
Abstract
In this work, a facile solvothermal method with the assistance of chitosan (CS) has been developed to prepare well-dispersed ceria core–shell nanospheres. The effects of CS on the growth mechanism and the catalytic performance of CeO2 and Au/CeO2 nanocomposites for CO oxidation are investigated in detail. CS leads to the formation of core–shell nanospheres as soft template, changing the exposed lattice planes, and reducing Ce4+ to Ce3+ as reducing agent, resulting in the increasing of the oxygen vacancy, following the co-existence of Au3+ and Au0, which leads to the improvement of the catalytic activity for CO oxidation. As a result, the synthesized Au/CeO2 nanospheres with the assistance of CS exhibit a higher catalytic activity in CO oxidation than Au/CeO2 like-cube.
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Noble metals such as Ag, Pd, Au and Pt combined with some other nanomaterials have attracted intense attention in the past decade because of their unique chemical and physical properties [1, 2]. It has been verified that gold nanoparticles show high activity on selective oxidation catalysis [3–6]. Meanwhile, the supports strongly impacted the catalytic property of gold nanoparticles, especially, the morphology of supports play an important role in catalytic performance [7]. To our knowledge, gold nanoparticles are easy to grow larger and sinter at high temperature [8]. In addition, conventional methods easily lead to the separation of gold nanoparticles from the support. Hence, it is still a great challenge to prepare ideal nanocatalyst supports for enhancing the catalytic performance.
Ceria has attracted comprehensive attention due to its wide applications in various fields such as physics [9], biomedical science [10], UV shielding [11, 12], environment protection [13] and chemistry [14], especially in catalytic oxidation of CO. In the Au/CeO2, CeO2 is beneficial to form surface and bulk vacancies which resulted from the redox properties of the Ce3+/Ce4+ pair [15]. Moreover, it can improve gold dispersion and stability against thermal sintering. Recently, gold nanoparticles supported on mesoporous CeO2 [16–18] have been reported for efficiently catalyze selective aerobic oxidation of aldehydes by Corma and Domine [19]. Yang’s group has done a lot of research work about Au/CeO2 for CO oxidation [11, 20–23].
CS, as a naturally generating polysaccharide, shows remarkable biodegradable, biocompatible characteristic and low toxicity [24], giving rise to the widely application in drug delivery systems, wastewater treatment, gene delivery, cell culture, antimicrobial agents and novel fibers as an absorbent. It is noticed that CS has been widely used to integrate with Au, Ag, Pt and Pd nanoparticles to form functional composites due to its different functional groups [25–28]. For instance, Esumi et al. [29] used CS as a protecting agent in the preparation of gold nanoparticles, and gold salt was reduced to Au0 nanoparticles by CS without any additional reducing agent. As a stabilizer and reducing agent, CS does not produce any environmental toxicity and biohazard, which is a kind of green materials [30, 31]. Xie’s group [32] synthesized Au/CS–CeO2 composite film for hydrogen peroxide sensor and the sample has shown superior conductibility and biocompatibility. However, the utilization of CS to prepare cerium oxide nanocomposites for catalyzing CO conversion was rarely reported.
Herein, we prepared CeO2 core–shell nanospheres (S0) by CS assisted solvothermal method, which were used for CO catalytic oxidation. Results show that CS plays an important role in the morphology change of ceria. Meanwhile, CS has great influence on the exposed lattice planes of ceria and the number of Ce3+, which further affects the species of Au on the samples surface. As a result, catalysts, prepared with the assistance of CS, exhibit highly efficient catalytic properties towards CO conversion.
2 Experimental
2.1 Preparation of CS–CeO2 Core–Shell Nanospheres
Cerium nitrate hexahydrate (Ce(NO3)3·6H2O, 0.2 g) and PVP (0.08 g) were dissolved in 10 mL mixed solution, which was consist of absolute methanol (CH3OH) and deionized water in the volume ratio of 1 to 1 under vigorous stirring. Then a mixture solution of CS and acetic acid was added into the above-mentioned solution with continuous stirring for 30 min. Finally, the solution was transferred into a Teflon-lined autoclave of 20 mL capacity and solvothermally heated for 24 h at 200 °C. After the autoclave was cooled down to room temperature naturally, the precipitates were washed with deionized water and absolute ethanol, dried at 60 °C overnight and then calcined at 600 °C for 2 h in air with a heating rate of 2 °C min−1. CS–CeO2 core–shell mesoporous nanospheres powders were obtained and were denoted as S0.
2.2 Preparation of CeO2 Like-Cube
Experiments without adding CS solution have also been carried out under the same conditions in order to examine the effect of CS on the formation of the core–shell products. Finally, CeO2 like-cube nanocomposites were obtained instead of core–shell structures and were expressed to H0.
2.3 Preparation of CeO2 with Different Amount of CS
CS concentration gradient experiments have been carried out under the same conditions except the amount of CS solution to investigate the effect of CS in the formation process of core–shell nanospheres. The amount of CS is 0.5, 2, 3 and 4 mL, separately. The samples were obtained for further study.
2.4 The Synthesis of Au/CS–CeO2 and Au/CeO2
According to previous literature [33]. CS–CeO2 (0.08 g) was suspended in 40 mL deionized water under vigorous stirring and ultrasonic shaking for 1 min. Then the solution was transformed into water bath at 60 °C and the pH was adjusted to the range of 7.5–8 by Na2CO3 (0.16 M) solution. An appropriate amount of HAuCl4·4H2O (0.024 M) was added into the mixture under continuous stirring at constant temperature. After 30 min of continuous stirring, the precipitate was cooled down to room temperature, filtered and washed with deionized water and absolute ethanol, dried at 60 °C overnight. The preparation procedure of Au/CeO2 was similar to that of Au/CS–CeO2. The Au concentrations were 1, 2 and 5% on the CS–CeO2 and the corresponding structures were represented as Sn (n = 1, 2 and 5); the same Au concentrations on the CeO2 were expressed to Hn (n = 1, 2 and 5).
2.5 Characterization
The phase purity of the sample was examined by a Bruker D8 Avance X-ray diffractometer with Cu–Kα radiation (γ = 0.15418 nm) in the 2θ range from 10° to 80°. The microstructure and morphology of the products were characterized using a transmission electron microscope (TEM, JEM-1011), a scanning electron microscope (SEM, SU8010) equipped with an energy-dispersive X-ray spectrometer (EDS) with an accelerating voltage of 200 kV, and a high-resolution transmission electron microscope (HRTEM, JEM-2100, 200 kV). The Raman data were obtained using a LabRAM HR4800 spectrometer while a 514 nm laser line was used as an excitation source. The X-ray photoelectron spectrometer (XPS) analysis was performed on an ESCALAB 250 X-ray photoelectron spectrometer using Al Kα radiation. Temperature-programmed reduction by hydrogen (H2-TPR) was carried out on a PCA-1200 instrument equipped with a thermal conductivity detector (TCD). Typically 50 mg catalyst was pretreated under 5% O2–Ar stream at 300 °C for 30 min (heating rate: 10 °C min−1). After cooled down to room temperature, a flow of 5% H2–Ar was introduced into the sample with a flow rate of 30 mL min−1, and then the temperature was increased to 1000 °C at a rate of 10 °C min−1. The surface areas were detected by nitrogen adsorption–desorption isotherm measurement at −196 °C via Brunauer–Emmett–Teller (BET) method and the pore-size distribution was calculated from the desorption branch using the Barrett–Joyner–Halenda (BJH) theory. Thermal gravimetric analysis (TGA) was carried out to monitor the mass loss of products at a heating rate of 10 °C min−1 from 25 to 800 °C under an air atmosphere (Metter Toledo, TGA/SDTA851).
2.6 Measurement of Catalytic Activity
The catalytic activity of the catalyst was measured by a continuous flow fixed-bed microreactor operating under atmospheric pressure. In a typical experiment, catalyst powders (25 mg) with silica sand (300 mg) were placed in the reactor. The reactant gases (1% CO, 10% O2, and 89% N2) passed through the reactor at a rate of 60 mL min−1. The composition of the gas exiting the reactor was analyzed with an online infrared gas analyzer (Gasboard, China Wuhan Cube Co.), which simultaneously detects CO and CO2 with a resolution of 10 ppm. We choose the catalytic data of the second circle as the evaluating indicator of catalytic property. Because the first circle is an activated process, especially for the catalysts loaded gold.
In-situ diffuse reflectance infrared Fourier transform (in-situ DRIFTS) spectroscopy characterization was measured in a diffuse reflectance cell (Harrick system) equipped with CaF2 windows on a Bruker Vertex 70 spectrometer using a mercury-cadmium-telluride (MCT) detector cooled by liquid nitrogen. The catalysts (ca. 40 mg) were pretreated in reactant gas (1%CO/10%O2/89%N2) at 80 °C for 30 min and cooled down to room temperature under pure N2 (30 mL min−1). Then a background spectrum was collected via 32 scans at 4 cm−1 resolution. The reaction gas (1%CO/10%O2/89%N2) was introduced into the in-situ chamber (30 mL min−1) and heated in a stepped way. DRIFTS spectra were obtained by subtracting the background spectrum from subsequent spectra. The IR spectra for every step were recorded continuously for 30 min to reach the equilibrium. And then the temperature was increasing at a rate of 2 °C min−1 and dealt at 20, 40 and 80 °C for 30 min, respectively. One DRIFT spectrum was recorded every 3 min, following the surface changes produced in the catalyst by the CO adsorption, and the curves started from 3 min, so we can obtain 9 curves at every temperature. To make the graphs clearly, we choose 3 typical curves at every temperature. They are the 3rd, 15th, 30th min, respectively.
3 Results and Discussion
3.1 Physical Characterization
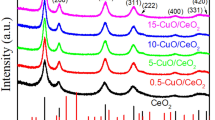
Figure 1 shows the powder XRD patterns of the samples, which are assigned to fluorite CeO2 phase (JCPDS No. 34-0394). These diffraction peaks are attributed to (111), (200), (220), (311), (222), (400), (331) and (420) of the face-centered cube structure. There are no any diffraction peaks corresponding to Au crystallite phase after loading of gold, which suggests that gold particles exist as well-dispersed state on the samples. The XRD patterns of H0 and S0 all show the diffraction peaks of CeO2 phase whether before or after the calcination at 600 °C (Fig. S1). But CS can be burned away after the calcination at 600 °C (Fig. S2).

XRD patterns of S0 (a), S1 (b), S2 (c), S5 (d), H0 (e), H1 (f), H2 (g), and H5 (h)
Figure 2 provides the morphology and structure information of S0 and H0. As can be seen in the Table S1, the pore diameter of S0 and H0 is 3.714 and 3.768 nm respectively, which indicates that the two samples are all mesoporous structures. The SEM and TEM images of S0 (Fig. 2a, b) show that the product is mesoporous core–shell nanostructures of 300 nm, assembled by many small nanocrystals, while H0 prepared without assistance of chitosan are like-cube which are also composed of small nanocrystals (Fig. 2e, f). Interestingly, it seems that the introduction of chitosan also increases the surface area of S0 which is 65.043 m2 g−1, while the one of H0 is only 42.628 m2 g−1 (listed in Table 1 ). The HRTEM image (Fig. 2c) of S0 shows that the lattice fringes are 0.27 and 0.31 nm, which correspond to the lattice planes (200) and (111) [34], respectively. From the HRTEM image (Fig. 2g), the lattice fringes of H0 is 0.19 nm, which matches well with the spacing of the well-defined plane (220) of ceria [35]. The selected area electron diffraction (SAED) patterns (Fig. 2d, h) shows that S0 is poly-crystal structure while H0 is signal-crystal structures. The above results indicate that like-cube (H0) were formed by oriented attachment of nanocrystals, while CS coats on the surface of nanocrystals, it prevents the directed self-assembly of nanocrystals, which leads to the formation of CS–CeO2 core–shell nanospheres. Therefore, CS not only induces the formation of the core–shell nanospheres, but also affects the exposed lattice planes when crystallization occurs.

The morphology characterizations (a–d) are responding to CS–CeO2 (S0), and those (e–h) are responding to CeO2 (H0): SEM (a, e), TEM (b, f), HRTEM (c, g) and the SAED patterns (d, h). The insets in a, b, e and f are the SEM and TEM images of an individual core–shell nanospheres and like-cube, respectively. The SAED pattern and HRTEM image of CS–CeO2 and CeO2 taken from a single core–shell nanosphere and a like-cube, which are shown as an inset in b and f
To further investigate the effect of CS in the formation process of core–shell nanospheres, we carried out a series of experiments by varying the amount of CS solution and found some noticeable differences in the structure of nanocatalysts shown in Figs. 2 and 3. With the amount of CS solution increasing to 2.0 mL, the morphology of ceria has changed from like-cube to solid nanospheres and the number of solid nanospheres increased. Moreover, the solid nanospheres gradually became core–shell nanospheres and the shells of core–shell structure are more and more obvious with the amount of CS solution up to 3.5 mL. When the amount of CS solution increases to 4 mL, there is no obvious difference in the morphology and structure. It suggests that certain amount of CS is crucial in the formation of core–shell nanospheres.

The TEM images of the as-obtained ceria by adding different amount of CS solution: a 0.5 mL, b 2.0 mL, c 3.0 mL and d 4.0 mL
In order to understand the formation mechanism better, we conducted a series of time-dependent experiments on the morphology evolution. Figure 4a shows the TEM images of ceria obtained at 200 °C for different solvothermal time: 4, 8, 12 and 24 h. When the solvothermal reaction time was 4 h, the samples were solid spheres with size of 300 nm. As reaction time increased to 8 h, the core–shell spheres began to appear but the shells were not well defined. The core–shell structure became more obvious when the reaction time reached 12 h, while some of them were still not core–shell nanospheres. When the reaction time was prolonged to 24 h, all the spheres almost became core–shell mesoporous spheres and the shells were distinct. As a result, both the introduction of CS and reaction time are the key factors in the formation process of CeO2 core–shell nanospheres.

a The TEM images of ceria obtained at 200 °C for different solvothermal time: 4, 8, 12 and 24 h; b the scheme illustration of the morphology formation of S0 and H0
Based on these results, we assume the formation mechanism for core–shell structures as follows (shown in Fig. 4b): at first, with the aid of surfactant PVP, small CeO2 nanocrystals are formed. Then CS also coats on the surface of each nanocrystal, which leads to non-directed self-assembly of nanocrystals, resulting to the superstructures as shown in Fig. 4a–h. As described before, S0 shows larger surface area compared with H0 without adding CS and the SAED patterns show that S0 is poly-crystal structure while H0 without assistance of CS is single-crystal structure, which means CS functions as soft template during reaction, inducing more porous structures in the superstructures. At last, some Ce4+ ions in CeO2 nanocrystals are reduced by CS to Ce3+ ions, especially the surface Ce4+ions, which cause the partial dissolution of small CeO2 nanocrystals inside superstructures. The core–shell structures begin to appear as the partial disappearance of the nanocrystals. Overall during the whole formation process of core–shell structure, CS, as soft template, firstly prevents the directed self-assembly of ceria nanocrystal and leads to the formation of the solid nanospheres, and then reduces some Ce4+ ions in CeO2 nanocrystals to Ce3+ ions, which results in the dissolution of CeO2 nanocrystals and the formation of core–shell structure.
After the loading of gold, the morphologies of the samples remain the original morphology of the pure ceria (Figs. 2a, b, 5a, b). The pore diameters of the samples change little, but the surface areas of them decrease dramatically with the deposition of Au (Table S1). As shown in Fig. 5c, g, Au nanoparticles (dark points in the white circles) are uniform and well dispersed on the surface of H0 and S0. Au nanoparticles supported on H0 and S0 have no difference in size and morphology from HRTEM images. As shown in Fig. S4, the sizes of gold particles are similar with different loading Au concentration. Elemental distributions of the samples can be detected by the elemental mapping analysis, which confirm the homogeneous distribution of Ce (purple), Au (yellow) and O (green) atom in H2 and S2. The elemental analysis shows that the Au nanoparticles are distributed uniformly on the supports.

SEM, TEM and HRTEM images of S2 (a–d) and H2 (e–h): a and e, the low-magnification SEM images; b and f, the high-magnification SEM image taken from the area marked in a and f; c and g, the TEM images; d and h, the HRTEM images of a single like-cube and a single core–shell nanosphere, respectively. Pictures under a and b are the corresponding SEM-EDS elemental mapping images of b; under c and d are the SEM-EDS elemental mapping images of f
3.2 Catalytic Properties
Because of high oxygen storage capacity [36–38], pure ceria has unique properties in the field of three-way catalysis and CO oxidation. The CO oxidation is chosen as the model catalytic reaction. H0 and S0 show different CO oxidation performance because of their different exposed facets and different morphologies. Figure 6 shows CO conversion of pure ceria and ceria series loaded with Au (1%, 2%, 5% molar ratio). The CO conversion rate reaches to 6% at 165 °C for H0, while 125 °C for S0. Moreover, the CO conversion rate is only 87% at 320 °C for H0 while 100% for S0. The catalytic performance of S0 is obviously better than that of H0, which shows that the addition of CS into the solvothermal reaction improved the catalytic properties of pure ceria. After Au loading, the catalytic performances of Sn are also better than that of Hn at same gold concentration, which shows that the addition of CS improves the catalytic properties of pure ceria. It is obviously that the samples with different Au loading concentration show higher catalytic activity when compared with pure ceria. The catalytic activities of H2 and S2 are the highest in their respective samples. The initial temperatures of H2 and S2 are all 25 °C, but T100 (the temperature at 100% conversion) is 85 °C for S2, which is lower 40 °C than that of H2. Further, the catalytic stability of S2 is high in our test of 12 h, which is similar with H2 (Fig. S3). All results indicate that the introduction of CS improves the catalytic activity of ceria and Au/CeO2 for CO oxidation.

CO conversion curves of Sn and Hn
3.3 Raman Spectra Analysis
To further investigate the relationship between the catalytic activity and the structure, Raman spectra analysis was performed. The main bands around 440–460 cm−1 are observed in Fig. 7, possibly due to the asymmetric breathing mode of the oxygen species [23]. However, the intensity of the band decreases after the loading of gold. Another noticed feature of the Raman spectra is that the band intensity decreases with the increase of the Au content. Both results can be attributed to the formation of Au–O–Ce. Besides, the other two weak bands at 260 and 600 cm−1 can be observed, corresponding to the second-order transverse acoustic (2TA) mode and defect-induced (D) mode [39–41], respectively. Compared with Hn, the position and intensity of the main band has little change for Sn. Therefore, the difference of oxygen vacancy between Hn and Sn is not observed from the Raman spectra analysis.

Raman spectra of (a) Hn and (b) Sn
3.4 H2-TPR
To clarify the reducibility of Au/CeO2 catalysts, the H2-TPR measurements are also performed (Fig. 8) in the temperature range of 30–650 °C. The peak (called α peak), appears around 530 °C for H0 and S0, are corresponded to the reduction of surface oxygen species attached to surface Ce4+ ions [20, 42–46]. The broad peak (α peak shown in Fig. 8) located at about 100 °C can be attributed to the reduction of surface oxygen on the gold particles. The weak peak (called β peak) appears at 250 °C, which can be attributed to the reduction of surface oxygen species attached to surface Ce4+ ions. The reduction behavior of the catalysts has a dramatic change by the addition of a small amount of Au. The α and β peaks of Hn and Sn (n = 1, 2 and 5) have little change on the position, which indicates that the species are quite similar in the catalysts. However, the peak areas show big differences (see Table 1). The α peak areas of S0 is larger than that of H0 [e.g., S0 (1103) > H0 (987)]. After the deposition of gold, the α peak areas became smaller. It can be attributed to the occupation of surface oxygen by Au for the formation of Au–O–Ce [47]. It should be pointed out that the α peak areas of Sn and Hn constantly decreased with the gold content up to 5%, which can be interpreted by that more and more surface oxygen are occupied by the increasing gold species. According to the following equation:

H2-TPR profiles of Hn (a) and Sn (b)
\(4{{\text{C}}_{\text{e}}}^{4+}+{{\text{O}}^{2 - }} \to 4{{\text{C}}_{\text{e}}}^{4+}+2{{\text{e}}^ - }/\upgamma +0.5{{\text{O}}_2} \to 2{{\text{C}}_{\text{e}}}^{4+}+2{{\text{C}}_{\text{e}}}^{3+}+\upgamma +0.5{{\text{O}}_2}\), γ represent an oxygen vacancy.
The generation of surface oxygen comes with the appearance of oxygen vacancy and Ce3+. The α and β peak areas of Sn are larger than that of Hn at the same gold content, indicating that the number of surface oxygen on the Sn is more than that of Hn, which is consistent with the results of the CO conversion. The results indicate that the loading of Au nanoparticles forms the Au–O–Ce structures on the surface, which weaken the Ce–O bands vastly [48]. The reduction of several forms of oxygen species on samples can interpret why the reduction signal appear below 200 °C is broaden and asymmetric [47, 49].
3.5 XPS Study
The surface elements and their valence states are detected by the X-ray photoelectron spectroscopy (XPS). The wide scanning XPS results of the two kinds of catalysts are illustrated in Fig. S6. It shows that the elements Ce, O and Au exist in the catalysts, as the XPS peaks of Ce3d, Au4f and O1s are observed clearly. According to literatures [43, 50–52] and as can be seen in Fig. 9a, the Au peaks of S2 at 83.9 eV (Au4f7/2), 86.3 eV (Au4f7/2), 87.7 eV (Au4f5/2) and 89.6 eV (Au4f5/2) are corresponded to Au0, Au3+, Au0, Au3+, respectively; while the Au peaks of H2 at 84.4 eV (Au4f7/2), 87.7 eV (Au4f5/2)indicate that Au element exists only as Au0, respectively. After use for CO oxidation, the XPS analysis of H2 and S2 are carried out which shown a chang in gold valence state for S2. The peaks of Au3+ disappeared, and only two peaks located at 83.9 and 87.7 eV indexed to Au4f7/2 and Au4f5/2 of Au0 are detectable (Fig. S7). It indicates the transformation of Au3+ to Au0 during the catalytic oxidation process. However, the gold state in the catalyst H2 has no changes and still exists as Au0. The mixed valence states of Au3+ and Au0 in S2 are believed to enhance catalytic activity, which can be verified by the higher catalytic performance of S2 than that of H2. The co-existence of Au3+ and Au0 in the catalyst can enhance the catalytic activity more effectively than the only existence of Au0 in the catalyst [50].

XPS spectra and fitting curves for H2 and S2: a Au 4f, b Ce 3d, c O 1s
The Ce 3d XPS spectra are shown in Fig. 9b. Peaks located at 917.3, 908.2, 903.6 and 901.9 eV correspond to the α1, α2, α3 and α4, which are components of Ce3d3/2; the other four peaks at 899.3, 889.5, 886.4 and 883.2 eV correspond to β1, β2, β3 and β4 of Ce3d5/2, respectively [17, 53, 54]. The α3 and β3 peaks are characteristic of Ce3+, and other peaks are attributed to Ce4+. The presence of Ce3+ reveals the defect structure of CeO2, which can cause oxygen vacancies. From the deconvolution of these spectra, the atomic ratio of Ce3+/Ce4+ present in the catalysts can be figured out and are 0.10 and 0.15 for H2 and S2 (listed in the Table 2), respectively. The results can be explained by the fact that CS is a kind of reducing agent and can reduce Ce4+ to Ce3+ [29]. This also further confirms our assumed formation mechanism of core–shell structures described before. From the Table 2, it also can be obtained that the mass ratio of Au/Ce is 1.5% and 1.7% for S2 and H2, separately, which is very close to the mass ratio of Au/Ce put into the reaction.
Figure 9c shows the O 1s spectra of H2 and S2. The O 1s could be fitted into two peaks: one at about 529–530 eV is attributed to the lattice oxygen and the other one at 531–532 eV is assigned to the chemisorbed oxygen species (Au–O–Ce) [55]. The species ratio is calculated by comparing the peaks areas of the oxygen species. The ratio of chemisorbed oxygen to the lattice oxygen is 0.70 for S2 and 0.29 for H2, which indicates that the relatively number of oxygen vacancies on the S2 is larger than that of H2. The results can be interpreted by the fact that the existence of Ce3+ could lead to the vacancies, a charge imbalance and unsaturated chemical bands on the sample surface. The increasing of Ce3+ peak areas represents more oxygen vacancies on the sample surface. The relative concentration of oxygen vacancies on the S2 surface is higher than H2, which is verified by H2-TPR and CO conversion.
Hence, for S2, the higher catalytic activity is ascribed to the co-existence of mixed valence states of Au and the more oxygen vacancies resulting from the more Ce3+ number, which is reduced by CS from Ce4+.
3.6 In-situ DRIFTS Spectra
DRIFT spectra of H2 and S2 at 20, 40 and 80 °C for 3td, 15th and 30th min are shown in Fig. 10 during the process of CO adsorption, respectively. The presence of a band at 2143 cm−1 is corresponding to CO adsorption on metallic gold nanoparticles [56–58]; while the band of the gaseous CO2 molecule appears at 2335 and 2360 cm−1 [59]. But in our data, the band of CO adsorption on metallic gold appears a red shift from 2143 to 2110 cm−1 and the peak position of CO2 is consistent with the data in the literature. The band of CO adsorption on metallic gold shifts to lower wavenumbers because the catalysts exposes electron-rich surface [56], which is attributed to the formation of the oxygen vacancies. For S2, the signal of CO appears from the start and the intensity of band increases with the time and the temperature increasing. But it begins to decrease with CO2 appearing at 80 °C from 3td min. In general case, once CO concentration begins to decline, CO2 starts to appear. But for H2, the appearance of CO2 is later than the decline of CO. The reasons are that CO is filling of the cell and adsorbed by the catalyst to a certain degree at the beginning of the reaction [56]. The CO2 signals are still weak for H2, while they are increasing for S2. The signals of CO2 still exist because CO cannot be transformed to CO2 completely at 80 °C. The band at 2155 cm−1 is also observed, which is assigned to CO adsorbed on positively charged species. However, the intensity of the band is very weak, which can be explained to the rapid reduction of positive charged species [59], which can promote CO convert to CO2 quickly, which is in good consistent with the XPS and CO conversion result. The result shows that the catalytic performance of S2 is higher than that of H2.

In-situ DRIFT spectra obtained under 1% CO, 10% O2 and 89% N2 for H2 (a) and S2 (b) after a 30 min activation in the reaction gas. There are three curves at every temperature and they are at the 3rd, 15th, 30th min, respectively
4 Discussion
Although XRD data has only detected the existence of CeO2 with well-defined crystal plane, TEM, HRTEM, mapping, Raman, H2-TPR and XPS data have confirmed that gold species have been successfully introduced into the system for both Hn and Sn. It is noticed that the morphology and exposed lattice planes have obvious differences between Hn and Sn. The differences are attributed to the introduction of CS during the solvothermal process. Both Raman spectra and H2-TPR results show that the number of the oxygen vacancy on the pure ceria is more than that on the samples after the loading of Au, and it decreases with the increase of the Au content, which can be attributed to the formation of Au–O–Ce. It is important to highlight that the oxygen vacancy of Sn is larger than that of Hn at the same gold content, resulting in the higher catalytic activity of Sn than Hn. As calculated from the XPS data, the atomic ratio of Ce3+/Ce4+ on the S2 are larger than that on the H2, which can be put down to the more oxygen vacancies resulting from more Ce3+, reduced by CS from Ce4+. Besides, because of the assistance of CS, the co-existence of Au3+ and Au0 in S2 also can enhance the catalytic activity. Overall, CS is the root of all the differences between Hn and Sn.
5 Conclusion
Ceria core–shell nanospheres and like-cube were synthesized via solvothermal method by adding CS solution and no-adding CS solution, respectively. It was found that the introduction of CS can not only bring on the generation of the core–shell structure and different exposed lattice planes, but also reduce some Ce4+ ions to Ce3+ ions, causing more oxygen vacancies in the Au/CS–CeO2, which has been proved by the XPS results. Furthermore, the catalytic activity of Au/CS–CeO2 was verified higher than that of Au/CeO2 at the same Au concentration by CO oxidation test, H2-TPR and in-situ DRIFTS. We hope these findings could provide a rational preparation way for other functional nanomaterials.
References
Zhu M, Qian H, Jin R (2009) J Am Chem Soc 131:7220
Hong X, Wang D, Cai S, Ron H, Li Y (2012) J Am Chem Soc 134:18165
Wang Z, Li L, Han D, Gu F (2014) Mater Lett 137:188
Chen Y, Zhu B, Yao M, Wang S, Zhang S (2010) CatalCommun 11:1003
Fabris S, Camellone MF (2009) J Am Chem Soc 131:10473
Zhang Q, Lee I, Joo J, Zaera F, Yin Y (2013) Acc Chem Res 46:1816
Chong H, Li P, XiangJ, Fu F, Zhang D, Ran X, Zhu M (2013) Nanoscale 5:7622
Arnal PM, Comotti M, Schüth F (2006) Angew Chem Int Ed 45:8224
Koski KJ, Cui Y (2013) ACS Nano 7:3739
Walkey CD, Chan WCW (2012) Chem Soc Rev 41:2780
Tang K, Zhang J, Wang W, Wang S, Guo J, Yang Y (2015) CrystEngComm 17:2690
García-Melchor M, Braga AA, Lledós A, Ujaque G, Masesras F (2013) Acc Chem Res 46:2626
Das SK, Khan MMR, Parandhaman T, Laffir F, Guha AK, Sekaran G, Mandal AB (2013) Nanoscale 5:5549
Patzke GR, Zhou Y, Kontic R, Conrad F (2011) Angew Chem Int Ed 50:826
Ma Z, Dai S (2010) Nano Res 4:3
Li Q, Xie W, Chen G, Li Y, Huang Y, Chen X (2015) Nano Res 8:3075
He B, Zhao Q, Zeng Z, Wang X, Han S (2015) J Mater Sci 50:6339
Hernández JA, Gómez SA, Zepeda TA, Fierro-González JC, Fuentes GA (2015) ACS Catal 5:4003
Corma A, Domine ME (2005) Chem Commun 32:4042
Liu W, Deng T, Feng L, Xie A, Zhang J, Wang S, Liu X, Yang Y, Guo J (2015) CrystEngComm 17:4850
Liu W, Feng L, Zhang C, Yang H, Guo J, Liu X, Zhang X, Yang Y (2013) J Mater Chem A 1:6942
Liu W, Tang K, Lin M, June LTO, Bai SQ, Young DJ, Li X, Yang Y, Hor TSA (2016) Nanoscale 8:9521
Tang K, Liu W, Li J, Guo J, Zhang J, Wang S, Niu S, Yang Y (2015) ACS Appl Mater Interfaces 7:26839
Li Y, Liu Z, Liu Y, Yang, Y, Shen G, Yu R (2006) Anal Biochem 349:33
Huang H, Yang X (2004) Biomacromolecules 5:2340
Huang H, Yuan Q, Yang X (2004) Colloids Surf B 39:31
Huang H, Yang X (2004) Carbohydr Res 339:2627
Ma Y, Li N, Yang C, Yang X (2005) Anal Bioanal Chem 382:1044
Esumi K, Takei N, Yoshimura T (2003) Colloids Surf B 32:117
Qiu Y, Ma Z, Hu P (2014) J Mater Chem 2:13471
Panahi-Kalamuei M, Alizadeh S, Mousavi-Kamazani M, Salavati-Niasari M (2015) J Ind EngChem 21:1301
Zhang W, Xie G, Li S, Lu L, Liu B (2012) Appl Surf Sci 258:8222
Schumacher B, Plzak V, Kinne M, Behm R (2003) Catal Lett 89:109
Mai H, Sun D, Zhang Y, Si H, Feng W, Zhang H, Liu H, Yan C (2005) J Phys Chem 109:24380
Wang Z, Feng X (2003) J Phys Chem B 107:13563
Gorte RJ (2010) AIChE J 56:1126
Perdomo C, Pérez A, Molina R, Moreno S (2016) Appl Surf Sci 383:42
Sun C, Chen L (2009) Eur J Inorg Chem 2009:3883
Wu Z, Li M, Howe J, Meyer HM, Overbury SH (2010) Langmuir 26:16595
Lee Y, He G, Akey AJ, Si R, Flytzani-Stephanopoulos M, Herman IP (2011) JAmChemSoc 133:12952
Chang S, Li M, Hua Q, Zhang L, Ma Y, Ye B, Huang W (2012) J Catal 293:195
Carabineiro SAS, Bastos SST, Órfão JJM, Pereira MFR, Delgado JJ, Figueiredo JL (2010) Appl Catal A 381:150
López JM, Arenal R, Puértolas B, Mayoral Á, Taylor SH, Solsona B, García T (2014) J Catal 317:167
Yen H, Seo Y, Kaliaguine S, Kleitz F (2012) Angew Chem Int Ed 51:12032
Andreeva D, Idakiev V, Tabakova T, Ilieva L, Falaras P, Bourlinos A, Travlos A (2002) Catal Today 7:51
Qi F, Adam W, Flytzani-Stephanopoulos M (2001) Catal Lett 7:87
Sasirekha N, Sangeetha P, Chen Y (2014) J Phys Chem C 118:15226
Si R, Flytzani-Stephanopoulos M (2008) Angew Chem 120:2926
Zhang J, Li L, Huang X, Li G (2012) J Mater Chem 22:10480
Zhang J, Jin Y, Li C, Shen Y, Han L, Hu Z, Di X, Liu Z (2009) Appl Catal B 91:11
Han M, Wang X, Shen Y, Tang C, Li G, Smith RL (2010) J Phys Chem C 114:793
Leppelt R, Schumacher B, Plzak V, KinneM, Behm R (2006) J Catal 244:137
Wang Q, Jia W, Liu B, Dong A, Gong X, Li C, Jing P, Li Y, Xu G, Zhang J (2013) J Mater Chem A 1:12732
Liu Y, Liu B, Liu Y, Wang Q, Hu W, Jing P, Liu L, Yu S, Zhang J (2013) Appl Catal B 142–143, 615
Ji P, Zhang J, Chen F, Anpo M (2008) J Phys Chem C 112:17809
Francisca RS, Leidy MMT, Miguel AC, Jose´ AO (2007) J Phys Chem C 111:14469
Manzoli M, Boccuzzi F, Chiorino A, Vindigni F, Deng W, Flytzanistephanopoulos M (2007) J Catal, 245:308
Tabakova T, Boccuzzi F, Manzoli M, Andreeva D (2003) Appl Catal A 252:385
Abd El-Moemen A, Abdel-Mageed AM, Bansmann J, Parlinska-Wojtan M, Behm RJ, Kučerová G (2016) J Catal 341:160
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 21476129 and 21506072), the Natural Science Foundation of Shandong Province (Grant Nos. ZR2013BM026 and BS2015CL010) and the Science & Technology Development Projects of Shandong Province (Grant No. 2014GSF117024).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ma, X., Tang, K., Liu, W. et al. Influence of Chitosan on the Microstructured Au/CeO2 Catalyst: An Enhanced Catalytic Performance for CO Oxidation. Catal Lett 147, 1322–1332 (2017). https://doi.org/10.1007/s10562-017-2039-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-017-2039-z