenoxy)ethyl)imidazoline (2) was isolated as yellowish gel in 96% yield.
Yield 96%, 7.733 g (yellowish gel); bp: 100–105 °C (under ~ 50 Torr pressure); FT-IR (vC-N): 1039 and 1250 cm−1; (vC=N): 1603 cm−1. 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 1.36 (t, J = 7.0 Hz, 3H, NCH2CH2OC6H4(OCH2CH3)-2); 3.29 (t, J = 9.8 Hz, 2H, NCH2CH2N); 3.44 (t, J = 5.2 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 3.73 (td, J = 9.8, 1.6 Hz, 2H, NCH2CH2N); 3.97 (q, J = 7.0 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 4.02 (t, J = 5.2 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 6.79–6.87 (m, 4H, arom. Hs of NCH2CH2OC6H4(OCH2CH3)-2); 6.89 (s, 1H, NCHN). 13C NMR (101 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 14.94 (NCH2CH2OC6H4(OCH2CH3)-2); 46.95 and 49.22 (NCH2CH2N); 55.12 (NCH2CH2OC6H4(OCH2CH3)-2); 64.32 (NCH2CH2OC6H4(OCH2CH3)-2); 67.80 (NCH2CH2OC6H4(OCH2CH3)-2); 113.90, 114.46, 120.85, 121.95, 148.24 and 149.20 (arom. Cs of NCH2CH2OC6H4(OCH2CH3)-2); 157.85 (NCHN). Elemental analysis calcd. (%) for C13H17N2O2: C 66.93, H 7.35, N 12.01; found (%): C 67.28, H 7.78, N 12.21. (For the 1H NMR, 13C NMR and FT-IR spectrum of the 1-(2-(2-ethoxyphenoxy)ethyl)imidazoline (2), see ESI file, pages S3-S4).
4.4 General Procedure for the Preparation of Imidazolinium Salts as Carbene Precursors (3a-3d)
The imidazolinium salts (3a-3d) were prepared by interaction of the 1-(2-(2-ethoxyphenoxy)ethyl)imidazoline (2) with para-substituted benzyl halides. The 1-(2-(2-ethoxyphenoxy)ethyl)imidazoline (1,17 g; 5,0 mmol) and para-substituted benzyl halide (5.0 mmol) were dissolved in degassed DMF (5 mL), and the solution was stirred at 80 °C for 16 h. After completion of the reaction, the mixture was allowed to cool to room temperature, and diethyl ether (15 mL) was added to solution. Then, obtained white solid was filtered off, was washed with diethyl ether (3 × 10 mL), and dried under vacuum. The crude product was recrystallized from EtOH/Et2O solvent system (1:5, v/v) at room temperature, and completely dried under vacuum. The new imidazolinium salts (3a-3d) were isolated as air- and moisture-stable white crystalline solids in moderate to high yields.
4.4.1 1-(2-(2-Ethoxyphenoxy)Ethyl)-3-(4-Methylbenzyl)Imidazolinium Bromide, 3a
Yield 67%, 1.413 g (white solid); mp: 99–100 °C; FT-IR (vC-N): 1040 and 1254 cm−1; (vC(2)-N): 1644 cm−1. 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 1.36 (t, J = 7.0 Hz, 3H, NCH2CH2OC6H4(OCH2CH3)-2); 2.35 (s, 3H, NCH2C6H4(CH3)-4); 3.79 (t, J = 11.5 Hz, 2H, NCH2CH2N); 4.02 (q, J = 7.0 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 4.17 (t, J = 4.4 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 4.27 (t, J = 10.6 Hz, 2H, NCH2CH2N); 4.29 (t, J = 4.6 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 4.79 (s, 2H, NCH2C6H4(CH3)-4); 6.86–7.00 (m, 4H, arom. Hs of NCH2CH2OC6H4(OCH2CH3)-2); 7.19 (d, J = 7.9 Hz, 2H, arom. Hs of NCH2C6H4(CH3)-4); 7.28 (d, J = 8.0 Hz, 2H, arom. Hs of NCH2C6H4(CH3)-4); 10.03 (s, 1H, NCHN). 13C NMR (101 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 15.02 (NCH2CH2OC6H4(OCH2CH3)-2); 21.18 (NCH2C6H4(CH3)-4); 47.82 and 48.05 (NCH2CH2N); 50.31 (NCH2C6H4(CH3)-4); 52.23 (NCH2CH2OC6H4(OCH2CH3)-2); 63.96 (NCH2CH2OC6H4(OCH2CH3)-2); 67.82 (NCH2CH2OC6H4(OCH2CH3)-2); 113.02, 114.79, 121.08, 122.58, 147.26 and 148.84 (arom. Cs of NCH2CH2OC6H4(OCH2CH3)-2); 128.84, 129.23, 129.95 and 139.12 (arom. Cs of NCH2C6H4(CH3)-4); 158.85 (NCHN). Elemental analysis calcd. (%) for C21H27BrN2O2: C 60.15, H 6.49, N 6.68; found (%): C 60.25, H 6.67, N 6.92. (For the 1H NMR, 13C NMR and FT-IR spectrum of the imidazolinium salt 3a, see ESI file, pages S5-S6).
4.4.2 1-(2-(2-Ethoxyphenoxy)Ethyl)-3-(4-Chlorobenzyl)Imidazolinium Bromide, 3b
Yield 67%, 1.482 g (white solid); mp: 110–111 °C; FT-IR (vC-N): 1040 and 1255 cm−1; (vC(2)-N): 1644 cm−1. 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 1.35 (t, J = 7.0 Hz, 3H, NCH2CH2OC6H4(OCH2CH3)-2); 3.81 (t, J = 11.3 Hz, 2H, NCH2CH2N); 4.01 (q, J = 6.9 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 4.13 (t, J = 4.5 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 4.26 (t, J = 4.6 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 4.28 (t, J = 11.8 Hz, 2H, NCH2CH2N); 4.88 (s, 2H, NCH2C6H4(Cl)-4); 6.85–6.99 (m, 4H, arom. Hs of NCH2CH2OC6H4(OCH2CH3)-2); 7.34 (d, J = 8.3 Hz, 2H, arom. Hs of NCH2C6H4(Cl)-4); 7.41 (d, J = 8.4 Hz, 2H, arom. Hs of NCH2C6H4(Cl)-4); 10.00 (s, 1H, NCHN). 13C NMR (101 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 15.05 (NCH2CH2OC6H4(OCH2CH3)-2); 47.96 and 48.15 (NCH2CH2N); 50.31 (NCH2C6H4(Cl)-4); 51.64 (NCH2CH2OC6H4(OCH2CH3)-2); 63.97 (NCH2CH2OC6H4(OCH2CH3)-2); 67.61 (NCH2CH2OC6H4(OCH2CH3)-2); 113.02, 114.83, 121.06, 122.63, 147.21 and 148.84 (arom. Cs of NCH2CH2OC6H4(OCH2CH3)-2); 129.45, 130.42, 131.02 and 135.11 (arom. Cs of NCH2C6H4(Cl)-4); 159.14 (NCHN). Elemental analysis calcd. (%) for C20H24BrClN2O2: C 54.62, H 5.50, N 6.37; found (%): C 54.74, H 5.44, N 6.67. (For the 1H NMR, 13C NMR and FT-IR spectrum of the imidazolinium salt 3b, see ESI file, pages S7-S8).
4.4.3 1-(2-(2-Ethoxyphenoxy)Ethyl)-3-(4-Isopropylbenzyl)Imidazolinium Chloride, 3c
Yield 75%, 1.502 g (white solid); mp: 103–104 °C; FT-IR (vC-N): 1040 and 1251 cm−1; (vC(2)-N): 1644 cm−1. 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 1.14 (d, J = 6.9 Hz, 6H, NCH2C6H4(CH(CH3)2)-4); 1.26 (t, J = 7.0 Hz, 3H, NCH2CH2OC6H4(OCH2CH3)-2); 2.81 (hept, J = 6.9 Hz, 1H, NCH2C6H4(CH(CH3)2)-4); 3.73 (t, J = 12.0 Hz, 2H, NCH2CH2N); 3.92 (q, J = 7.0 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 4.09 (t, J = 4.8 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 4.17 (t, J = 4.6 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 4.18 (t, J = 11.6 Hz, 2H, NCH2CH2N); 4.72 (s, 2H, NCH2C6H4(CH(CH3)2)-4); 6.77–6.89 (m, 4H, arom. Hs of NCH2CH2OC6H4(OCH2CH3)-2); 7.13 and 7.23 (d, J = 8.1 Hz, 4H, arom. Hs of NCH2C6H4(CH(CH3)2)-4); 10.23 (s, 1H, NCHN). 13C NMR (101 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 14.96 (NCH2CH2OC6H4(OCH2CH3)-2); 23.82 (NCH2C6H4(CH(CH3)2)-4); 33.78 (NCH2C6H4(CH(CH3)2)-4); 47.72 and 47.86 (NCH2CH2N); 50.15 (NCH2C6H4(CH(CH3)2)-4); 51.99 (NCH2CH2OC6H4(OCH2CH3)-2); 63.90 (NCH2CH2OC6H4(OCH2CH3)-2); 67.87 (NCH2CH2OC6H4(OCH2CH3)-2); 112.97, 114.73, 120.95, 122.48, 147.26 and 148.81 (arom. Cs of NCH2CH2OC6H4(OCH2CH3)-2); 127.24, 128.84, 129.74 and 149.84 (arom. Cs of NCH2C6H4(CH(CH3)2)-4); 159.24 (NCHN). Elemental analysis calcd. (%) for C23H31ClN2O2: C 68.56, H 7.75, N 6.95; found (%): C 68.74, H 8.13, N 7.08. (For the 1H NMR, 13C NMR and FT-IR spectrum of the imidazolinium salt 3c, see ESI file, pages S9-S10).
4.4.4 1-(2-(2-Ethoxyphenoxy)Ethyl)-3-(4-tert-Butylbenzyl)Imidazolinium Bromide, 3d
Yield 76%, 1.751 g (white solid); mp: 96–97 °C; FT-IR (vC-N): 1050 and 1254 cm−1; (vC(2)-N): 1645 cm−1. 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 1.26 (s, 9H, NCH2C6H4(C(CH3)3)-4); 1.31 (t, J = 7.0 Hz, 3H, NCH2CH2OC6H4(OCH2CH3)-2); 3.80 (t, J = 12.1 Hz, 2H, NCH2CH2N); 3.97 (q, J = 7.0 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 4.13 (t, J = 4.8 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 4.23 (t, J = 4.6 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 4.25 (t, J = 12.0 Hz, 2H, NCH2CH2N); 4.77 (s, 2H, NCH2C6H4(C(CH3)3)-4); 6.82–6.94 (m, 4H, arom. Hs of NCH2CH2OC6H4(OCH2CH3)-2); 7.30 and 7.35 (d, J = 8.4 Hz, 4H, arom. Hs of NCH2C6H4(C(CH3)3)-4); 9.95 (s, 1H, NCHN). 13C NMR (101 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 15.02 (NCH2CH2OC6H4(OCH2CH3)-2); 31.21 (NCH2C6H4(C(CH3)3)-4); 34.63 (NCH2C6H4(C(CH3)3)-4); 47.93 and 47.98 (NCH2CH2N); 50.26 (NCH2C6H4(C(CH3)3)-4); 52.00 (NCH2CH2OC6H4(OCH2CH3)-2); 63.96 (NCH2CH2OC6H4(OCH2CH3)-2); 67.67 (NCH2CH2OC6H4(OCH2CH3)-2); 113.03, 114.86, 121.02, 122.54, 147.27 and 148.85 (arom. Cs of NCH2CH2OC6H4(OCH2CH3)-2); 126.16, 128.63, 129.30 and 152.18 (arom. Cs of NCH2C6H4(C(CH3)3)-4); 158.70 (NCHN). Elemental analysis calcd. (%) for C24H33BrN2O2: C 62.47, H 7.21, N 6.07; found (%): C 62.59, H 7.22, N 6.24. (For the 1H NMR, 13C NMR and FT-IR spectrum of the imidazolinium salt 3d, see ESI file, pages S11-S12).
4.5 General Procedure for the Preparation of Pd-PEPPSI-NHC Complexes (4a-4d)
An acetonitrile (10 mL) solution of imidazolinium salts 4a-4d (1 mmol), PdCl2 (0.177 g, 1 mmol), K2CO3 (0.691 g, 5 mmol), KBr (1.190 g, 10 mmol) and pyridine (0.119 g, 1.5 mmol) was stirred at 80 °C for 16 h. Then, all volatiles were removed under vacuum, and the solid residue was washed with n-pentane (2 × 5 mL). The crude product was purified by column chromatography using CH2Cl2 to afford the corresponding Pd-PEPPSI-NHC complex. The palladium complex was crystallized from CH2Cl2/n-pentane solvent mixture (1:5, v/v) at room temperature, and completely dried under vacuum. Imidazolidin-2-ylidene based Pd-PEPPSI-NHC complexes were isolated as air- and moisture-stable yellow solids in 39–50% yields.
4.5.1 Dibromo-[1-(2-(2-Ethoxyphenoxy)Ethyl)-3-(4-Methylbenzyl)Imidazolidin-2-Ylidene](Pyridine) Palladium(II), 4a
Yield 43%, 0.295 g (yellow solid); mp: 87–88 °C; FT-IR (vC-N): 1040 and 1251 cm−1; (vC(2)-N): 1592 cm−1. 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 1.37 (t, J = 7.0 Hz, 3H, NCH2CH2OC6H4(OCH2CH3)-2); 2.34 (s, 3H, NCH2C6H4(CH3)-4); 3.45 (dd, J = 11.2, 9.0 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 4.03 (dd, J = 11.1, 9.0 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 4.04 (q, J = 7.0 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 4.55 (dt, J = 26.5, 4.7 Hz, 4H, NCH2CH2N); 5.31 (s, 2H, NCH2C6H4(CH3)-4); 6.85–6.92 and 6.98–7.00 (m, 4H, arom. Hs of NCH2CH2OC6H4(OCH2CH3)-2); 7.17 (d, J = 7.9 Hz, 2H, arom. Hs of NCH2C6H4(CH3)-4); 7.32 (ddd, J = 7.8, 5.1, 1.4 Hz, 2H, arom. Hs of pyridine); 7.45 (d, J = 7.9 Hz, 2H, arom. Hs of NCH2C6H4(CH3)-4); 7.74 (tt, J = 7.8, 1.5 Hz, 1H, arom. H of pyridine); 9.00 (dd, J = 6.4, 1.5 Hz, 2H, arom. CHs of pyridine). 13C NMR (101 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 14.98 (NCH2CH2OC6H4(OCH2CH3)-2); 21.19 (NCH2C6H4(CH3)-4); 48.00 (NCH2C6H4(CH3)-4); 50.01 and 50.83 (NCH2CH2N); 54.61 (NCH2CH2OC6H4(OCH2CH3)-2); 64.04 (NCH2CH2OC6H4(OCH2CH3)-2); 68.77 (NCH2CH2OC6H4(OCH2CH3)-2); 112.96, 113.69, 120.96, 121.50, 148.08 and 148.67 (arom. Cs of NCH2CH2OC6H4(OCH2CH3)-2); 128.83, 129.40, 132.08 and 137.79 (arom. Cs of NCH2C6H4(CH3)-4); 124.49, 137.84 and 152.48 (arom. Cs of pyridine); 180.89 (Pd-Ccarbene). Elemental analysis calcd. (%) for C26H31Br2N3O2Pd: C 45.67, H 4.57, N 6.15; found (%): C 45.62, H 4.67, N 6.16. (For the 1H NMR, 13C NMR and FT-IR spectrum of the Pd-PEPPSI-NHC complex 4a, see ESI file, pages S13-S14).
4.5.2 Dibromo-[1-(2-(2-Ethoxyphenoxy)Ethyl)-3-(4-Chlorobenzyl)Imidazolidin-2-Ylidene](Pyridine) Palladium(II), 4b
Yield 44%, 0.307 g (yellow solid); mp: 149–150 °C; FT-IR (vC-N): 1045 and 1243 cm−1; (vC(2)-N): 1594 cm−1. 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 1.38 (t, J = 7.0 Hz, 3H, NCH2CH2OC6H4(OCH2CH3)-2); 3.45 (dd, J = 10.4, 9.0 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 4.03 (q, J = 7.1 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 4.07 (dd, J = 11.1, 9.0 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 4.56 (dt, J = 21.1, 4.7 Hz, 4H, NCH2CH2N); 5.32 (s, 2H, NCH2C6H4(Cl)-4); 6.85–6.92 and 6.97–7.00 (m, 4H, arom. Hs of NCH2CH2OC6H4(OCH2CH3)-2); 7.33 (ddd, J = 7.8, 5.1, 1.4 Hz, 2H, arom. Hs of pyridine); 7.34 (d, J = 8.4 Hz, 2H, arom. Hs of NCH2C6H4(Cl)-4); 7.52 (d, J = 8.4 Hz, 2H, arom. Hs of NCH2C6H4(Cl)-4); 7.74 (tt, J = 7.7, 1.5 Hz, 1H, arom. H of pyridine); 8.99 (dd, J = 6.4, 1.5 Hz, 2H, arom. CHs of pyridine). 13C NMR (101 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 14.98 (NCH2CH2OC6H4(OCH2CH3)-2); 48.03 (NCH2C6H4(Cl)-4); 50.03 and 50.97 (NCH2CH2N); 54.19 (NCH2CH2OC6H4(OCH2CH3)-2); 64.02 (NCH2CH2OC6H4(OCH2CH3)-2); 68.72 (NCH2CH2OC6H4(OCH2CH3)-2); 112.94, 113.68, 120.95, 121.57, 148.01 and 148.66 (arom. Cs of NCH2CH2OC6H4(OCH2CH3)-2); 128.93, 130.23, 133.71 and 133.96 (arom. Cs of NCH2C6H4(Cl)-4); 124.53, 137.92 and 152.45 (arom. Cs of pyridine); 181.51 (Pd-Ccarbene). Elemental analysis calcd. (%) for C25H28Br2ClN3O2Pd: C 42.64, H 4.01, N 5.97; found (%): C 42.78, H 4.04, N 6.09. (For the 1H NMR, 13C NMR and FT-IR spectrum of the Pd-PEPPSI-NHC complex 4b, see ESI file, pages S15-S16).
4.5.3 Dibromo-[1-(2-(2-Ethoxyphenoxy)Ethyl)-3-(4-Isopropyl-Benzyl)Imidazolidin-2-Ylidene](Pyridine) Palladium(II), 4c
Yield 50%, 0.358 g (yellow solid); mp: 111–112 °C; FT-IR (vC-N): 1039 and 1255 cm−1; (vC(2)-N): 1592 cm−1. 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 1.17 (d, J = 6.9 Hz, 6H, NCH2C6H4(CH(CH3)2)-4); 1.30 (t, J = 7.0 Hz, 3H, NCH2CH2OC6H4(OCH2CH3)-2); 2.83 (hept, J = 6.9, 1H, NCH2C6H4(CH(CH3)2)-4); 3.39 (dd, J = 11.2, 8.9 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 3.94 (dd, J = 10.7, 8.6 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 3.95 (q, J = 7.0 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 4.48 (dt, J = 9.1, 4.8 Hz, 4H, NCH2CH2N); 5.24 (s, 2H, NCH2C6H4(CH(CH3)2)-4); 6.78–6.84 and 6.90–6.93 (m, 4H, arom. Hs of NCH2CH2OC6H4(OCH2CH3)-2); 7.15 (d, J = 8.0 Hz, 2H, arom. Hs of NCH2C6H4(CH(CH3)2)-4); 7.25 (ddd, J = 7.8, 5.0, 1.5 Hz, 2H, arom. Hs of pyridine); 7.41 (d, J = 8.0 Hz, 2H, arom. Hs of NCH2C6H4(CH(CH3)2)-4); 7.66 (tt, J = 7.7, 1.6 Hz, 1H, arom. H of pyridine); 8.92 (dd, J = 6.5, 1.5 Hz, 2H, arom. CHs of pyridine). 13C NMR (101 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 15.01 (NCH2CH2OC6H4(OCH2CH3)-2); 24.02 (NCH2C6H4(CH(CH3)2)-4); 33.88 (NCH2C6H4(CH(CH3)2)-4); 48.09 (NCH2C6H4(CH(CH3)2)-4); 50.02 and 50.87 (NCH2CH2N); 54.62 (NCH2CH2OC6H4(OCH2CH3)-2); 64.08 (NCH2CH2OC6H4(OCH2CH3)-2); 68.79 (NCH2CH2OC6H4(OCH2CH3)-2); 113.00, 113.71, 120.99, 121.52, 148.11 and 148.69 (arom. Cs of NCH2CH2OC6H4(OCH2CH3)-2); 126.80, 128.83, 132.49 and 148.78 (arom. Cs of NCH2C6H4(CH(CH3)2)-4); 124.51, 137.88 and 152.49 (arom. Cs of pyridine); 180.88 (Pd-Ccarbene). Elemental analysis calcd. (%) for C28H35Br2N3O2Pd: C 47.25, H 4.96, N 5.90; found (%): C 47.22, H 5.13, N 5.99. (For the 1H NMR, 13C NMR and FT-IR spectrum of the Pd-PEPPSI-NHC complex 4c, see ESI file, pages S17-S18).
4.5.4 Dibromo-[1-(2-(2-Ethoxyphenoxy)Ethyl)-3-(4-tert-Butylbenzyl)Imidazolidin-2-Ylidene](Pyridine) Palladium(II), 4d
Yield 39%, 0.279 g (yellow solid); mp: 150–151 °C; FT-IR (vC-N): 1041 and 1256 cm−1; (vC(2)-N): 1591 cm−1. 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 1.24 (s, 9H, NCH2C6H4(C(CH3)3)-4); 1.30 (t, J = 7.0 Hz, 3H, NCH2CH2OC6H4(OCH2CH3)-2); 3.40 (dd, J = 11.2, 9.0 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 3.96 (q, J = 7.0 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 3.97 (dd, J = 10.9, 9.1 Hz, 2H, NCH2CH2OC6H4(OCH2CH3)-2); 4.49 (dt, J = 9.4, 4.7 Hz, 4H, NCH2CH2N); 5.25 (s, 2H, NCH2C6H4(C(CH3)3)-4); 6.78–6.86 and 6.91–6.93 (m, 4H, arom. Hs of NCH2CH2OC6H4(OCH2CH3)-2); 7.24 (ddd, J = 7.5, 5.1, 1.3 Hz, 2H, arom. Hs of pyridine); 7.32 and 7.43 (d, J = 8.3 Hz, 4H, arom. Hs of NCH2C6H4(C(CH3)3)-4); 7.67 (tt, J = 7.7, 1.6 Hz, 1H, arom. H of pyridine); 8.93 (dd, J = 6.5, 1.5 Hz, 2H, arom. CHs of pyridine). 13C NMR (101 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 15.00 (NCH2CH2OC6H4(OCH2CH3)-2); 31.37 (NCH2C6H2(C(CH3)3)-4); 34.60 (NCH2C6H2(C(CH3)3)-4); 48.11 (NCH2C6H4(C(CH3)3)-4); 50.02 and 50.87 (NCH2CH2N); 54.52 (NCH2CH2OC6H4(OCH2CH3)-2); 64.07 (NCH2CH2OC6H4(OCH2CH3)-2); 68.80 (NCH2CH2OC6H4(OCH2CH3)-2); 112.98, 113.68, 120.99, 121.51, 148.11 and 148.68 (arom. Cs of NCH2CH2OC6H4(OCH2CH3)-2); 125.66, 128.57, 132.13 and 151.02 (arom. Cs of NCH2C6H4(C(CH3)3)-4); 124.51, 137.87 and 152.49 (arom. Cs of pyridine); 180.86 (Pd-Ccarbene). Elemental analysis calcd. (%) for C29H37Br2N3O2Pd: C 47.99, H 5.14, N 5.79; found (%): C 47.72, H 5.14, N 5.84. (For the 1H NMR, 13C NMR and FT-IR spectrum of the Pd-PEPPSI-NHC complex 4d, see ESI file, pages S19-S20).
4.6 General Procedure for the Pd-NHC Catalyzed Direct C5-Arylation of N-Methylpyrrole-2-Carboxaldehyde with Aryl Halides
An oven dried Schlenk flask was charged with Pd-NHC precatalyst (0.01 equiv., 1 mol%), N-methylpyrrole-2-carboxaldehyde (0.5 mmol, 2 equiv.), (hetero)aryl halide derivative (0.25 mmol, 1 equiv.), KOAc (0.5 mmol, 2 equiv.) and DMA (2 mL) under argon atmosphere. Then, the reaction mixture was stirred at 120 °C for different durations, as given in Table 2. Completion of the reaction, the solution cooled to room temperature, and CH2Cl2 (1 mL) was added to Schlenk tube to dilute the solution. The solution filtered through a pad of celite to remove the solid particles, then, was used for GC analysis. The yields (%) were calculated according to (hetero)aryl halide by GC analysis with dodecane as internal standard. Chemical characterizations of some products were made by NMR.
References
Jones RA, Bean GP (1977). In: Blomquist AT, Wasserman HH (eds) The chemistry of pyrroles, vol 34. Academic Press, London
Jones RA (ed) (1990) Pyrroles: the synthesis and the physical and chemical aspects of the pyrrole ring. Wiley, New York
Pudleiner H, Laatsch H (1990) Liebigs Ann Chem 5:423
Ho L, Péra MH, Taillandier G, Fatome M, Laval JD, Leclerc G (1993) Eur J Med Chem 28:703
Yokoyama A, Kato A, Miyakoshi R, Yokozawa T (2008) Macromolecules 41:7271
Tamilavan V, Sakthivel P, Li Y, Song M, Kim CH, Jin SH, Hyun MH (2010) J Polym Sci 48:3169
Anderson WK, Halat MJ, Rick AC (1980) J Med Chem 23:87
Portevin B, Tordjman C, Pastoureau P, Bonnet J, De Nanteuil G (2000) J Med Chem 43:4582
Bellina F, Rossi R (2006) Tetrahedron 62:7213
Roth BD (2002) Prog Med Chem 40:1
Chong PH, Seeger JD (1997) Pharmacotherapy 17:1157
Zhang Z, Olland AM, Zhu Y (2005) J Biol Chem 280:28468
Xiao G, Kumar A, Li K, Rigl CT, Bajic M, Davis TM, Boykin DW, Wilson WD (2001) Bioorg Med Chem 9:1097
Thaher BA, Koch P, Schattel V, Laufer S (2009) J Med Chem 52:2613
Wilkerson WW, Copeland RA, Covington M, Trzaskos JM (1995) J Med Chem 38:3895
de Laszlo SE, Hacker C, Li B, Kim D, MacCoss M, Mantlo N, Pivnichny JV, Colwell L, Koch GE, Cascieri MA, Hagmann WK (1999) Bioorg Med Chem Lett 9:641
Rieth RD, Mankad NP, Calimano E, Sadighi JP (2004) Org Lett 6:3981
Wang X, Lane BS, Sames D (2005) J Am Chem Soc 127:4996
Dang TT, Ahmad R, Dang TT, Reinke H, Langer P (2008) Tetrahedron Lett 49:1698
Lavallo V, Frey GD, Donnadieu B, Soleilhavoup M, Betrand G (2008) Angew Chem 120:5302; Angew Chem Int Ed 47:5224
Wen J, Qin S, Ma LF, Dong L, Zhang J, Liu SS, Duan YS, Chen SY, Hu CW, Yu YQ (2010) Org Lett 12:2694
Vakuliuk O, Koszarna B, Gryko DT (2011) Adv Synth Catal 353:925
Toguem SMT, Fatunsin O, Villinger A, Langer P (2011) Tetrahedron Lett 52:3732
Kaloğlu N, Özdemir İ (2019) Tetrahedron 75:2306
İmik F, Yaşar S, Özdemir İ (2019) Inorg Chim Acta 495:118969
Karataş MO, Özdemir N, Alıcı B, Özdemir İ (2020) Polyhedron 176:114271
Li JJ, Gribble GW (2000) Palladium in heterocyclic chemistry. Pergamon, Amsterdam
Negishi E (ed) (2002) Handbook of organopalladium chemistry for organic synthesis. Wiley, New York, p 213
Bheeter CB, Chen L, Soulé JF, Doucet H (2016) Catal Sci Technol 6:2005
Bellina F, Rossi R (2009) Tetrahedron 65:10259
Bernhammer JC, Singh H, Huynh HV (2014) Organometallics 33:4295
Akita Y, Inoue A, Yamamoto K, Ohta A, Kurihara T, Shimizu M (1985) Heterocycles 23:2327
Ohta A, Akita Y, Ohkuwa T, Chiba M, Fukunaga R, Miyafuji A, Nakata T, Tani N, Aoyagi Y (1990) Heterocycles 31:1951
Aoyagi Y, Inoue A, Koizumi I, Hashimoto R, Tokunaga K, Gohma K, Komatsu J, Sekine K, Miyafuji A, Kunoh J, Honma R, Akita Y, Ohta A (1992) Heterocycles 33:257
Deprez NR, Kalyani D, Krause A, Sanford MS (2006) J Am Chem Soc 128:4972
Gryko DT, Vakuliuk O, Gryko D, Koszarna B (2009) J Org Chem 74:9517
Liégault B, Petrov I, Gorelsky SI, Fagnou K (2010) J Org Chem 75:1047
Jafarpour F, Rahiminejadan S, Hazrati H (2010) J Org Chem 75:3109
Lazareva A, Daugulis O (2011) J Org Chem 76:471
Bheeter CB, Bera JK, Doucet H (2012) Tetrahedron Lett 53:509
Bernhammer JC, Huynh HV (2014) Organometallics 33:1266
Romero M, Harrak Y, Basset J, Ginet L, Constans P, Pujol MD (2006) Tetrahedron 62:9010
Touré BB, Lane BS, Sames D (2006) Org Lett 8:1979
Wang X, Gribkov DV, Sames D (2007) J Org Chem 72:1476
Liégaut B, Lapointe D, Caron L, Vlassova A, Fagnou K (2009) J Org Chem 74:1826
Dong JJ, Roger J, Verrier C, Martin T, Le Goff R, Hoarau C, Doucet H (2010) Green Chem 12:2053
René O, Fagnou K (2010) Adv Synth Catal 352:2116
Laidaoui N, Roger J, Miloudi A, El Abed D, Doucet H (2011) Eur J Org Chem 21:4373
Ghosh D, Lee HM (2012) Org Lett 14:5534
Özdemir İ, Gürbüz N, Kaloğlu N, Doğan Ö, Kaloğlu M, Bruneau C, Doucet H (2013) Beilstein J Org Chem 9:303
Yiğit B, Gübüz N, Yiğit M, Dağdeviren Z, Özdemir İ (2017) Inorg Chim Acta 465:44
Kaloğlu N, Kaloğlu M, Tahir MN, Arıcı C, Bruneau C, Doucet H, Dixneuf PH, Çetinkaya B, Özdemir İ (2018) J Organomet Chem 867:404
Kaloğlu M, Kaloğlu N, Özdemir İ (2018) ChemistrySelect 3:5600
Kaloğlu M, Demir Düşünceli S, Özdemir İ (2020) J Organomet Chem 915:121236
Özdemir İ, Gök Y, Özeroğlu Ö, Kaloğlu M, Doucet H, Bruneau C (2010) Eur J Inorg Chem 12:1798
Coy ED, Cuca LE, Sefkow M (2010) Synth Commun 41:41
Akkoç S, Gök Y, Özer İİ, Kayser V (2016) Beilstein J Org Chem 12:81
He X-X, Li Y, Ma B-B, Ke Z, Liu F-S (2016) Organometallics 35:2655
Hu L-Q, Deng R-L, Li Y-F, Zeng C-J, Shen D-S, Liu F-S (2018) Organometallics 37:214
Karthika S, Gandhi T (2018) New J Chem 42:15811
Song A-X, Zeng X-X, Ma B-B, Xu C, Liu FS (2020) Organometallics 39:3524
Zhao Q, Meng G, Nolan SP, Szostak M (2020) Chem Rev 120:1981
Zhao L, Bruneau C, Doucet H (2013) ChemCatChem 5:255
Xu K, Li W, Sun R, Luo L, Chen X, Zhang C, Zheng X, Yuan M, Fu H, Li R, Chen H (2020) Org Lett 22:6107
Roger J, Doucet H (2009) Adv Synth Catal 351:1977
Castro MCR, Belsley M, Raposo MMM (2016) Dyes Pigm 131:333
Martin AR, Chartoire A, Slawin AMZ, Nolan SP (2012) Beilstein J Org Chem 8:1637
Acknowledgements
This study was supported by the Technological and Scientific Research Council of Turkey TÜBİTAK (Project No: 119R003).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kaloğlu, M., Kaloğlu, N. & Özdemir, İ. Palladium-PEPPSI-NHC Complexes Bearing Imidazolidin-2-Ylidene Ligand: Efficient Precatalysts for the Direct C5-Arylation of N-Methylpyrrole-2-Carboxaldehyde. Catal Lett 151, 3197–3212 (2021). https://doi.org/10.1007/s10562-021-03561-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-021-03561-4