
Abstract
A series of Pt/CeO2 catalysts were prepared by impregnation (IM), deposition-precipitation (DP) and impregnation-reduction (IR) methods, and their catalytic activities for the oxidation of CO were evaluated at room temperature. Pt/CeO2 (IR) catalyst presented the best catalytic activity, over which CO could be completely oxidized at room temperature. Based on the characterization, it was found that on Pt/CeO2 (IR) catalyst more negatively charged metallic Pt species existed, which could efficiently absorb atmospheric oxygen and thus enhance the CO oxidation.
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Carbon monoxide (CO), as a typical inflammable and explosive gaseous contaminant, seriously threatens the safety of human. Moreover, long-term exposure to CO leads to harmful effects on human health [1]. Therefore, elimination of CO has attracted more and more attention in many aspects, such as CO2 lasers, fuel cells, indoor air cleaning, automotive exhaust treatment and so on [2]. Different methods have been put into the removal of CO, for example, the installation of ventilation equipment and adsorbents with large pores. Among them the catalytic oxidation has been regarded as a potential and green technology for the removal of CO [3].
Recently, great progress has been made in the catalytic oxidation of CO and two types of catalyst, including metal oxide and noble metal catalyst, have been applied for the oxidation of CO. Previous studies have demonstrated that CuxO/CeO2 catalyst is a potential candidate for the elimination of CO. 100 % CO conversion can be achieved in the temperature range of 100–140 °C [4–9]. Over Ag/CeO2 [10] and CuO/TixZr1 − xO2 [11] catalysts, CO can be completely removed at 200 and 150 °C, respectively. These facts indicate that high temperature is needed for the complete conversion of CO on metal oxide catalysts, which limited the application of this type of catalyst [1]. In contrast, noble catalysts could effectively eliminate CO at low temperature [12–18] and Au catalyst has been proved to be the most active one. CO could be completely removed at room temperature over Au/γ-Al2O3 [12], Au/TiO2 [14], Au/Fe2O3 [15] and Au/CeO2-TiO2 [18], respectively.
Different from Au, Pt catalyst was not active for the CO oxidation due to the CO self-poisoning effect. Pt active sites are occupied by the presence of CO due to its high sticking coefficient [19, 20]. In that case, O2 could not be adsorbed on Pt active sites according to the Langmuir–Hinshelwood mechanism [21, 22], leading to the deactivation of Pt catalyst for the CO oxidation. Bera et al. have reported that CO could be removed over 2 % Pt/CeO2 at 150 °C with a GHSV of 30,000 h−1 [23]. Liu et al. have found that Pt group metal catalysts operated well in a wide temperature window (80–180 °C) for the preferential oxidation of CO [24]. Pt39Ni22Co39 nanoalloy supported on three different supports have been investigated and CO could be completely removed at 55 °C on Pt39Ni22Co39/TiO2 catalyst [25]. This research showed that the activity is dependent on the type of the support. CeO2 has been recognized as an attractive support due to its unique redox properties. Ce3+ ions could facilitate the adsorption and activation of O2 [26], and also enhance oxygen transfer to active species [27]. Chen et al. reported that Au/CeO2 catalyst with more Ce3+ ions presented higher activity for the oxidation of formaldehyde [28].
Preparation method could significantly influence on the catalytic properties of catalysts. It has been found that precipitant affected the size and chemical state of Au species [28]. Na et al. [29] have found that compared with impregnation method and deposition-precipitation method, impregnation deposition-precipitation method could afford separated Pt active sites and Au active sites, thus promoting the activity of the catalyst. Huang et al. found that the Pt catalyst prepared by reduction method showed excellent catalytic activity for the oxidation of formaldehyde [30]. Herein, Pt/CeO2 catalysts were prepared by impregnation (IM), deposition-precipitation (DP) and impregnation-reduction (IR) methods, and their catalytic activities for the oxidation of CO were evaluated. Physicochemical properties of the catalysts have been characterized and subsequently correlated with their catalytic performance to elucidate the origin of the high CO oxidation activity.
2 Experimental
2.1 Catalyst Preparation
A series of Pt/CeO2 catalysts were prepared by impregnation method [donated as Pt/CeO2 (IM)], deposition-precipitation method [donated as Pt/CeO2 (DP)] and impregnation-reduction method [donated as Pt/CeO2 (IR)].The theoretical content of Pt in all the catalysts is 1 wt%. Pt/CeO2 (IM) catalyst was prepared as follows: CeO2 support was added into H2PtCl6 solution and the suspension was stirred for 2 h. After impregnation the suspension was dried at 60 °C by rotary evaporator under vacuum. The sample was dried at 120 °C for 12 h and then treated at 400 °C in 5 % H2/N2 for 6 h to obtain Pt/CeO2 (IM) catalyst.
Pt/CeO2 (DP) catalyst was prepared as follows: Ce(NO3)2 was added to H2PtCl6 solution to form a homogeneous system and the pH value of the system was adjusted to 10 by NaOH solution with vigorously being stirred. The system was aged at 70 °C for 2 h and then filtered and washed with distilled water to remove chlorine anion. The residue was dried at 120 °C for 12 h and then treated at 400 °C in 5 % H2/N2 for 6 h to obtain Pt/CeO2 (DP) catalyst.
Pt/CeO2 (IR) catalyst was prepared as follows: Ce(NO3)2 was added to H2PtCl6 solution to form a suspension, then NaBH4 solution was quickly added into the suspension (NaBH4/Pt = 20, molar ration) with vigorously being stirred. After being stirred for 2 h, the suspension was filtered and washed with distilled water to remove chlorine anion. The residue was dried at 80 °C for 12 h under vacuum to obtain Pt/CeO2 (IR) catalyst.
2.2 Catalyst Characterization
Surface areas of the catalysts were determined by the BET method by a Micromeritics ASAP 2000 instrument. The surface chemical states of Pt/CeO2 catalysts were investigated by XPS (PHI Quantro SXM ULVAC-PHI, Japan) using an Al Kα X-ray source (1486.7 eV) at 15 kV and 25 W with the binding energy calibrated by C1s at 284.8 eV. X-ray powder diffraction (XRD) patterns were recorded with a Shimadzu XRD-6000 diffractometer operated at 40 kV and 40 mA, using nickel-filtered Cu Kα (λ = 0.1542 nm) radiation. High-resolution transmission electron microscopy (HRTEM) micrographs were obtained with a Tecnai G2 20 S-TWIN microscope and operated at 200 kV. CO chemisorption measurements were measured by an Autochem II 2920 automated chemisorption analyzer. The reoxidation ability of the Pt/CeO2 catalysts was investigated through an O2-reoxidation experiment. Catalyst was pretreated by Ar at 120 °C for 30 min. After the catalyst was cooled down to room temperature, 1 % O2/N2 was introduced continuously and the concentration of O2 was monitored by the Thermal Conductivity Detector (TCD).
In situ Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFTS) spectra were recorded in a Nicolet 6700 FTIR spectrometer equipped with a MCT detector. Before experiments, catalysts were placed in a DRIFT cell equipped with ZnSe windows and were pretreated at 120 °C for 1 h by Ar at a flow rate of 100 mL min−1 and then cooled to 25 °C. Subsequently, the reactant gas mixture (100 ppm CO and N2) was introduced into the DRIFT cell for 50 min at room temperature via separate mass flow controllers at a flow rate of 100 mL min−1. After that air was introduced into the DRIFT cell for 50 min at room temperature. All spectra were recorded by accumulating 32 scans with a resolution of 4 cm−1.
2.3 Evaluation of Catalytic Performance
The activity evaluation for the oxidation of CO was performed in a continuous flow fixed-bed quartz reactor (i.d. = 10 mm) by using 0.36 g catalyst at 25 °C. Before the activity evaluation, the Pt/CeO2 catalysts were pretreated at 120 °C for 1 h by Ar at a flow rate of 100 mL min−1. After cooled to 25 °C, the gas was switched to the reaction gas consisted of 200 ppm CO and room air (50 % relative humidity, RH) from air compressor as the balance gas. The total flow rate was fixed at 350 mL min−1, corresponding to a GHSV of 60,000 h−1. The concentrations of CO and CO2 were monitored by a gas chromatograph (GC) equipped with Ni catalytic converter.
3 Results and Discussion
Figure 1 shows the activities of Pt/CeO2 catalysts for the oxidation of CO under the different preparation methods. For Pt/CeO2 (IM) catalyst the CO conversion is only 27 % at 25 °C. The conversion increases with the increase of temperature, which could be due to the fact that the promoted desorption of adsorbed CO leaves more active sites available for the adsorption of O2. When the reaction temperature is at 70 °C, the CO can be completely removed. The Pt/CeO2 (IR) catalyst presents higher active than Pt/CeO2 (IM) catalyst. For Pt/CeO2 (IM) catalyst, the CO conversion at 25 °C is 54 % and then 100 % conversion was obtained at 60 °C. In contrast, over Pt/CeO2 (IR) catalyst 100 % CO conversion can be achieved at room temperature. Therefore, the Pt/CeO2 (IR) catalyst possesses the highest catalytic activity for the oxidation of CO at room temperature.

The activities of Pt/CeO2 catalysts for the catalytic oxidation of CO
Figure 2 presents the effect of RH on the oxidation of CO over Pt/CeO2 (IR) catalyst at room temperature. CO conversion is 84 % when the reaction gas is dry gas. CO can be completely removed when the RH is 10 %. It indicates that the little H2O can improve the catalytic activity by generating OH species on the active sites, which can adsorb and oxidize the reactant. However, when the RH increases to 70 %, the catalytic activity of Pt/CeO2 (IR) catalyst is decreased. Previous studies have also demonstrated the negative effect of H2O on the oxidation of CO. The oxidation of CO would be suppressed by H2O if the H2O content is over 200 ppm [31]. The inhibiting effect of H2O is due to the competitive adsorption between H2O and CO molecules on the surface twofold coordinated oxygen site [32]. The effect of Pt content on the oxidation of CO has been investigated and the results are shown in Fig. 3. It is evident that the activity of Pt/CeO2 (IR) catalyst is significantly enhanced as the Pt content increased from 0.1 to 0.6 wt%. When the Pt content reaches 0.6 wt%, CO can be completely eliminated.

Effect of RH on the activity of Pt/CeO2 (IR) catalyst for the catalytic oxidation of CO

Effect of Pt content on the activity of Pt/CeO2 catalyst for catalytic oxidation of CO
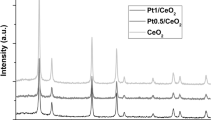
The XRD patterns Pt/CeO2 catalysts are illustrated in the Fig. 4. It is obvious that all the catalysts present typical cubic CeO2 diffraction peaks (JCPDS 43-1002). The diffraction peaks attributed to Pt species is absent, which indicates that Pt species are highly dispersed on the CeO2 support. Moreover, the intensity of the diffraction peaks of Pt/CeO2 (DP) catalyst becomes weaker, suggesting a decline in the crystallite size of CeO2. The dispersions of Pt species are 40.3, 46.7 and 45.8 % on the Pt/CeO2 (DP) catalyst, Pt/CeO2 (IR) catalyst and Pt/CeO2 (IM) catalyst, respectively. The Fig. 5 show that particle sizes of Pt species are 4–6, 2 and 2–6 nm on the Pt/CeO2 (DP) catalyst, Pt/CeO2 (IR) catalyst and Pt/CeO2 (IM) catalyst, respectively. It can be included that the preparation methods influence the dispersions and particle size of Pt species in Pt/CeO2 catalysts.

XRD patterns of Pt/CeO2 catalysts: a Pt/CeO2 (IR) catalyst, b Pt/CeO2 (IM) catalyst, c Pt/CeO2 (DP) catalyst

HRTEM images of Pt/CeO2 catalysts
The BET surface area of CeO2 support, Pt/CeO2 (IR) catalyst, Pt/CeO2 (IM) catalyst and Pt/CeO2 (DP) catalyst is 41, 33, 35 and 84 m2 g−1, respectively. It’s worth noting that the Pt/CeO2 (DP) catalyst possesses much higher BET surface area than those of Pt/CeO2 (IR) and Pt/CeO2 (IM) catalysts, however, the activity of Pt/CeO2 (DP) catalyst is poorer than that of Pt/CeO2 (IR) and Pt/CeO2 (IM) catalysts. Therefore, the activity of Pt/CeO2 catalyst is not mainly determined by the BET surface area and there exist other dominative factors for the activity of Pt/CeO2 catalyst. In addition, the BET surface area decrease of the Pt/CeO2 (IM) catalyst and Pt/CeO2 (IR) catalyst is caused by the addition of Pt species. To reveal the relationship between chemical states of the Pt species and the activity of catalysts, XPS characteristics for fresh Pt/CeO2 catalyst were carried out, as shown Fig. 6, and the corresponding data analyses are shown in Table 1. The BE values of Pt 4f7/2 at 71.1, 72.4 and 74.2 eV are ascribed to Pt0, Pt2+ and Pt4+, respectively [33]. There are no BE values of Pt 4f7/2 attributed to Pt2+ and Pt4+ over all Pt/CeO2 catalysts in Table 1, which suggests that all the Pt species have been reduced into metallic Pt species. For Pt/CeO2 (IM) catalyst, there are two peaks at 69.6 and 71.1 eV as shown in Fig. 6b. The BE value of Pt 4f7/2 at 71.1 eV is ascribed to Pt0 species, however BE value of Pt 4f7/2 at 69.6 eV is far from 71.1 eV, which is defined as Pt0′species [30, 34]. The negative shift of Pt0′specie from 71.1 to 69.6 eV is due to the electron transfer from CeO2 to Pt core [35]. The assignment of Pt species over Pt/CeO2 (DP) catalyst and Pt/CeO2 (IR) catalyst is similar to that of Pt/CeO2 (IM) catalyst. The ratios of Pt0′species to the total Pt species over Pt/CeO2 (DP) catalyst, Pt/CeO2 (IM) catalyst and Pt/CeO2 (IR) catalyst are 35.3, 51.5 and 94.5 %,respectively. Therefore the Pt/CeO2 (IR) catalyst possesses the highest content of Pt0′species. It has been proved that the negatively charged Pt0′species contributes to O2 adsorption and chemisorbed oxygen activation [30, 34]. The chemisorbed oxygen should be highly active and deeply involved in the redox cycles of CO oxidation. The charge was further transferred from negatively charged Pt to the chemisorbed oxygen. Meanwhile, the chemisorbed oxygen was activated during the charge transfer. The unique feature due to negatively charged Pt nanoparticles can facilitate the electron transfer and contribute to the activation and transfer of oxygen species. This probably accounts for the high activity for catalytic oxidation of CO at room temperature. In addition, higher electron density on Pt species due to electron transfer from CeO2 to Pt core can weaken the Pt–CO bonds, and thus enhancing its activity for the CO oxidation [35]. So high content of Pt0′specie over Pt/CeO2 (IR) catalyst should be responsible for the high catalytic activity of the CO oxidation at room temperature. It indicates that the impregnation-reduction (IR) method is superior to the impregnation (IM) and deposition-precipitation (DP) methods.

Pt 4f XPS spectra of Pt/CeO2 catalysts: a Pt/CeO2 (DP) catalyst, b Pt/CeO2 (IM) catalyst, c Pt/CeO2 (IR) catalyst
To better study the relationship between catalyst activity and catalyst physicochemical performance, in situ DRIFT spectra of the Pt/CeO2 catalysts were obtained upon exposure to CO for 50 min, and then upon exposure to O2 for another 50 min. The peak at 2084 cm−1 is ascribed to chemisorbed CO over Pt species [23]. Figure 7a shows the in situ DRIFTS spectra of the Pt/CeO2 (IR) catalyst and the intensity of peak at 2084 cm−1 increases with the reaction proceeding. It can be observed that the intensity of peak at 2084 cm−1 seriously decreases in the first 10 min in Fig. 7b, which indicates that most of the chemisorbed CO over Pt species is rapidly oxidized into CO2. Then the residual chemisorbed CO is nearly removed in the first 20 min. For Pt/CeO2 (IM) catalyst in Fig. 8, it takes 50 min to completely remove the chemisorbed CO. However, from Fig. 9, it can be seen that the peaks ascribed to the chemisorbed CO is still observed even after the 50 min reaction. Therefore, the chemisorbed CO can be efficiently removed over the Pt/CeO2 (IR) catalyst, which is accordance with the results of activity test.

In situ DRIFTS of the Pt/CeO2 (IR) catalyst as a function of time at room temperature. a 100 ppm CO/N2 reaction and b O2 reaction

In situ DRIFTS of the Pt/CeO2 (IM) catalyst as a function of time at room temperature. a 100 ppm CO and N2 reaction and b O2 reaction

In situ DRIFTS of the Pt/CeO2 (DP) catalyst as a function of time at room temperature. a 100 ppm CO and N2 reaction and b O2 reaction
To evaluate the ability of the catalyst to adsorb atmospheric oxygen, an O2-reoxidation experiment was carried out. The consumed oxygen peaks were measured by infusing 1.5 % O2 and Ar gas monitored with a TCD at room temperature. O2-reoxidation experiment results are shown in Fig. 10. The peak area of Pt/CeO2 (DP), Pt/CeO2 (IM) and Pt/CeO2 (IR) catalyst is 0.041, 0.065 and 0.091, respectively. The peak area of Pt/CeO2 (IR) catalyst is the biggest, indicating that over Pt/CeO2 (IR) catalyst more atmospheric oxygen can be adsorbed, which plays an important role for the oxidation reaction [36]. It suggests that high content of Pt0′species over Pt/CeO2 (IR) catalyst can enhance the oxygen adsorption ability, which contributes to the oxidation of CO at room temperature.

O2-reoxidaion test over Pt/CeO2 catalysts
4 Conclusion
The present research shows that the preparation method significantly affect the activity of Pt/CeO2 catalyst for the oxidation of CO. Pt/CeO2 catalyst prepared by the impregnation-reduction method presents excellent activity for the removal of CO at room temperature. Over this catalyst, more Pt0′ species could enhance the adsorption and activation of oxygen species, resulting in the formation of more chemisorbed oxygen (OII). The abundant more Pt0′ species are mainly responsible for the high activity of Pt/CeO2 (IR) catalyst for the removal of CO.
References
Seo PW, Choi HJ, Hong SI, Hong SC (2010) J Hazard Mater 178:917
Pillai UR, Deevi S (2006) Appl Catal B 64:146
Tang XF, Chen JL, Huang XM, Xu Y, Shen WJ (2008) Appl Catal B 81:115
Wang WW, Du PP, Zou SH, He HY, Wang RX, Jin Z, Shi S, Huang YY, Si R, Song QS, Jia CJ, Yan CH (2015) ACS Catal 5:2088
Caputo T, Lisi L, Pirone R, Russo G (2008) Appl Catal A 348:42
Dong G, Jia CJ, Bongard H, Spliethoff B, Weidenthaler C, Schmidt W, Schüth F (2014) Appl Catal B 152:11–18
Pérez NC, Miró EE, Zamaro JM (2013) Appl Catal B 129:416
Moretti E, Lenarda M, Riello P, Storaro L, Talon A, Frattini R, Reyes-Carmona A, Jiménez-López A, Rodríguez-Castellón E (2013) Appl Catal B 129:556
Wang ZH, Li R, Chen QW (2015) Chemphyschem 16:2415
Yang F, Huang JL, Odoom-Wubah T, Hong YL, Du MM, Sun DH, Jia LS, Li QB (2015) Chem Eng J 269:105
Huang J, Kang YF, Wang LW, Yang TL, Wang Y, Wang SR (2011) Catal Commun 15:41
Wang J, Hu ZH, Miao YX, Li WC (2014) Gold Bull 47:95
Liu LQ, Qiao BT, He YD, Zhou F, Yang BQ, Deng YQ (2012) J Catal 294:29
Liu H, Kozlov AI, Kozlova AP, Shido T, Asakura K, Iwasawa Y (1999) J Catal 185:252
Daniells ST, Overweg AR, Makkee M, Moulijn JA (2005) J Catal 230:52
Dobrosz-Gómez I, Kocembaa I, Rynkowski JM (2008) Appl Catal B 83:240
Qian K, Luo LF, Bao HZ, Hua Q, Jiang ZQ, Huang WX (2013) Catal Sci Technol 3:679
Li S, Zhu H, Qin Z, Wang G, Zhang Y, Wu Z, Li Z, Gang C, Dong W, Wu Z (2014) Appl Catal B 144:498
Chatterjee D, Deutschmann O, Warnatz J (2001) Faraday Discuss 119:371
Engel T (1978) J Chem Phys 69:373
Engel T, Ertl G (1978) J Chem Phys 69:1267
Freyer N, Kiskinova M, Pirug G, Bonzel HP (1986) Surf Sci 166:206
Bera P, Gayen A, Hegde MS, Lalla NP, Spadaro L, Frusteri F, Arena F (2003) J Phys Chem B 107:6122
Liu K, Wang AQ, Zhang T (2012) ACS Catal 2:1165
Lefu Y, Shiyao S, Rameshwori L, Valeri P, Yang R, Wanjala BN, Engelhard MH, Jin L, Jun Y, Yongsheng C (2012) J Am Chem Soc 134:15048
Carrettin S, Concepcion P, Corma A, Lopez Nieto JM, Puntes VF (2004) Angew Chem Int Ed 43:2538
Vayssilov GN, Lykhach Y, Migani A, Staudt T, Petrova GP, Tsud N, Skála T, Bruix A, Illas F, Prince KC (2011) Nat Mater 10:310
Chen BB, Shi C, Crocker M, Wang Y, Zhu AM (2013) Appl Catal B 132:245
Na H, Zhu T, Liu Z (2014) Cata Sci Technol 4:2051
Huang H, Leung DYC, Ye D (2011) J Mater Chem 21:9647
Daté M, Haruta M (2001) J Catal 201:221
Xu XL, Li JQ (2011) Surf Sci 605:1962
Tiernan MJ, Finlayson OE (1998) Appl Catal B 19:23
Li ZH, Yang K, Liu G, Deng GF, Li JQ, Li G, Yue RL, Yang J, Chen YF (2014) Catal Lett 144:1080
Alexeev OS, Chin SY, Engelhard MH, Ortiz-Soto L, Amiridis MD (2006) J Phys Chem B 109:23430
Hong X, Sun Y, Zhu T, Liu Z (2016) Catal Sci Technol 6:3606
Acknowledgments
This work has been financially supported by Special Foundation for Environmental Public Sector Research of Ministry of Environmental Protection of People’s Republic of China (No. 201409080).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hong, X., Sun, Y. Effect of Preparation Methods on the Performance of Pt/CeO2 Catalysts for the Catalytic Oxidation of Carbon Monoxide. Catal Lett 146, 2001–2008 (2016). https://doi.org/10.1007/s10562-016-1828-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-016-1828-0