
Abstract
Cu–ZrO2–MgO catalysts with varying Cu and ZrO2 weight ratios were prepared by co-precipitation method. The catalysts were characterized by X-ray diffraction, temperature programmed reduction, BET surface area, TPD of CO2 and N2O chemisorption. The catalysts were studied for selective hydrogenolysis of glycerol. The catalysts hydrogenolysis activity was related to the number available Cu sites on the support and moderate basicity of the catalysts. The catalyst with 20 %Cu–10 %ZrO2–MgO gave about 62 % of glycerol conversion with 97 % selectivity towards 1,2-propanediol. The effects of reaction temperature, hydrogen pressure, glycerol concentration and reaction time were also studied for the optimization of reaction conditions. A detailed kinetic study was also elucidated.
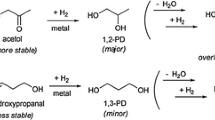
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Glycerol is a major byproduct from biodiesel industry, produced during the transesterification of vegetable oils with methanol [1]. Due to the increasing demand for green fuels, biodiesel production has been increased significantly in recent years, resulting large quantities of glycerol, whose separation and utilization has become a major concern [2]. Glycerol is a highly functional compound, an attractive reactant for the production of a large number of valuable compounds [3]. Among the various possible chemicals from glycerol, 1,2-propanediol (1,2-PD) is a important product which can be produced by the selective hydrogenolysis of glycerol. 1,2-PD is an industrially important product, which can be widely used in the production of unsaturated polyester resins, functional fluids, pharmaceuticals, cosmetics, tobacco humectants, flavors, fragrances, personal care and paints etc. [4]. Catalytic hydrogenolysis of glycerol is an attractive alternative for the preparation of 1,2-PD, due to easy availability of glycerol and eco-friendliness of the process [5].
In general, noble metal catalysts are used for the hydrogenolysis of glycerol. These noble metal catalysts are not selective due to their high activity towards C–C bond cleavage rather than C–O bond to yield lower alcohols. Non-noble metal catalysts are more selective compared to noble metal catalysts [6]. Non-noble catalysts such as Cu [7–12], Ni [13, 14], and Co [15, 16] are reported as catalysts for the selective hydrogenolysis of glycerol. Though, the activity of these catalysts are generally lower than that of noble metals, the factors such as low price and high resistance to catalyst poisoning make these catalysts more competitive [17]. Among all the above catalysts, copper is well known for its lower ability to cleave the C–C bonds of glycerol molecule, resulting in less undesired degradation products [18]. Various Cu based catalysts are reported in literature including Cu/Al2O3 [19], Cu/SiO2 [20], Cu/ZrO2 [21], Cu/ZnO [22],Cu–MgO [23] etc. However, the conversion of glycerol and yield of 1,2-PD with those catalysts was found to be limited. In addition, the studies reported with Cu based catalysts such as Cu–MgO/USY, Cu–Zn–Cr–Zr, Cu–ZnO–Al2O3 etc. also resulted in lower conversions and yields [24–26]. Though, improved yields of 1,2-PD has been achieved in few studies, they are disadvantageous inters of requiring high reaction times [5, 27], high catalyst to glycerol ratio [28], and high reaction pressure [29]. Further there are not many of the above studies cover the kinetic modeling of hydrogenolysis reaction. Therefore, it is essential to develop efficient catalysts for the hydrogenolysis of glycerol to 1,2-PD. It is also important to carry out kinetic studies to quantify the reaction progress with respect to time.
In the present work, Cu–ZrO2–MgO catalysts were prepared and studied for hydrogenolysis of glycerol to 1,2-propanediol with the main objective of enhancing conversion of glycerol and selectivity towards 1,2-PD. These catalysts are thoroughly characterized by various techniques in order to understand the relation between catalyst activity and its surface-structural characteristics. Further, studies were performed to evaluate the kinetic model for glycerol hydrogenolysis over these catalysts.
2 Experimental
2.1 Catalyst Preparation
A series of Cu–ZrO2–MgO catalysts were prepared with varied Cu to ZrO2 weight ratios by co-precipitation method. Required amounts of aqueous solutions of Cu (NO3)2·3H2O, Mg (NO3)2·6H2O and ZrO (NO3)2·H2O were taken and precipitated with 0.1 M aqueous potassium carbonate solution. The precipitate was then washed with hot water at 80 °C until the filtrate was found to be neutral pH in order to ensure the complete removal of base. Further, after cooling the precipitate was separated by filtration. The obtained precipitate was then dried overnight at 120 °C and finally calcinated at 400 °C for 3 h. The resulted catalysts were referred as xCu–yZrO2–MgO, where x and y are the weight ratios of Cu and ZrO2 respectively.
2.2 Catalyst Characterization
The catalysts were thoroughly characterized by various techniques in order to derive its surface-structural characteristics. The BET surface area of the catalyst samples were calculated from N2 adsorption–desorption data acquired on an Autosorb-1 instrument (Quantachrome, USA) at liquid N2 temperature.
The exposed copper surface area and dispersion of catalysts were evaluated by conducting dissociative N2O adsorption-H2 temperature programmed reduction (TPR) reverse titration experiments as described in the literature using BELCAT II equipment [30]. In a typical experiment, TPRI is carried out to reduce the CuO phase to Cu(0) with a 5 % H2–Ar mixture with a heating rate of 10 °C/min. Then the sample, which is in the form of Cu(0) phase is exposed to continuous N2O flow for 1 h at 100 °C to oxidize Cu(0) to Cu2O by adsorptive decomposition. Then, again TPRII is carried out for the second time on the re-oxidized Cu2O surface in order to reduce Cu2O to Cu(0). The thermal conductivity detector (TCD) was used to measure the amount of H2 uptake in TPR experiments.
X-ray powder diffraction (XRD) patterns of the catalysts were recorded on a Rigaku Miniflex (Rigaku Corporation, Japan) X-ray diffractometer using Ni filtered Cu Kα radiation (λ = 1.5406 Å) with a scan speed of 2° min−1 with a scan range of 10°–80° at 30 kV and 15 mA.
Temperature programmed reduction of the catalysts were carried out in the presence of gaseous mixture of 5 % H2 in Ar at a flow rate of 60 ml/min with a temperature ramp of 10 °C/min. Before the TPR run the catalysts were pretreated in Ar at 300 °C for 2 h. The hydrogen consumption was monitored using a thermal conductivity detector.
Basicity of the catalysts was studied by temperature programmed desorption using CO2 as probe molecule. In a typical experiment of TPD, 0.1 g of catalyst was loaded in a quartz reactor between two quartz plugs. Prior to CO2 adsorption, the catalyst was pretreated in He gas at 300 °C for 2 h and then cooled to 80 °C. The adsorption of CO2 was carried out by passing a mixture of 10 % CO2-balanced He gas over the catalyst for 1 h. Before the TPD run, the catalyst surface was flushed in He gas at 100 °C for 2 h to flush off the physisorbed CO2. TPD of the catalyst was carried out in a He gas flow at a flow rate of 30 ml/min with a temperature ramp of 10 °C/min. The CO2 desorption was monitored using the thermal conductivity detector (TCD) of a gas chromatograph (Varian, 8301).
X-ray photoelectron spectroscopy (XPS) is one of the important techniques to determine the oxidation state of transition metal. XPS measurements are conducted on a KRATOS AXIS 165 with a DUAL anode (Mg and Al) apparatus using the Mg Kα anode. The non-monochromatized Al–Kα X-ray source (hν = 1486.6 eV) is operated at 12.5 kV and 16 mA. Before acquisition of the data, the sample is out-gassed for about 3 h at 100 °C under a vacuum of 1.0 × 10−7 torr to minimize the surface contamination. The XPS instrument was calibrated using Au as standard. For energy calibration, carbon 1s photoelectron line has been used and the respective binding energy is taken as 285 eV. Charge neutralization of 2 eV was used to balance the charge up of the sample. The spectra is deconvoluted using Sun Solaris based Vision-2 curve resolver. The location and the full width at half maximum (FWHM) value for the species is first determined using the spectrum of pure sample. Symmetric Gaussian shapes were used in all cases. Binding energies for identical samples can be, in general, reproducible within ±0.1 eV.
2.3 Reaction Procedure
Glycerol hydrogenolysis experiments were carried out in 80 mL Haste alloy PARR 4843 autoclave. Prior to the experiment, the catalyst was reduced at 400 °C for 2 h with H2 flow (30 ml/min). In a typical run, 50 g of the aqueous glycerol solution (20 wt%) and 0.6 g of catalyst was loaded into the reactor. The reactor was then sealed and flushed with flowing H2 for 10 min to remove the air present in autoclave. After the purge, the reaction temperature and hydrogen pressure was raised to the required levels. The stirring speed was set at 500 rpm and maintained constant throughout the reaction. During the reaction, the decrease in hydrogen pressure was compensated by passing additional H2 gas to maintain the required pressure.
2.4 Analysis of Glycerol Hydrogenolysis Products
After completion of the reaction, the system allowed to cool down to room temperature. The gas products were collected in a gasbag and the liquid products were separated from the catalyst by filtration. The liquid products were analyzed using a gas chromatograph (Shimadzu 2010) equipped with a flame ionization detector (FID) by separating them on Inno wax capillary column (diameter 0.25 mm, length 30 m). The products were identified by using GC–MS (Shimadzu, GCMS-QP2010S) analysis. The gas phase products were analyzed by a gas chromatograph equipped with Porapak Q column and thermal conductivity detector. The products identified during glycerol hydrogenolysis include 1,2-PD, acetol, 1-propanol, and 2-propanol as hydrogenolysis products and ethylene glycol, ethanol, methanol, ethane and methane are as degradation products.
Conversion of the glycerol and selectivity to 1,2-PD was calculated on the basis of the following equations.
3 Results and Discussion
3.1 Catalysts Characterization
The physico-chemical properties of the Cu–ZrO2–MgO catalysts with varied Cu and ZrO2 content were presented in Table 1. Considering the values given in Table 1, it was observed that the individual Cu–MgO and Cu–ZrO2 catalysts have shown relatively low surface area as compared to Cu–ZrO2–MgO catalysts. In the case of Cu–ZrO2–MgO catalysts, with increase in Cu content from 5 to 20 wt% (decrease in ZrO2 loading from 25 to 10 wt%), the surface area of the catalysts was found to be increased and showed a maximum for the catalyst with 20 wt% Cu loading. Further increase in Cu content led to a decrease in surface area.
Surface active sites of Cu were measured by N2O chemisorption and the results were summarized in Table 1. The Cu surface active sites were found to exhibit an increasing trend with the increase in Cu loading from 5 to 25 wt%. These results reveal that the maximum BET surface area and surface active sites were obtained for 20 %Cu–10 %ZrO2–MgO catalyst.
XRD patterns of the calcined catalysts were shown in Fig. 1. The characteristic peaks corresponding to CuO phases were not observed for the catalysts with 5 wt% Cu loading. The peaks correspond to CuO at 2θ values of 35.5°, 38.8° and 48.5° were clearly seen for the catalysts with copper loadings above 10 wt%. The presence of peaks at 2θ values of 42.8° and 62.1° suggests the crystalline phases of MgO. In the case of catalyst with 20 wt% Cu loading, the MgO peak at 42.8° intensity decreased. This may be due to the participation of MgO in the formation of MgO–ZrO2 solid solution. The absence of ZrO2 peaks for the catalyst with 20 wt% Cu loading indicates that ZrO2 might be involved in the formation of solid solution. The crystalline phases related to tetragonal ZrO2 were detected at 2θ of 30.3° and 50.4° for the 15 % Cu–15 %ZrO2–MgO catalyst. This can be justified by the fact that at this particular composition Cu stabilizes ZrO2, both in the cubic and tetragonal phases [31].

X-ray diffraction patterns of calcined Cu–ZrO2–MgO catalysts. (a ) 5 %Cu–25 %ZrO2–MgO, (b) 10 %Cu–20 %ZrO2–MgO, (c) 15 %Cu–15 %ZrO2–MgO, (d) 20 %Cu–10 %ZrO2–MgO, (e) 25 %Cu–5 %ZrO2–MgO (ampersand symbol) CuO (asterisk symbol) MgO (hash symbol) t-ZrO2 phases
Figure 2 shows the XRD patterns of the catalysts reduced at 400 °C. The peaks related to metallic Cu were clearly observed for all the catalysts at 2θ values of 42.6°, 50.3° and 74.1°. However the peak at 42.6° is found to be merged with MgO peak. The intensity of the peaks related to Cu were increased with increase in copper loadings except for the catalysts with 20 wt% Cu content. The low intensity of the peak related to 20 wt% Cu might be due to the presence of highly dispersed copper particles on the support. With the increase in Cu loading the peak at 42.6° shifted towards higher 2θ value. This shift indicates the possible formation of solid solution, as well as incorporation of some amount of Cu into the ZrO2–MgO solid solution. The high area surface observed for this catalyst indicates the formation of solid solution.

X-ray diffraction patterns of reduced Cu–ZrO2–MgO catalysts. (a) 5 %Cu–25 %ZrO2–MgO, (b) 10 %Cu–20 %ZrO2–MgO, (c) 15 %Cu–15 %ZrO2–MgO, (d) 20 %Cu–10 %ZrO2–MgO, (e) 25 %Cu–5 %ZrO2–MgO (dollar symbol) Cu (asterisk symbol) MgO (hash symbol) t-ZrO2 phases
TPR profiles of Cu–ZrO2–MgO catalysts were shown in Fig. 3. All these catalysts showed one main reduction peak in the temperature range of 415–467 °C related to the single stage reduction of CuO to Cu which is in well agreement with the studies reported elsewhere [32]. With increase in Cu content from 5 to 15 wt% the reduction peak was marginally shifted towards lower temperature. This might be due to decrease in the interaction between Cu and MgO–ZrO2. Further increase in Cu content to 20 wt% led to shift in the reduction peak to higher temperature. This was mainly because of the reduction of strongly interacted/diffused Cu with the MgO–ZrO2 solid solution [33]. Formation of solid solution has also been evidenced by XRD results. It was observed that the hydrogen consumption rate of these catalysts was increased from 164.2 to 450.1 µmol/g with the respective increase in Cu loading from 5 to 25 wt%.

Temperature programmed reduction profiles of Cu–ZrO2–MgO catalysts. (a) 5 %Cu–25 %ZrO2–MgO, (b) 10 %Cu–20 %ZrO2–MgO, (c) 15 %Cu–15 %ZrO2–MgO, (d) 20 %Cu–10 %ZrO2–MgO, (e) 25 %Cu–5 %ZrO2–MgO
Temperatures programmed desorption of CO2 was studied to verify the basic characteristics of Cu–ZrO2–MgO catalysts. The amount of basic sites of the samples obtained from CO2 desorption peak area are listed in Table 1. The number of basic sites per gram of catalyst increased with the increase in Cu loading from 5 to 20 wt% and further increase in Cu loading the number of basic sites were marginally decreased. In the case of 15 wt% and 20 %Cu catalysts, the amount of moderate basic sites were more or less same.
X-ray photoelectron spectrum of the calcined, reduced and used 20 %Cu–10 %ZrO2–MgO catalysts were presented in Fig. 4. In the case of calcined catalyst two distinct peaks corresponding to Cu 2p 3/2 and Cu 2p 1/2 were present at the binding energies of 934.5 and 954.2 eV respectively. The main peaks were followed by strong satellite peaks around 944.1 and 963.5 eV confirming the presence of Cu2+ species. However, in the case of reduced sample the binding energy values at 932.7 and 952.7 eV demonstrates the presence of Cu0 state. The Mg 2p binding energy value of 49.5 eV was same for calcined and reduced samples. The binding energy of the Zr 3d 5/2 at 182.7 eV was attributed to Zr4+ of ZrO2. The O 1s spectra showed two bands in the binding range of 530–533 eV for calcined and reduced catalysts. XPS results suggests that copper was in metallic state during the reaction. The XPS spectrum of used catalyst suggests the presence of Cu in metallic state in the used catalyst.

X-ray photo electron spectroscopy profiles of calcined, reduced and used 20 %Cu–10 %ZrO2–MgO catalysts
3.2 Glycerol Hydrogenolysis Activity on Cu–ZrO2–MgO Catalysts
Hydrogenolysis of glycerol reaction was carried over Cu–ZrO2–MgO catalysts and results were presented in Table 2. For comparison, 20 %Cu–MgO and 20 %Cu–ZrO2 catalysts were also studied. The 20 %Cu/MgO and 20 %Cu/ZrO2 catalysts have exhibited lower glycerol conversions of 15 and 10.8 % respectively. However, the Cu–ZrO2–MgO catalysts have shown better activity than the Cu supported on ZrO2 and MgO catalysts. From the Table 2, it is important to note that with increase in the Cu loading up to 20 wt%, the glycerol conversion and 1,2-PD selectivity were found to be increased. Maximum conversion of 62 % with 97 % selectivity to 1,2-PD was obtained for the catalyst with 20 %Cu–10 %ZrO2–MgO. Further increase in Cu loading, the conversion of glycerol decreased to 54.7 %. The high activity of 20 %Cu–10 %ZrO2–MgO catalyst can be explained based on its characteristics.
The formation of 1,2-PD from glycerol proceeds in two ways. In the first one glycerol undergo dehydration to acetol over acidic sites or metallic sites of the catalyst [9, 34]. In another route glycerol undergo dehydrogenation to glyceraldehydes under basic conditions [35]. These intermediates hydrogenated on Cu sites to form 1,2-PD. The second route was favored when the reaction carried in basic medium. In neutral or acidic reaction conditions dehydration-hydrogenation is the main route. In the present catalysts it was observed the formation of acetol and no traces of glyceraldehyde was noticed. This indicates that the glycerol hydrogenolysis is following dehydration-hydrogenation route over the present catalysts. It is reported that the formation of acetol is also possible to form in both the routes [36]. In the present case the second route dehydrogenation–hydrogenation can be over ruled because of two reasons. Firstly the reaction was not carried in basic conditions. Secondly under basic conditions or on basic sites of the catalyst acetol undergoes dehydrogenation into pyruvaldehyde followed by a Cannizzaro reaction to lactic acid. It was not observed any formation of pyruvaldehyde or lactic acid [36]. The formation of acetol and absence of glyceroldehyde suggests the glycerol hydrogenolysis reaction might be proceeded dehydration-hydrogenation route over Cu–ZrO2–MgO catalysts. The 15 %Cu–15 %ZrO2–MgO catalyst showed less activity due to stabilization of ZrO2 phase on the surface and might be hindering the participation of active sites in the reaction. The number of surface active sites was increased with increase in Cu content and the catalyst with 20 wt% Cu showed high amount of available Cu active sites. The 20 %Cu–10 %ZrO2–MgO catalyst with high amount of surface area, active Cu sites and basicity might be responsible for high activity towards glycerol hydrogenolysis. These results indicate that the metallic Cu particles and basic sites are required for selective hydrogenolysis of glycerol. The 20 %Cu–10 %ZrO2–MgO catalyst is stable under present reaction conditions. XPS analysis of used catalyst suggests the presence of Cu metallic sites (Fig. 4).
3.2.1 Effect of Reaction Temperature
The hydrogenolysis of glycerol was carried out at different temperatures ranging from 160 to 220 °C using the most active 20 %Cu–10 %ZrO2–MgO catalyst and the results were presented in Fig. 5. The increase in reaction temperature from 160 to 220 °C led to substantial increment in the glycerol conversion from 41 to 82 %. At the same time the selectivity to 1,2-PD was marginally decreased from 97 to 90 %. It was observed from the results that, increase in reaction temperature leads to excessive hydrogenolysis of 1,2-PD to lower alcohols such as methanol, ethanol etc. [22, 37]. The optimum reaction temperature is 180 °C for the present catalysts to obtain maximum activity.

Influence of reaction temperature during glycerol hydrogenolysis over 20 %Cu–10 %ZrO2–MgO catalyst. Reaction conditions: glycerol conc.: 20 wt%, H2 pressure: 40 bar, reaction time: 8 h, catalyst weight: 0.6 g
3.2.2 Influence of Hydrogen Pressure
The effect of H2 pressure on glycerol hydrogenolysis has been studied by conducting the reaction with varying pressures from 20 to 50 bar at constant temperature (180 °C) and reaction time (8 h). Figure 6 shows the effect of H2 pressure on glycerol hydrogenolysis activity. The results showed that, glycerol conversion increased with increase in H2 pressure and reached to a maximum of 61.7 % at a hydrogen pressure of 40 bar. Further increase in H2 pressure, a marginal decrease in glycerol conversion was observed. This may be due to the fact that high H2 pressure conditions were thermodynamically unfavourable for initial dehydrogenation step of glycerol. Further it was observed that the selectivity to 1,2-PD was insensitive to pressure over the present catalyst.

Role of H2 pressure on the hydrogenolysis of glycerol over 20 %Cu–10 %ZrO2–MgO catalyst. Reaction conditions: glycerol conc.: 20 wt%, reaction time: 8 h, reaction temperature: 180 °C, catalyst weight: 0.6 g
3.2.3 Effect of Reaction Time
The effect of reaction time on the conversion of glycerol over 20 %Cu–10 %ZrO2–MgO catalyst was studied and the results were shown in Fig. 7. Glycerol conversion was increased with increase in reaction time from 2 to 10 h. The initial conversion of glycerol was 18 % after second hour and increased up to a maximum of 69.5 % within 10 h. If the trend of selectivity toward 1,2-PD was analyzed, it remained more or less constant. This suggests that the present catalyst is highly selective compared to any other noble metal catalysts.

Effect of reaction time on glycerol hydrogenolysis over 20 %Cu–10 %ZrO2–MgO catalyst. Reaction conditions: glycerol conc.: 20 wt%, H2 pressure: 40 bar, reaction temperature: 180 °C, catalyst weight: 0.6 g
3.2.4 Effect of Glycerol Concentration
Figure 8 shows the effect of glycerol concentration on the glycerol conversion and selectivity to 1,2-PD. The formation rate of hydrogenolysis products was high when the concentration of glycerol was low. Previous studies also suggested that the glycerol conversion was high at low glycerol concentration [38, 39]. Glycerol conversion has been decreased as the concentration of glycerol was increased. The decreasing trend in glycerol conversion with increased concentrations was expected, because the number of active catalytic sites may not be sufficient at high glycerol concentrations. It was important to mention that the overall selectivity has not changed with glycerol concentration suggesting the selective nature of the catalyst.

Effect of glycerol concentration on the hydrogenolysis of glycerol over 20 %Cu–10 %ZrO2–MgO catalyst. Reaction conditions: H2 pressure: 40 bar, reaction time: 8 h, reaction temperature: 180 °C, catalyst weight: 0.6 g
3.2.5 Effect of Catalyst Amount
Figure 9 shows the influence of catalyst quantity on the hydrogenolysis of glycerol. These results suggest that the conversion of glycerol has been increased with the increase in catalyst amount from 0.2 to 0.8 g. The increase in activity was expected as there will be more number of active sites available with increased catalyst quantities. With catalyst weight more than 0.6 g, no considerable increase in glycerol conversion was observed. A maximum of 96.5 % selectivity towards 1,2-PD was achieved at a catalyst loading of 0.6 g. Further increase in catalyst amount, the selectivity was found to be dropped slightly due to the formation of degradation products. These results suggest that 0.6 g of catalyst weight was optimum for glycerol hydrogenolysis.

Influence of catalyst loading on the hydrogenolysis of glycerol over 20 %Cu–10 %ZrO2–MgO catalyst. Reaction conditions: glycerol conc.: 20 wt%, H2 pressure: 40 bar, reaction time: 8 h, reaction temperature: 180 °C
3.3 Kinetic Studies
The present glycerol hydrogenolysis reaction falls into the category of typical gas–liquid–solid reaction with glycerol being in liquid phase, H2 in gaseous phase and catalyst in solid suspended form within the reactor. For such systems, prior to derive models representing the actual kinetics of reaction through experimentation, it is essential to ensure the absence of mass and heat transfer resistances within the system. Gas phase transfer resistance was considered to be insignificant, as the gas phase in the glycerol hydrogenolysis reactor consists of mainly hydrogen and also gas–liquid interphase has been instantaneously saturated with Hydrogen. Further, the catalyst considered in the present study was in the powdered form, reaction occurs on the surface and therefore pore diffusion resistances were treated as negligible. Uniform mixing of the reactants is ensured by studying the effect of stirring on the reaction rate within the range of 400–700 rpm. It was observed that there is no significant change in reaction rate with the change in stirrer speed beyond 500 rpm. Also the same fact is noted from the findings of Lahr et al. [40] that above 500 rpm there was no significant change in the rate of glycerol hydrogenolysis over Ru-based catalyst. In addition, the heat transfer resistances were also considered to be negligible as there was no temperature gradient existing within the reactor.
A power law type of kinetic model was considered for the study and the model parameters were evaluated by collecting the data from batch experimentation carried out for glycerol hydrogenolysis reaction using the catalyst with optimal composition 20 %Cu–10 %ZrO2–MgO as suggested by the results given in Table 2. Similar type of models were reported in recent literature for the kinetics of glycerol hydrogenolysis with some other catalysts [16, 41, 42]. The rate expression representing the glycerol hydrogenolysis reaction is given by,
where, r G rate of glycerol consumption, k is the overall reaction rate constant, C G , C H are the respective molar concentrations of glycerol and hydrogen, and α, β are the apparent reaction orders with respect to glycerol and hydrogen respectively. Different batch experiments were conducted, with varied glycerol concentrations in the range of 10–40 wt% while maintaining all other reaction parameters such as temperature, time of reaction, catalyst quantity and H2 pressure at their optimal values. Rate of glycerol consumption was computed and the ln(−r G ) versus ln(C G ) was plotted in Fig. 10a. The resulted apparent reaction order w.r.t glycerol was 0.6955. Further experiments were conducted with varying H2 pressure from 20 to 50 bars, while keeping all other reaction parameters at their optimal values. The resulted and the ln(−r G ) versus ln(C H ) was plotted in Fig. 10b. From the slope of the graph, the apparent reaction order w.r.t. H2 was found to be 0.6069. Further, the temperature dependency of the rate constant k can be evaluated by using Arrhenius law given as,
where, k o and E are the Arrhenius constants, R is the universal gas constant and T is the temperature. The reactions were conducted at different temperatures in the range of 160–220 °C and the corresponding rate constants have been evaluated by using equation Eq. (2). The Arrhenius law, ln(k) versus 1/T plotted in Fig. 11 has resulted a straight line. The activation energy obtained for the present catalyst 20 %Cu–10 %ZrO2–MgO is 22.78 kJ/mol, whose value is nearly comparable with the activation energies reported in literature for the same glycerol hydrogenolysis using some other catalysts [41, 42]. Further the effectiveness of the kinetic model developed has been evaluated by validating the model developed using the experimental data other than that used for model development. The validation data is generated by conducting additional experiments within the same window of operating conditions but at different conditions. Figure 12 compares the values of glycerol dissipation rates obtained experimentally with the values calculated using the kinetic model developed. Out of the seven experimental points considered for the analysis in the figure, four data points that are denoted as actual were the data points used in model development, where as the other three points, which are denoted as validation points are the ones that were not considered in model development. It is observed that almost all of the data points fall within the lines of ±10 % error, which demonstrate that the calculated reaction rates were in good agreement with the experimental values both in case of actual and validation data points.

Glycerol consumption rate: a apparent reaction order with respect to glycerol, b apparent reaction order with respect to hydrogen

Arrhenius plot for overall reaction of glycerol hydrogenolysis

Plot for experimental and predicted reaction rates of glycerol hydrogenolysis
4 Conclusions
Cu–ZrO2–MgO catalysts were highly active and selective for hydrogenolysis of glycerol. The catalyst with 20 %Cu–10 %ZrO2–MgO showed 62 % glycerol conversion with 97 % selectivity for 1,2-propanediol. The activity of the catalysts related to the Cu active sites present on the surface which intern related on composition of Cu–ZrO2 in the catalyst. The reaction conditions were optimized to improve the conversion and selectivity of glycerol to 1,2-propanediol. A detailed kinetics was evaluated and the activation energy for the present catalysts for glycerol hydrogenolysis is 22.78 kJ/mol.
References
Foo GS, Wei D, Sholl DS, Sievers C (2014) ACS Catal 4:3180
Li Y, Ma L, Liu H, He D (2014) Chin J Catal 35:677
Brandner A, Lehnert K, Bienholz A, Lucas M, Claus P (2009) Top Catal 52:278
Balaraju M, Rekha V, Devi BLAP, Prasad RBN, Prasad PSS, Lingaiah N (2010) Appl Catal A Gen 384:107
Yun YS, Park DS, Yi J (2014) Catal Sci Technol 4:3191
Wang S, Liu H (2007) Catal Lett 117:62
Dasari MA, Kiatsimkul P-P, Sutterlin WR, Suppes GJ (2005) Appl Catal A Gen 281:225
Yuan Z, Wang L, Wang J, Xia S, Chen P, Hou Z, Zheng X (2011) Appl Catal B Environ 101:431
Vasiliadou ES, Lemonidou AA (2011) Appl Catal A Gen 396:177
Bienholz A, Schwab F, Claus P (2010) Green Chem 12:290
Wu Z, Mao Y, Wang X, Zhang M (2011) Green Chem 13:1311
Yue C-J, Gu L-P, Su Y, Zhu S-P (2014) React Kinet Mech Catal 111:633
Yu W, Zhao J, Ma H, Miao H, Song Q, Xu J (2010) Appl Catal A Gen 383:73
Yin A-Y, Guo X-Y, Dai W-L, Fan K-N (2009) Green Chem 11:1514
Guo X, Li Y, Shi R, Liu Q, Zhan E, Shen W (2009) Appl Catal A Gen 371:108
Rekha V, Sumana C, Douglas SP, Lingaiah N (2015) Appl Catal A Gen 491:155
Nakagawa Y, Tomishige K (2011) Catal Sci Technol 1:179
Wu Z, Mao Y, Song M, Yin X, Zhang M (2013) Catal Commun 32:52
Guo L, Zhou J, Mao J, Guo X, Zhang S (2009) Appl Catal A Gen 367:93
Huang Z, Cui F, Kang H, Chen J, Xia C (2009) Appl Catal A Gen 366:288
Durán-Martín D, Ojeda M, Granados ML, Fierro JLG, Mariscal R (2013) Catal Today 210:98
Balaraju M, Rekha V, Sai Prasad PS, Prasad RBN, Lingaiah N (2008) Catal Lett 126:119
Yuan Z, Wang J, Wang L, Xie W, Chen P, Hou Z, Zheng X (2010) Bioresour Technol 101:7088
Niu L, Wei R, Yang H, Li X, Jiang F, Xiao G (2013) Chin J Catal 34:2230
Sharma RV, Kumar P, Dalai AK (2014) Appl Catal A Gen 477:147
Wang S, Liu H (2014) Chin J Catal 35:631
Meher LC, Gopinath R, Naik SN, Dalai AK (2009) Ind Eng Chem Res 48:1840
Xia S, Nie R, Lu X, Wang L, Chen P, Hou Z (2012) J Catal 296:1
Liu H, Liang S, Jiang T, Han B, Zhou Y (2012) Clean Soil Air Water 40:318
Balaraju M, Jagadeeswaraiah K, Prasad PSS, Lingaiah N (2012) Catal Sci Technol 2:1967
Ramaswamy V, Bhagwat M, Srinivas D, Ramaswamy AV (2004) Catal Today 97:63
Van Der Grift CJG, Wielers AFH, Jogh BPJ, Van Beunum J, De Boer M, Versluijs-Helder M, Geus JW (1991) J Catal 131:178
Bellido JDA, Assaf EM (2009) Appl Catal A Gen 352:179
Xia S, Yuan Z, Wang L, Chen P, Hou Z (2011) Appl Catal A Gen 403:173
Gandarias I, Arias PL, Requies J, El Doukkali M, Güemez MB (2011) J Catal 282:237
Auneau J, Michel C, Delbecq F, Pinel C, Sautet P (2011) Chem Eur J 17:14288
Alhanash A, Kozhevnikova E, Kozhevnikov I (2008) Catal Lett 120:307
Miyazawa T, Koso S, Kunimori K, Tomishige K (2007) Appl Catal A Gen 318:244
Kusunoki Y, Miyazawa T, Kunimori K, Tomishige K (2005) Catal Commun 6:645
Lahr DG, Shanks BH (2005) J Catal 232:386
Vasiliadou ES, Lemonidou AA (2013) Chem Eng J 231:103
Torres A, Roy D, Subramaniam B, Chaudhari RV (2010) Ind Eng Chem Res 49:10826
Acknowledgments
The authors VR and NR thank CSIR, New Delhi for the award of Senior Research Fellowship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Rekha, V., Raju, N., Sumana, C. et al. Selective Hydrogenolysis of Glycerol Over Cu–ZrO2–MgO Catalysts. Catal Lett 146, 1487–1496 (2016). https://doi.org/10.1007/s10562-016-1775-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-016-1775-9