
Abstract
Ag3PO4/g-C3N4 nanocomposite photocatalysts were synthesized by calcining the mixture of urea and trace amount of Ag3PO4 microcrystals at 600 °C, in which Ag3PO4 nanoparticles with size of 20–30 nm were wrapped with g-C3N4 layers. The obtained Ag3PO4/g-C3N4 composites exhibited efficient utilization of visible light and separation of photoinduced electron–hole pairs, resulting in enhanced photocatalytic activity for H2 evolution. The optimum photocatalytic H2 evolution rate under visible light without Pt co-catalyst for 0.04AP-CN composite was 89.4 µmol/h, which is 3.34 times of that of g-C3N4 (26.7 µmol/h). The H2 evolution rate for 0.04AP-CN with 3 wt% Pt co-catalyst was up to 273.6 µmol/h, which is 2.39 times of that g-C3N4 (114.4 µmol/h). The performance of Ag3PO4/g-C3N4 composite was enhanced significantly due to the synergistic effect of the interface formed between Ag3PO4 and g-C3N4.
Graphical Abstract

Photocatalytic mechanism of the Ag3PO4/g-C3N4 composite
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Resource deficiency, energy shortage and environmental deterioration pressed even more grave conditions for human to confront with. Hydrogen energy was generally thought as the ultimately energy resource for its advantages of high combustion calorific value, abundant resources and clean combustion products. Photocatalysis provided certain promising approaches for H2 evolution via water splitting and pollutant degradation using solar energy directly and aroused considerable interests for research experts in materials science and chemistry [1,2,3,4]. The graphitic carbon nitride (g-C3N4) is a new metal-free semiconductor with layered structure and suitable band gap of 2.7 eV allowing it to serve as a promising photocatalyst for realizing the hydrogen evolution via water splitting using solar resource conversion [5,6,7,8,9]. Besides its advantages of chemical stability, low-cost precursors and nonmetal photocatalyst, some shortcomings restricted its practical application, such as small specific surface area and rapid recombination of the photogenerated electron–hole pairs. Ever since its emergence as a photocatalyst, lots of research strategies have been proposed to improve its photocatalytic performance by doping metal or non-metal elements, designing appropriate molecular structures, supporting metal nanoparticles, and forming heterojunction with other semiconductors [10,11,12,13,14,15]. Among all those methods, forming heterojunction with other semiconductors is one of the most effective strategy to improve the separation of photoinduced charge carriers, and enhance the visible light absorption, thus improve the photocatalytic activities. Those reported semiconductors included metal oxide,[16,17,18,19,20,21,22,23,24] sulfide,[19] halide,[19, 25] carbide, and phosphate, etc.[21,22,23,24,25,26,27,28,29,30].
The Ag3PO4 nanocrystals were studied as a visible light driven photocatalyst for having good oxidation ability to degradate organic pollutants and O2 evolution via water splitting, but lacking the reduction ability enough to reduce water to hydrogen. Silver halide and phosphate are not stable under light irradiation, and Ag nanoparticles may be formed during the photocatalysis process. The surface plasma effect induced by Ag nanoparticles can effectively improve the visible light absorption on the thin surface layer (10 nm), and the charge carrier separation of the photoinduced electron–hole pairs. The size of Ag3PO4 particles can be reduced significantly through the preparation process of forming the Ag3PO4/g-C3N4 composite, enhancing the migration of photoinduced charge carriers. Therefore, the photocatalytic performance was improved by forming Ag3PO4/g-C3N4 nanocomposite [28,29,30,31,32,33]. The reported literatures usually involved the Ag3PO4/g-C3N4 composites with Ag3PO4 fraction of more than 50%, in which Ag3PO4 acts as a matrix and g-C3N4 as a solute. Those composite photocatalysts have enhanced performance in degradation of Rhodaming B (RhB), Methyl orange (MO) and conversion of CO2 with g-C3N4 hybrids with Ag3PO4. The enhanced photocatalytic activity can be explained using the Z-scheme via Ag3PO4 and g-C3N4 heterojunction. We explore the possibility that the Ag3PO4/g-C3N4 composite with lower ratio of Ag3PO4 may have different photocatalytic activities using g-C3N4 as a matrix since the performance of g-C3N4 can be enhanced significantly by small amount of compound semiconductors such as NiC and Ag2O [12, 14].
In this paper, we reported a facile calcination technique to prepare the Ag3PO4/g-C3N4 nanocomposites by thermal annealing the mixture of urea and trace amount of Ag3PO4. The H2 evolution rate of g-C3N4 was enhanced significantly by introducing a trace amount of Ag3PO4, and the H2 evolution process was in progress without co-catalyst. The Ag nanoparticles formed under solar light irradiation may act as the combination center for photoinduced electrons and holes, thus enhanced the charge separation in the photocatalyst by forming an efficient Z-scheme photocatalysis system for H2 evolution via water splitting.
2 Experimental Details
2.1 Synthesis of the Photocatalysts
All the chemicals were of analytical grade and used without further purification.
Pure Ag3PO4 was prepared via an ion exchange method. 0.4 M aqueous solution of silver nitrate was added to a beaker and 0.15 M aqueous solution of sodium acetate was added dropwise under continuous stirring for 2 h at room temperature. Then 0.1 M solution of KH2PO4 added to above-mentioned solution under stirring for 3 h. The solution was centrifuged and the solid product was washed three times with distilled water and ethanol and then dried in an oven at 60 °C. The obtained Ag3PO4 microcrystal was marked as AP.
For the preparation of the Ag3PO4/g-C3N4 nanocomposite photocatalysts, an appropriate amount (0.01 g, 0.02 g, 0.04 g, 0.06 g, and 0.08 g) of Ag3PO4 was mixed with 8 g urea, and calcined at 600 °C for 90 min. The obtained yellow products were noted as 0.01AP-CN, 0.02AP-CN, 0.04AP-CN, 0.06AP-CN, and 0.08AP-CN, respectively. The pure g-C3N4 was prepared by calcified 8 g urea at 600 °C for 90 min, and noted as CN as reference sample.
2.2 Characterization
The morphologies of the prepared samples were observed by scanning electron microscope (SEM, JSE-7800F, Jeol), transmission electron microscope (TEM, Libra 200FE, Zeiss), and tapping mode atomic force microscope (AFM, CSPM5500, Being, china). The X-ray diffraction (XRD) patterns were performed to characterized the crystalline structures of the photocatalysts by a Shimadzu XRD7000 instrument with Cu Kα radiation (λ = 1.5418 Å). Fourier transform infrared (FTIR) was measured using a Perkin Elmer (Frontier) spectroscopy instrument in KBr pellets. X-ray photoelectron spectroscopy (XPS) was carried out to analyze the chemical state and composition on a VG ESCALAB 250 spectrometer with Al Kα radiation (hν = 1486.8 eV). Ultraviolet–visible (UV–vis) absorption spectra were performed on U-3310 spectrophotometer (Hitachi, Japan) in the wavelength range of 300 nm to 800 nm. Photoluminescence (PL) spectra were carried out on an F-7000 fluorescence spectrophotometer (Hitachi, Japan) with an excitation wavelength at 273 nm using a 150 W Xe lamp as the excitation source. The Brunauer-Emmett-Teller (BET) specific surface area was characterized by nitrogen adsorption at 77 K with Quadrasorbevo 2QDS-MP-30 (Quantachrome Instruments, USA). The photocurrent measurements were conducted on an Autolab electrochemical workstation (Aut86039) in a standard three-electrode system, using a platinum wire and the saturated Ag/AgCl electrode as the counter electrode and reference electrode, respectively. The working electrode was prepared by coating the catalysts on a 1.8 cm × 1.2 cm fluorine-doped tin oxide (FTO) glass substrate. A 500 W Xe lamp with a light intensity of 113.8 mW/cm2 was used as a light source.
2.3 Photocatalytic Hydrogen Evolution
Photocatalytic hydrogen evolution was performed by a full spectrum photocatalysts performance evaluation system (Suncat Instruments Co., China). The reactor was made up a quartz container with highly transparent window in a diameter of 2.5 cm at the bottom. A 500 W Xe lamp (Zolix, Gloria-X500A) with adjustable intensity was used as solar simulator with a 400 nm cutoff filter for the photocatalytic reactions. The reactor with volume of 130 ml was kept at 20 °C and the light intensity was 113.8 mW/cm2. 10 mg of photocatalyst was dispersed in 30 ml water solution with 20 vol% triethanolamine (TEOA) aqueous solution as an electron donor. Prior to irradiation, the reactor was evacuated to high vacuum up to 10−8 Torr to remove air and filled with argon. The reaction suspension was stirred with a magnetic stirrer under solar irradiation. During the photocatalytic process, 1 ml of the gas sample was withdrawn from the reaction cell at 30 min intervals and the pressure was automatically compensated with argon. The amount of hydrogen evolved was measured with a Shimadzu gas chromatography (GC-2018; Molecular sieve TDX-01, TCD detector, Ar carrier gas) and calibrated with an Agilent 5 ml syringe. The apparent quantum yield (AQY) of the photocatalysts was calculated according to the light absorption and H2 evolution rate as follows:
3 Results and Discussion
3.1 Structural and Composition Characterization
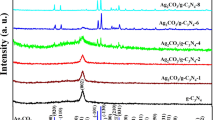
Figure 1a showed the XRD patterns of the as-synthesized Ag3PO4/g-C3N4 photocalysts with different amount of Ag3PO4, and the reference samples of the pure g-C3N4 and Ag3PO4 particles. The XRD pattern indicated that Ag3PO4 particles were indexed as body-centered cubic phase (JCPDS No. 06-0505) with good crystallinity. For the pure g-C3N4 sample, there existed two broad peaks around 27.4° and 13.0°, corresponding to the (002) and (100) diffraction planes, respectively. The (002) plane is attributed to the long-range interplanar stacking of aromatic units with interlayer distance of 0.326 nm; while the (100) plane with weak intensity is associated with the in-plane trigonal nitrogen linked tri-s-triazine motifs with distance of 0.681 nm. For the Ag3PO4/g-C3N4 composites, the intrinsic diffraction peaks of g-C3N4 were observed clearly, indicating that g-C3N4 structure remained intact via hybridization with Ag3PO4 nanoparticles. No Ag3PO4 diffraction peaks could be discerned for the Ag3PO4/g-C3N4 composites owing to the low Ag3PO4 fraction and small particle size. With the increase of Ag3PO4 component ratio, the peak for (002) plane is shifted to higher 2θ position, which indicated the decreased interlayer distance due to the enhanced interaction of g-C3N4 layer by intercalating Ag3PO4. In addition, the diffraction intensity for peak (002) increased and full width at half maxima (FWHM) decreased a little with the increase of Ag3PO4 amount, indicating improved crystallinity of Ag3PO4/g-C3N4 composites. The XRD results showed that small amount of Ag3PO4 nanoparticles increased the interlayer interaction, facilitating the compact g-C3N4 with better crystallinity.

XRD patterns (a) and FTIR spectra (b) of Ag3PO4, the pure g-C3N4, and the Ag3PO4/g-C3N4 composites with variable Ag3PO4 contents (0.01 g, 0.02 g, 0.04 g, 0.06 g, and 0.08 g) mixed with 8 g urea
The FTIR spectra of Ag3PO4, g-C3N4, and Ag3PO4/g-C3N4 photocatalysts were shown in Fig. 1b. In the spectrum of g-C3N4, there exist the broad bands around 3000–3500 cm−1, assigning to the stretching mode of N–H. The peak at 810 cm−1 is related to the bending mode of the s-triazine ring. The peaks at 1233, 1315, 1400, 1460, 1540 cm−1 correspond to the stretching vibrations of the aromatic CN heterocycles. In the spectrum of Ag3PO4, there exist two peaks at 930 cm−1 and 570 cm−1, corresponding to the P–O stretching vibrations of PO43−. No obvious change can be detected for the characteristic IR vibration peaks of g-C3N4 by adding small amount of Ag3PO4, revealing that the chemical structure of g-C3N4 remains intact in the Ag3PO4/g-C3N4 composites.
The morphologies of Ag3PO4 nanocrystals, pure g-C3N4, and Ag3PO4/g-C3N4 composites were observed by SEM and TEM images, as shown in Fig. 2. Figure 2a showed the SEM image of Ag3PO4 crystals with diameter around 1 micrometer. SEM image for the pure g-C3N4, as shown in Fig. 2b, displayed the pure g-C3N4 are cotton-like flocculus aggregated by irregular nanosheets. The SEM image in Fig. 2c showed that the 0.04AP-CN sample remains a similar appearance compared with the pure g-C3N4. TEM images in Fig. 2d and e showed the overview of the Ag3PO4/g-C3N4 composite. The Ag3PO4 nanoparticles were randomly distributed among the g-C3N4 layers. A high magnification TEM image in Fig. 2f showed the AP particles with size around 20–30 nm were wrapped by graphitic carbon nitride layers. The size of Ag3PO4 particle is obvious smaller than that of pure Ag3PO4 microcystals, which split to smaller particle during the thermal annealing process with urea.

The SEM images of Ag3PO4 crystals (a), pure g-C3N4 (b), and 0.04AP-CN composite (c). TEM images of 0.04AP-CN nanocomposite with different magnifications (d)–(f)
AFM was employed to further investigate the surface morphologies of the pure g-C3N4 and 0.04AP-CN. Samples were dispersed in ethanol by ultrasonic treatment and then dropped onto a silicon surface. AFM image, as shown in Fig. 3a, demonstrated that the pure g-C3N4 is composed of triangle plate with side length of around 500–800 nm. AFM image for 0.04AP-CN showed that Ag3PO4 nanoparticles distributed randomly on the surface of g-C3N4 layers. The line profile inserted in Fig. 3b showed that the thickness of g-C3N4 plates is about 6.5 nm, roughly about 20 layers of g-C3N4, and the Ag3PO4 particle size is around 20–30 nm, which is consistent with TEM images.

AFM images for the pure g-C3N4 and the Ag3PO4/g-C3N4 (0.04AP-CN) nanocomposites
Figure 4 showed the scan survey and the high-resolution XPS spectra of C, N, Ag, P and O elements for Ag3PO4, the pure g-C3N4, and the Ag3PO4/g-C3N4 composite. For the pure g-C3N4 sample, there are three sharp peaks at 286, 398, and 530 eV, which are assigned to C 1 s, N 1 s, and O 1 s signals, respectively. The Ag3PO4/g-C3N4 composite formed after mixing with Ag3PO4 appears one new peak positioned at 367 eV assigning to Ag 3d and another at 133 eV assigning to P 2p, as shown in the survey scan in Fig. 4a. The high-resolution C 1 s XPS spectra of the three samples were shown in Fig. 4b. For the pure g-C3N4 nanosheets, the peaks centered at about 287.9 eV (C1) is typically attributed to the sp2 hybrid C atoms bonded to N-containing aromatic skeleton rings (N–C=N) coordination. The peak at 285.8 eV (C2) is attributed to C–NH2 bonded in the triazine ring of the reactant intermediary product. The peak at 284.4 eV (C3) could be attributed to the graphite C = C and adventitious carbon. The N 1 s spectra for the pure g-C3N4 in Fig. 4c can be mainly decomposed to three typical peaks located at about 398.3 eV (N1), 399.6 eV (N2), 400.7 eV (N3), which could be attributed to the sp2-hybridized aromatic N atoms bonded to carbon atoms (C–N=C), and sp3-hybridized N atoms of N(–C)3 and terminal amino functions (C–NH2), respectively [18,19,20]. The high resolution Ag 3d spectrum for Ag3PO4 in Fig. 4d showed that the peaks at 367.5 eV and 373.5 eV are attributed to 3d5/2 and 3d3/2 of Ag+ in Ag3PO4. After forming the Ag3PO4/g-C3N4 composite, the peaks for Ag+ still exist and shift to higher binding energy compared with the pure Ag3PO4, suggesting the presence of Ag3PO4 in the Ag3PO4/g-C3N4 composite [34]. The high resolution P 2p showed that the P 2p peak for the Ag3PO4/g-C3N4 composite shifts to higher binding energy side compared with that of pure Ag3PO4, implying that there exists electron interaction between Ag3PO4 and g-C3N4. The O 1 s spectrum for the pure g-C3N4 in Fig. 4f was fitted to two peaks at 531.7 and 532.9 eV, corresponding to the adsorbed –OH group and the adventitious O2. After forming the Ag3PO4/g-C3N4 composite, a new peak at 530.3 eV appears, which is attributed to the O signal arising from Ag3PO4. Therefore, XPS results confirmed that there were both Ag3PO4 and g-C3N4 species in the composite structure. The binding energy shifts of those elements imply that there exists interaction in the interface by attracting electrons of both Ag3PO4 and g-C3N4 layer.

XPS survey scan (a) and high resolution spectra for C 1 s (b) and N 1 s (c), Ag 3d (d), P 2p (e), and O 1 s (f) for the pure g-C3N4 nanosheets (CN), Ag3PO4 nanoparticles (AP) and the Ag3PO4/g-C3N4 (0.04AP-CN) composites
3.2 Photogenerated Charge Separation
To identify the electronic structure and photoelectric properties of the pure g-C3N4 and the Ag3PO4/g-C3N4 nanocomposite photocatalysts, UV–vis diffusion reflectance spectroscopy and PL spectra were measured, as shown in Fig. 5a and b. The UV–vis absorbance spectra, as shown in Fig. 5a, demonstrated that the pure g-C3N4 absorbs light from UV through the visible range up to 465 nm, corresponding to band gap of 2.67 eV of the intrinsic absorption from valence to conduction band. The absorption edge of pure Ag3PO4 has a broader absorption in the visible region with an absorption edge at about 550 nm. The absorption edge of Ag3PO4/g-C3N4 composites displays a slightly red shift (~ 475.5 to ~ 485 nm) as the of Ag3PO4 content increased. The absorption edge for 0.01AP-CN, 0.02AP-CN, 0.04AP-CN, 0.06AP-CN, and 0.08AP-CN is 475.5 nm, 475.5 nm, 485.0 nm, 481.5 nm, and 485.0 nm, respectively. The corresponding band gap is 2.61 eV, 2.61 eV, 2.56 eV, 2.58 eV, and 2.56 eV, which is smaller than that of g-C3N4 and larger than that of Ag3PO4 (2.09 eV). Especially, the light absorption in the visible light range was enhanced significantly. The enhanced visible light absorption indicated that the composite photocatalysts could utilize more visible light, thus facilitate the photocatalytic reaction. Figure 5b showed the photoluminescence spectra of the pure g-C3N4 and Ag3PO4/g-C3N4 composites with different amount of Ag3PO4 under an excitation wavelength of 375 nm. Pure g-C3N4 shows a strong intrinsic emission band with a peak at 473.2 nm, which attributed the intrinsic emission of direct electron–hole recombination. With the increase of Ag3PO4 content, the PL intensity for AP-CN decreases at first, and 0.04AP-CN with lowest PL intensity. After that, the PL intensity increases a little with increase of Ag3PO4 content.

a UV–vis absorbance spectra, b PL spectra of Ag3PO4 (AP), the pure g-C3N4 nanosheets (CN) and the Ag3PO4/g-C3N4 (0.01AP-CN, 0.02AP-CN, 0.04AP-CN, 0.06AP-CN, and 0.08AP-CN) nanocomposites
The specific surface area and pore size distribution of the prepared samples were obtained from N2 absorption–desorption measurements, shown in Fig. 6. The specific surface area of 0.04AP-CN was calculated to be 137.1 m2/g, larger than the 109.0 m2/g of g-C3N4. The larger surface area for the 0.04AP-CN composite provides more active reaction sites during photocatalytic process, thus improves the photocatalytic performance. The pore distribution (Fig. 6b) showed the size of mesopores is in the range of 1–6 nm. The small pores existed in the 0.04AP-CN composite contribute to improve the photocatalytic performance.

Nitrogen adsorption–desorption isotherms (a) and the corresponding pore-size distribution curves (b) for the pure g-C3N4 and 0.04AP-CN composite
The transient photocurrent responses of the pure g-C3N4 and the Ag3PO4/g-C3N4 composites under visible light were recorded in Fig. 7a. The photocurrent for the Ag3PO4/g-C3N4 composite showed a gradual attenuation at the initial 400 s period upon switching on light, which demonstrated the composites showed obvious photocorrossion phenomenon. The photocurrent tends to level stable for prolonged light irradiation. The spike-like shape of photocurrent transients is indicating of surface recombination process going on. A rapid decay is observed after initial rise, which is ascribed to the accumulation of photoinduced holes, rendering its combination with the photoinduced electrons [35,36,37]. The pure g-C3N4 photocatalyst reached at a stable photocurrent under visible light irradiation without photocorrosion occuring. The current density for 0.04AP-CN is the highest among all the composites, which is about 3 times of that observed for the pure g-C3N4 sample, revealing the most effective separation and transition of photoinduced electron/hole pairs, which is consistent with PL spectra.

Transient photocurrents (a) and EIS (b) spectra of the pure g-C3N4 nanosheets (CN) and the Ag3PO4/g-C3N4 (0.01AP-CN, 0.02AP-CN, 0.04AP-CN, 0.06AP-CN, and 0.08AP-CN) nanocomposites
The electrochemical impedance spectra (EIS) of the pure g-C3N4 and the Ag3PO4/g-C3N4 composites were measured to understand the photocatalytic mechanism. The arc on the EIS Nyquist plot indicates the charge transfer resistance. Generally speaking, the smaller of arc radius is, the lower of the charge transfer resistance will be [21, 24]. As shown in Fig. 7b, the Nyquist plots of all the Ag3PO4/g-C3N4 samples demonstrate smaller arc radius attributing to the reduced electronic resistance and increased electronic conductivity by hybridization Ag3PO4 with the pure g-C3N4. Similarly, the arc radius for 0.04AP-CN is smallest in all samples, which is associated with the highest efficiency of the charge separation [38,39,40]. Moreover, it is vital that the change trend of the arc radius for g-C3N4 samples is roughly consistent with the results displayed in PL spectra.
3.3 Photocatalytic Activity for H2 Evolution
The photocatalytic performance of the prepared samples was evaluated with H2 production with 20 wt% TEOA as sacrificial electron donor and without Pt ion co-catalyst under visible light irradiation, as shown in Fig. 8a. The photocatalytic H2 evolution rate for 0.01AP-CN, 0.02AP-CN, 0.04AP-CN, 0.06AP-CN, and 0.08AP-CN is recorded as 4.63, 5.21, 8.93, 6.08, 3.27 µmol/h, respectively, which are all higher than that of pure g-C3N4 with 2.67 µmol/h. These results clearly demonstrate the photocatalytic activity of the Ag3PO4/g-C3N4 composites have been significantly improved by introducing trace amount of Ag3PO4 nanoparticles to form the Ag3PO4/g-C3N4 composites. The 0.04AP-CN photocatalyst has the most favorable photocatalytic activity with an average H2 evolution rate of among all Ag3PO4/g-C3N4 composites, which is 3.3 times as high as that of pure g-C3N4. The composites loading Ag3PO4 beyond that of the 0.04AP-CN sample nevertheless exhibit weaker photocatalytic performance, which is attributed that loading excessive Ag3PO4 nanoparticles may shield the g-C3N4 surface active sites. It is interesting that Ag3PO4/g-C3N4 composites have good photocatalytic H2 evolution rate even at a condition of without Pt ion co-catalyst. The stability of H2 evolution for 0.04AP-CN was tested four round photocatalytic experiments under same condition (Fig. 8b). The H2 evolution remains unchanged under four runs of photocatalytic reaction, which demonstrated the composite photocatalyst are basically stable. In order to explore the effect of different sacrificial hole reagents on the photocatalytic performance, the photocatalytic H2 evolution rate for 0.04AP-CN was measured using TEOA, ethanol (CH3CH2OH), methanol (CH3OH), and ethanolamine (C2H7NO), as shown in Fig. 8c. Only TEOA can act as an efficient sacrificial reagent in the water splitting process, and the H2 evolution is very stable.

Photocatalutic H2 evolution of the Ag3PO4/g-C3N4 composites with different Ag3PO4 contents (a) and stability of 0.04AP-CN (b) with 20 vol% TEOA, 0 wt% Pt, and 10 mg photocatalysts under visible light irradiation (λ > 400 nm). (c) Photocatalytic H2 evolution of 0.04AP-CN with different sacrificial electron donors
In the condition with 20 vol% TEOA and 3 wt% Pt, the photocatalytic H2 evolution rate increased dramatically, but in different degrees, as shown in Fig. 9a, compared with that of without Pt co-catalyst. The H2 evolution rate for the pure g-C3N4, 0.01AP-CN, 0.02AP-CN, 0.04AP-CN, 0.06AP-CN, and 0.08AP-CN is 11.44, 10.70, 13.80, 27.36, 14.73, and 4.84 µmol/h respectively. The 0.04AP-CN has the highest increase rate among all the Ag3PO4/g-C3N4 composite photocatalysts, and the pure g-C3N4 has the second highest increase rate compared with the values in Fig. 8a. For the AP-CN composites, Ag particles acted as co-catalyst may have occupied some active sites of g-C3N4. In contrast, the pure g-C3N4 have more active sites for Pt co-catalyst. Therefore, g-C3N4 has higher hydrogen evolution rate than other AP-CN composites. The photocatalytic performance for AP-CN composites is consistent with the charge transfer properties. The photocatalytic H2 evolution rate for 0.04AP-CN is as 2.14 times as that of the pure g-C3N4. The H2 evolution tests in Figs. 8a and 9a showed that Ag3PO4 cannot produce H2 under visible light irradiation. The Ag3PO4/g-C3N4 composite formed by wrapping Ag3PO4 nanopaticles with g-C3N4 layer can produce H2 via water splitting by improve the separation of photoinduced charge carriers and increase the visible light absorption. Figure 9b showed the photocatalytic performance of 0.04AP-CN with 0 wt% and 3 wt% Pt co-catalyst. The 0.04AP-CN sample can produce H2 continually during 7 h visible light irradiation. Even if at the condition of without Pt co-catalyst, 0.04AP-CN keeps photocatalytic vitality and produce H2 under visible light irradiation for 7 h. The H2 production increases dramatically with 3 wt% Pt co-catalyst. Table 1 shows the H2 evolution rate (HER) and AQY (%) of the Ag3PO4/g-C3N4 composites with TEOA as sacrificial regent and 0 wt%, 3 wt% Pt under visible light irradiation.

a Photocatalutic H2 evolution of the Ag3PO4/g-C3N4 composites with different Ag3PO4 contents with 20 vol% TEOA, 3 wt% Pt, and 10 mg photocatalysts under visible light irradiation (λ > 400 nm). b Photocatalytic H2 evolution rates of 0.04AP-CN with 0 wt% and 3 wt% Pt co-catalyst
3.4 Discussion on Mechanism
After 150 min of visible light irradiation, the 0.04AP-CN catalyst was retrieved by centrifugation and a subsequent drying process. XPS was used to analyze its chemical composition changes under visible light irradiation. The XPS analysis in Fig. 10 showed that the spectra of C 1 s, N 1 s, and P 2p remain unchanged. In the Ag 3d spectrum, two new peaks at binding energy of 366.8 eV and 372.7 eV can be ascribed to Ag 3d5/2 and 3d3/2 of Ag0 atoms, while the other two peaks at 367.8 eV and 373.8 eV corresponded to Ag 3d5/2 and 3d3/2 of Ag+ in Ag3PO4. A new peak at 529.3 eV in the O 1 s spectrum can be ascribed to the oxygen bonded to the surface of Ag particles. The XPS results demonstrated that Ag3PO4 particles underwent partially decomposing to Ag particles under visible light irradiation, nevertheless, the g-C3N4 remained intact.

XPS spectra for C 1 s, N 1 s, Ag 3d, O 1 s, and P 2p for the 0.04AP-CN photocatalyst after visible light irradiation of 150 min
Ultraviolet photoelectron spectroscopy (UPS) was employed to measure the valence band and the conduction band of the Ag3PO4/g-C3N4 composite, shown in Fig. 11a. The valence band (Ev) energy of g-C3N4, Ag3PO4, and the Ag3PO4/g-C3N4 composite is determined to be 6.57 eV, 6.67 eV, and 6.97 eV by substracting the width of the He I UPS spectra from the excitation energy (21.22 eV). The conduction band (Ec) energy is thus estimated at 3.90 eV, 4.58 eV, and 4.41 eV for the pure g-C3N4, Ag3PO4, and 0.04AP-CN, respectively. The Eg, Ev, and Ec values of g-C3N4, Ag3PO4, and 0.04AP-CN are converted to electrochemical potentials in volts according to the reference standard for which 0 V versus reversible hydrogen electrode (RHE) equals − 4.44 eV versus vacuum level. Figure 11b showed the proposed band structure diagram of heterojunction for the Ag3PO4/g-C3N4 composite. The reduction level for H2 is positioned below the conduction band of the Ag3PO4/g-C3N4 composite. These bands are properly positioned to permit the transfer of electrons and holes for water splitting. The enhanced photocatalytic performance could be attributed to the improved separation of photoinduced charge carriers at the interface between Ag3PO4 and g-C3N4. The PL, EIS and photocurrent results showed that the separation of photoinduced electron–hole pairs was improved obviously after introducing a trace amount of Ag3PO4 to form the Ag3PO4/g-C3N4 hybrid composite, in which Ag nanoparticles at the interface of Ag3PO4/g-C3N4 act as the charge separation center to form the visible light driven Z-scheme system. With energy higher than the band gap in g-C3N4 and Ag3PO4, photoinduced electrons jump into CB and leave holes in VB. Ag nanoparticles were formed under visible light irradiation. The CB potential of Ag3PO4 is more negative than the Fermi level of metallic Ag, and the VB potential of g-C3N4 is more positive than the Fermi level of Ag. The phtotoinduced electrons in CB of Ag3PO4 could easily shift into Ag nanoparticles because of the more positive Fermi energy of Ag than the CB level of Ag3PO4. Synchronously, the holes in the VB of g-C3N4 can also shift into Ag nanoparticles due to the more negative Fermi energy of Ag than the VB level of g-C3N4. In the photocatalytic process, Ag nanoparticles at the interface act as the recombination center for the electrons from the CB of Ag3PO4 and holes from VB of g-C3N4. Thus the photoinduced electrons hold back at the CB of g-C3N4, and photoinduced holes at the VB of Ag3PO4, enhancing the separation of the photoinduced electrons and holes. The electrons in CB of g-C3N4 is more negative, with strong reduction power, which is responsible for the enhanced photocatalytic efficiency of water splitting [30,31,32].

a UPS spectra of Ag3PO4, pure g-C3N4, and 0.04AP-CN nanocomposite. b Proposed band structure diagram of heterojunction for the Ag3PO4/g-C3N4 composite
4 Conclusion
In summary, the Ag3PO4/g-C3N4 nanocomposites were successfully prepared by a facile calcination method by one-pot calcining the mixture of Ag3PO4 and urea. Trace Ag3PO4 introduced in the composite photocatalysts enhanced the photocatalytic H2 evolution rate under visible light irradiation. 0.04AP-CN photocatalyst showed highest photocatalytic H2 evolution rate of 27.36 µmol/h with 3 wt% Pt ion and 8.93 µmol/h without co-catalyst. Compared to the pure g-C3N4 nanosheets, the photocatalytic H2 evolution rate increases 3.34 times without Pt co-catalyst, and 2.39 times with 3 wt% Pt co-catalyst. The experimental results showed that the synergistic effect of the interface between Ag3PO4 and g-C3N4 resulted in the improved separation of photoinduced charge carriers and enhanced absorption in the visible light region.
References
Liu C, Tang J, Chen HM, Liu B, Yang P (2013) A fully integrated nanosystem of semiconductor nanowires for direct solar water splitting. Nano Lett 13:2989–2992
Hisatomi T, Kubota J, Domen K (2014) Recent advances in semiconductors for photocatalytic and photoelectrochemical water splitting. Chem Soc Rev 43:7520–7535
Liu B, Wu CH, Miao JW, Yang PD (2014) All inorganic semiconductor nanowire mesh for direct solar water splitting. ACS Nano 8:11739–11744
Cui YJ (2015) In-situ synthesis of C3N4/CdS composites with enhanced photocatalytic properties. Chin J Catal 36:372–379
Liu G, Wang T, Zhang H, Meng X, Hao D, Chang K, Li P, Kako T, Ye J (2015) Nature-Inspired environmental “phosphorylation” boosts photocatalytic H2 production over carbon nitride nanosheets under visible-light irradiation. Angew Chem Int Ed Engl 54:13561–13565
Wu P, Wang J, Zhao J, Guo L, Osterloh FE (2014) High alkalinity boosts visible light driven H2 evolution activity of g-C3N4 in aqueous methanol. Chem Commun 50:15521–15524
Niu P, Zhang L, Liu G, Cheng H-M (2012) Graphene-like carbon nitride nanosheets for improved photocatalytic activities. Adv Funct Mater 22:4763–4770
Dou H, Chen L, Zheng S, Zhang Y, Xu GQ (2018) Band structure engineering of graphitic carbon nitride via Cu2+/Cu+ doping for enhanced visible light photoactivity. Mate Chem Phys 214:482–488
Liu E, Chen J, Ma Y, Feng J, Jia J, Fan J, Hu X (2018) Fabrication of 2D SnS2/g-C3N4 heterojunction with enhanced H2 evolution during photocatalytic water splitting. J Colloid Interface Sci 524:313–324
Pan C, Xu J, Wang Y, Li D, Zhu Y (2012) Dramatic activity of C3N4/BiPO4 photocatalyst with core/shell structure formed by Self-assembly. Adv Funct Mater 22:1518–1524
Wang X, Zhang L, Lin H, Nong Q, Wu Y, Wu T, He Y (2014) Synthesis and characterization of a ZrO2/g-C3N4 composite with enhanced visible-light photoactivity for rhodamine degradation. RSC Adv 4:40029–40035
He K, Xie J, Liu ZQ, Li N, Chen X, Hu J, Li X (2018) Multi-functional Ni3C cocatalyst/g-C3N4 nanoheterojunctions for robust photocatalytic H2 evolution under visible light. J Mater Chem A 6:13110–13122
Bi L, Xu D, Zhang L, Lin Y, Wang D, Xie T (2015) Metal Ni-loaded g-C3N4 for enhanced photocatalytic H2 evolution activity: the change in surface band bending. Phys Chem Chem Phys 17:29899–29905
Ren HT, Jia SY, Wu Y, Wu SH, Zhang TH, Han X (2014) Improved photochemical reactivities of Ag2O/g-C3N4 in phenol degradation under UV and visible light. Ind Eng Chem Res 53:17645–17653
Yan H, Yang H (2011) TiO2-g-C3N4 composite materials for photocatalytic H2 evolution under visible light irradiation. J Alloys Compd 509:L26–L29
Wei H, McMaster WA, Tan JZY, Chen D, Caruso RA (2018) Tricomponent brookite/anatase TiO2/g-C3N4 heterojunction in mesoporous hollow microspheres for enhanced visible-light photocatalysis. J Mater Chem A 6:7236–7245
Chai B, Peng T, Mao J, Li K, Zan L (2012) Graphitic carbon nitride (g-C3N4)-Pt-TiO2 nanocomposite as an efficient photocatalyst for hydrogen production under visible light irradiation. Phys Chem Chem Phys 14:16745–16752
Xu H, Yan J, Xu Y, Song Y, Li H, Xia J, Huang C, Wan H (2013) Novel visible-light-driven AgX/graphite-like C3N4 (X = Br, I) hybrid materials with synergistic photocatalytic activity. Appl Catal B 129:182–193
Wang W, Yu JC, Xia D, Wong PK, Li Y (2013) Graphene and g-C3N4 nanosheets cowrapped elemental alpha-sulfur as a novel metal-free heterojunction photocatalyst for bacterial inactivation under visible-light. Environ Sci Technol 47:8724–8732
Hu S, Qu X, Li P, Wang F, Li Q, Song L, Zhao Y, Kang X (2018) Photocatalytic oxygen reduction to hydrogen peroxide over copper doped gaphitic carbon nitride hollow microsphere: the effect of Cu(I)-N active sites, Chem. Engineering J 334:410–418
Dou H, Zheng S, Zhang Y (2017) Graphitic carbon nitride with S and Fe(III) codoping for improved photodegradation performance. Catal Lett 148:601–611
Chen F, Yang Q, Wang Y, Zhao J, Wang D, Li X, Guo Z, Wang H, Deng Y, Niu C, Zeng G (2017) Novel ternary heterojunction photcocatalyst of Ag nanoparticles and g-C3N4 nanosheets co-modified BiVO4 for wider spectrum visible-light photocatalytic degradation of refractory pollutant. Appl Catal B 205:133–147
Wang X, Yan J, Ji H, Chen Z, Xu Y, Huang L, Zhang Q, Song Y, Xu H, Li H (2016) MO degradation by Ag-Ag2O/g-C3N4 composites under visible-light irradation. Springerplus 5:369
Zhou T, Xu Y, Xu H, Wang H, Da Z, Huang S, Ji H, Li H (2014) In situ oxidation synthesis of visible-light-driven plasmonic photocatalyst Ag/AgCl/g-C3N4 and its activity. Ceram Int 40:9293–9301
Bi Y, Ouyang S, Umezawa N, Cao J, Ye J (2011) Facet effect of single-crystalline Ag3PO4 sub-microcrystals on photocatalytic properties. J Am Chem Soc 133:6490–6492
Hou Y, Zuo F, Ma Q, Wang C, Bartels L, Feng P (2012) Ag3PO4 Oxygen Evolution Photocatalyst Employing Synergistic Action of Ag/AgBr Nanoparticles and Graphene Sheets. J Phy Chem C 116:20132–20139
He P, Song L, Zhang S, Wu X, Wei Q (2014) Synthesis of g-C3N4/Ag3PO4 heterojunction with enhanced photocatalytic performance. Mater Res Bull 51:432–437
Kumar S, Surendar T, Baruah A, Shanker V (2013) Synthesis of a novel and stable g-C3N4–Ag3PO4 hybrid nanocomposite photocatalyst and study of the photocatalytic activity under visible light irradiation. J Mater Chem A 1:5333
Ren Y, Zhao Q, Li X, Xiong W, Tade M, Liu L (2014) 2D Porous graphitic C3N4 nanosheets/Ag3PO4 nanocomposites for enhanced visible-light photocatalytic degradation of 4-chlorophenol. J Nanopart Res 16:2532
Jiang D, Zhu J, Chen M, Xie J (2014) Highly efficient heterojunction photocatalyst based on nanoporous g-C3N4 sheets modified by Ag3PO4 nanoparticles: synthesis and enhanced photocatalytic activity. J Colloid Interface Sci 417:115–120
He Y, Zhang L, Teng B, Fan M (2015) New application of Z-scheme Ag3PO4/g-C3N4 composite in converting CO2 to fuel. Environ Sci Technol 49:649–656
Yang X, Tang H, Xu J, Antonietti M, Shalom M (2015) Silver phosphate/graphitic carbon nitride as an efficient photocatalytic tandem system for oxygen evolution. Chem Sus Chem 8:1350–1358
Cummings CY, Marken F, Peter LM, Tahir AA, Wijayantha KG (2012) Kinetics and mechanism of light-driven oxygen evolution at thin film alpha-Fe2O3 electrodes. Chem Commun 48:2027–2029
Zhong DK, Gamelin DR (2010) Photoelectrochemical water oxidation by cobalt catalyst (“Co-Pi”)/alpha-Fe2O3 composite photoanodes: oxygen evolution and resolution of a kinetic bottleneck. J Am Chem Soc 132:4202–4207
Bledowski M, Wang L, Ramakrishnan A, Khavryuchenko OV, Khavryuchenko VD, Ricci PC, Strunk J, Cremer T, Kolbeck C, Beranek R (2011) Visible-light photocurrent response of TiO2-polyheptazine hybrids: evidence for interfacial charge-transfer absorption. Phys Chem Chem Phys 13:21511–21519
Li J, Shen B, Hong Z, Lin B, Gao B, Chen Y (2012) A facile approach to synthesize novel oxygen-doped g-C3N4 with superior visible-light photoreactivity. Chem Commun 48:12017–12019
Lin L, Ren W, Wang C, Asiri AM, Zhang J, Wang X (2018) Crystalline carbon nitride semiconductors prepared at different temperatures for photocatalytic hydrogen production. Appl Catal B 231:234–241
Zhu Y, Ren J, Yang X, Chang G, Bu Y, Wei G, Han W, Yang D (2017) Interface engineering of 3D BiVO4/Fe-based layered double hydroxide core/shell nanostructures for boosting photoelectrochemical water oxidation. J Mater Chem A 5:9952–9959
Dou H, Long D, Zheng S, Zhang Y (2018) A facile approach to synthesize graphitic carbon nitride microwires for enhanced photocatalytic H2 evolution from water splitting under full solar spectrum. Catal Sci Technol 8:3599–3609
Li T, He Y, Cai J, Lin H, Luo M, Zhao L (2013) Preparation and characterization of Ag-loaded SmVO4 for photocatalysis application. Photochem Photobiol 89:529–535
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant Nos. 21173170, 51801164).
Author information
Authors and Affiliations
Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Long, D., Dou, H., Rao, X. et al. Z-Scheme Ag3PO4/g-C3N4 Nanocomposites for Robust Cocatalyst-Free Photocatalytic H2 Evolution Under Visible Light Irradiation. Catal Lett 149, 1154–1166 (2019). https://doi.org/10.1007/s10562-019-02704-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-019-02704-y