
Abstract
Injured nerves cannot regenerate on their own, and a lack of engraftable human nerves has been a major obstacle in cell-based therapies for regenerating damaged nerves. A monolayer culture approach to obtain adherent neural stem cells from human embryonic stem cells (hESC-NSCs) was established, and the greatest number of stemness characteristics were achieved by the eighth generation of hESC-NSCs (P8 hESC-NSCs). To overcome deficits in cell therapy, we used microvesicles secreted from P8 hESC-NSCs (hESC-NSC-MVs) instead of entire hESC-NSCs. To investigate the therapeutic efficacy of hESC-NSC-MVs in vitro, hESC-NSC-MVs were cocultured with dorsal root ganglia to determine the length of axons. In vivo, we transected the sciatic nerve in SD rats and created a 5-mm gap. A sciatic nerve defect was bridged using a silicone tube filled with hESC-NSC-MVs (45 μg) in the MVs group, P8 hESC-NSCs (1 × 106 single cells) in the cell group and PBS in the control group. The hESC-NSC-MVs group showed better morphological recovery and a significantly greater number of regenerated axons than the hESC-NSCs group 12 weeks after nerve injury. These results indicated that the hESC-NSC-MVs group had the greatest ability to repair and reconstruct nerve structure and function. As a result, hESC-NSC-MVs may have potential for applications in the field of nerve regenerative repair.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autologous nerve graft is considered the clinical “golden standard” for repairing peripheral nerve defects. However, autologous nerve graft is limited due to the lack of donor resources (Houschyar et al. 2016). As technology advances, researchers have tried several experimental methods to restore injured peripheral nerves to pre-injury levels, such as supplementation with stem cells, neurotrophic factors and gene therapy (Hess et al. 2007; Zheng et al. 2016; Tsai et al. 2010). Among these methods, cell transplantation has been considered one of the most promising; many studies have shown that neural stem cells (NSCs), Schwann cells, and marrow stromal cells can regenerate peripheral nerves (Gonzalez-Perez et al. 2017; Wang et al. 2016; Xu et al. 2012). Neural stem cells possess the ability to self-renew and the potential to differentiate into various cell types, such as neurons, astrocytes, and oligodendroglia, in vitro. These characteristics have been widely used to generate a variety of differentiated neural cell types for a wide range of clinical applications that might lead to modifying therapeutic approaches for treating neuronal disease in the future (Li et al. 2014). With recent advances in stem cell culture techniques, NSCs are mainly derived from embryonic stem cells (ESCs), induced pluripotent stem cells (iPSCs) and fetal tissues in vitro(Svendsen et al. 1999). However, fetal tissues are difficult to acquire, and the acquired NSCs are non-standardized (Pollard 2013). Human embryonic stem cells (hESCs), derived from the inner cell mass of preimplantation mammalian embryos, are a unique population of pluripotent cells that could differentiate into the embryonic precursors of all corresponding adult tissues both in vitro and in vivo (Cai and Grabel 2007; Fu et al. 2014; Prajumwongs et al. 2016; Shroff et al. 2017). Human embryonic stem cell-derived neural stem cells (hESC-NSCs) are easier to standardize, are noninvasive and can greatly proliferate in vitro (Fournier et al. 2016). However, an important factor in cell transplantation is the low implantation rate after transplantation. In addition, an increasing number of studies have shown that NSCs play a role in repairing injured nerves not through direct cell integration but through the secretion of factors and that NSCs themselves are dangerous in terms of tumorigenesis (Zhang et al. 2017). Therefore, to improve poor engraftment, we tried to use microvesicles (MVs) from hESC-NSCs to restore cell–cell interactions without the need for additional cytokines. Microvesicles are active and tangible components secreted by cells into conditioned media and are the basis for material transmission and information exchange between cells and their surrounding cells or environment (Chen et al. 2014; Jie et al. 2015; Sun et al. 2012). Our group has previously shown that mesenchymal stem cell-derived microvesicles (MSC-MVs) can promote the regeneration of the sciatic nerve after crush injury. Therefore, we speculated that microvesicles released from hESC-NSCs (hESC-NSC-MVs) might promote axonal regeneration as well as MSC-MVs and chose to study them further.
Materials and methods
Human embryonic stem cell culture
We obtained a human embryonic stem cell line [10] (SHhES2, passage 49) from the stem cell bank of the Chinese Academy of Sciences under a Materials Transfer Agreement. The hESCs were cultured on Matrigel (Corning, USA) and grown in mTeSR™1 (STEMCELL Technologies, Inc., Vancouver, Canada). hESCs were ready to be passaged when the majority of colonies were large and compact and had formed centers that were dense compared to their edges. A 1 ml dispase solution (1 mg/ml, STEMCELL Technologies, Inc., Vancouver, Canada) was added to the well of hESCs and incubated for 5 min at 37 °C. When the colony edges began to curl up, we aspirated the dispase and gently washed the cells three times with Dulbecco’s Modified Eagle’s Medium/F-12 (DMEM/F-12) (Gibco, Grand Island, NY, USA). A sterile 1 ml pipette was used to scrape the detached colonies. When the cells were detached, fresh mTeSR™1 medium was used for resuspension, and then the cells were seeded on the prepared Matrigel-coated plates. Cells were cultured with a daily medium change.
Derivation of neural stem cells from hESCs
Upon reaching 70–80% confluence, hESCs were washed with DMEM/F12, and the mTeSR™1 medium was replaced by neural induction medium (DMEM/F12, 1 × B27 (Gibco), 20 ng/ml bFGF (Gibco), 20 ng/ml EGF (PeproTech), 1% nonessential amino acid (Gibco), and 1% penicillin–streptomycin). The cells were washed with DMEM/F12 after 18–21 days and dissociated by Accutase (STEMCELL Technologies, Inc., Vancouver, Canada) for 5 min at 37 °C. Finally, cells were resuspended in neural induction medium and seeded on the prepared Matrigel-coated plates (human embryonic stem cell derived-neural stem cells, P0 hESC-NSCs). The medium was changed every 2–3 days.
Flow cytometry analysis
The 1 × 105 cells were dissociated with Accutase, washed with 1X PBS, and fixed with 4% paraformaldehyde. Cells were stained with fluorochrome-conjugated mouse anti-human antibodies (Nestin (cat. no. 539843), GFAP (cat. no. 539892) and mouse IgG1 K Iso Control Alexa FluorTM488 Conjugate (cat. no. 534714); β-III tubulin (cat. no. 504510), O1 (cat. no. 506506) and mouse IgG1 K Iso Control eFluor 660 Conjugate (cat. no. 504724); CD44 (cat. no. 120441), CD133 (cat. no. 121338) and mouse IgG1 K Iso Control PE Conjugate (cat. no. 124714), all from eBioscience Thermo Fisher) for 30 min on ice and washed with 5% FBS (diluted using 1X PBS). Background staining for antibodies was determined with matched fluorochrome-conjugated isotype controls. After immunostaining, cells were resuspended in 0.3 ml 5% FBS and analyzed with FACSCalibur (BD Biosciences, USA). It did not need to be permeabilized if the membrane positively expressed proteins; otherwise, the cells were fixed with 4% paraformaldehyde and permeabilized (both from Multisites, China) with 5% FBS and then stained with fluorochrome-conjugated antibodies, as mentioned above.
Immunofluorescence staining
Upon reaching 70–80% confluence, the cells were fixed with 4% paraformaldehyde overnight at 4 °C. The cells were blocked and permeabilized for 1 h at 37 °C in PBS, 2% bovine serum albumin (BSA) and 0.1% Triton X‐100, followed by incubation with the primary antibodies at the appropriate dilution overnight at 4 °C: Nestin (cat. no. PB0920, dilution, 1:300, rabbit IgG, Boster, China), Pax6 (cat. no. PB0821, dilution, 1:300, rabbit IgG, Boster, China), and β-III tubulin (cat. no. #ab18207, dilution, 1:300, Abm, Canada). After washing with PBS, cells were incubated with the appropriate secondary antibodies at 37 °C for 2 h: CyTM3-conjugated AffiniPure Goat Anti-Rabbit IgG (H + L) (cat. no. 111-165003, dilution,1:600, Jackson, USA), Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, Boster, China). Images were captured using an Olympus inverted fluorescence microscope (IX73, Olympus Corporation of the Americas).
Polymerase chain reaction
Total RNA was isolated using the RNAprep pure Cell/Bacteria Kit (Tiangen Biotech, China), which contained a step for removing genomic DNA, according to the manufacturer’s procedures. The purity and integrity of total RNA were evaluated by OD260/280, OD260/230 and 1% (w/v) agarose gel electrophoresis. Absorption ratios were determined in NanoDrop 2000 (Thermo Fisher Scientific, USA). One microgram of total RNA from each sample was reverse transcribed into cDNA with a One-Step gDNA Removal and cDNA Synthesis kit (Transgen Biotech, China), using Oligo (dT)18 as the primer according to the manufacturer’s instructions. The cDNA was diluted tenfold for q-PCR. PCR amplification was carried out by standard procedures with 2 × Taq Master Mix (Generay, China) with initial denaturation at 94 °C for 2 min, followed by 35 cycles of denaturation at 94 °C for 15 s, annealing at 58 °C for 15 s, and extension at 72 °C for 20 s. The PCR products were separated on a 2% agarose gel and visualized by staining the prestained 5 × Loading buffer. qPCR was performed using 2 × EasyTaq PCR SuperMix (Transgen Biotech, China) and analyzed with the StepOnePlus™ Real-Time PCR system (Applied Biosystems). qPCR was performed in three steps with the following thermal setting: 2 min at 95 °C for initial enzyme activation followed by 40 amplification cycles (each 15 s at 95 °C, 15 s at 58 °C and 30 s at 72 °C with fluorescence detection) and a final step of melting curve analysis. All samples were analyzed in duplicate, and average values were used for quantification. A relative quantitative model was performed to calculate the expression of the target gene in comparison to GAPDH as the endogenous control. For PCR amplification and qPCR, genes and their related specific primers are represented in Table 1.
MVs isolation and characterization by NanoSight nanoparticle analysis instruments.
The supernatants (or conditioned medium) of the hESC-NSCs were collected to isolate microvesicles. hESC-MSC-MVs were purified following the standardized procedure reported by Bruno et al. (2013) and Gadkari et al. (2014). In brief, NSC conditioned medium was centrifuged at 2000×g for 30 min and then at 12,000×g for 15 min to remove cell fragments and impurities. Then, the conditioned medium was centrifuged at 100,000×g for at least 1 h (Beckman Coulter Optima L-90 K ultracentrifuge), washed in PBS containing 10 mM HEPES (Sigma) and followed by a second ultracentrifugation in the same way. We removed the excess fluid and added 100 μl PBS to resuspend the pellets. The protein content of the hESC-NSC-MVs was analyzed by the BCA Protein Assay Kit (CWBIO,China). hESC-NSC-MVs were diluted with PBS in a certain ratio to measure the optimum concentration of instrument detection (1.0 × 108–2.5 × 109/ml), and diluted MVs (approximately 500 μl) were injected into the sample chamber of NanoSight nanoparticle analysis instruments (LM10 instrument, NanoSight, Britain) and analyzed by the instrument's own NTA software (Version 2.3 Build 0006 BETA2).
Dorsal root ganglia isolation and culture
Dorsal root ganglia (DRG) explants were isolated from newborn Sprague–Dawley (SD) rats which were purchased from Jiangsu University Laboratory Animal Center. DRG were isolated following the standardized procedure reported by Zuchero, J Bradley (Zuchero 2014). Immediately following isolation, we seeded the DRG onto Matrigel-coated culture dishes and incubated them in DMEM supplemented with 2% FBS at 37 °C with 5% CO2. DRG were treated after 24 h with Neurobasal medium (Gibco) supplemented with 10 μM cytosine arabinoside. After 48 h, DRG were cultured with Neurobasal medium supplemented with 2% B27 supplement (Gibco), 10 ng/ml NGF (Gibco), and L-glutamine 2 mM (Gibco), and the medium was changed every other day. All experimental procedures were conducted in accordance with China’s legislation on animal protection and approved by the Ethics Committee of Jiangsu University.
Coculture of hESC-NSC-MVs with DRG
Purified DRG were replanted on a 24-well plate precoated with Matrigel, DRG were cultured with Neurobasal medium supplemented with 45 μg hESC-NSC-MVs (MV concentration measured with BCA of 90 μg/100 μl) as an experimental group, and no MVs were used as a blank control. After culturing for 36 h in the incubator, DRG neurons were fixed with 4% paraformaldehyde and then stained for β-III tubulin. Axon length was evaluated by ImageJ software.
Rat sciatic nerve truncation injury model
Twenty-seven SD male rats (190 ± 5 g) were purchased from the Jiangsu University Laboratory Animal Center. All experimental procedures were conducted in accordance with Chinese legislation on animal protection and were approved by the Ethics Committee of Jiangsu University. The experiments were divided into 3 groups: hESC-NSCs, hESC-NSC-MVs and controls. The animal experimental procedure is shown in Fig. 1a. The rats were anesthetized by intraperitoneal administration of 1% sodium pentobarbital solution (40 mg/kg, i.p.). After anesthesia, the hair on the lateral thigh of the rats was shaven, and the skin was treated with a 70% alcohol solution. The incision extended from the lateral femoral oblique, and the muscle tissue was split. Then, the sciatic nerve was exposed in the visual field. The nerve was cut with a sterile blade, leaving the damaged ends of the nerve approximately 3.0–4.0 mm apart. An 8 mm-length silicone tube was used to bridge the nerve gap using 9-0 nylon sutures, and the muscle and skin were stitched closed with 4-0 nylon sutures. Rats were housed 2–3% and maintained in a 12–12 h light–dark cycled room with ad libitum access to food and water. In the hESC-NSCs group, the conduit was filled with hESC-NSCs (P8 hESC-NSCs mixed with 20 μl DMEM/F12+ Matrigel, 1 × 106 single cell per animal); in the hESC-NSC-MVs group, the conduit was filled with hESC-NSC-MVs (45 μg hESC-NSC-MVs diluted with 20 μl DMEM/F12+ Matrigel, 20 μl per animal); and in the control group, the conduit was filled with PBS (20 μl per animal).

Rat animal model a Establishment of a 5 mm sciatic nerve defect injury model in rats. b Rat footprints in the walking box. c Rat hindlimb footprints
General observations and behavioral analysis
After surgery, we observed the mental state, diet, ulceration and autophagy of SD rats. Functional recovery was assessed by walking track analysis, and the sciatic functional index (SFI) was calculated. SFIs were calculated at 12 weeks after surgery (Varejao et al. 2001). In brief, the rats were trained to walk across a narrow wooden track (60 cm long and 10 cm wide) leading to a darkened box (Fig. 1b). Postoperatively, the rat’s hind paws were dipped in red dye (nontoxic) before walking on the track, and the recordings continued until five measurable footprints were collected (Fig. 1c). Then, the footprints were scanned, and the SFIs were measured and calculated using the following formula:
where print length (PL) is the distance from the heel to the top of the third toe; intermediary toe (IT) spread is the distance from the second to the fourth toe; and toe spread (TS) is the distance between the first and the fifth toe. Respectively, the NPL, NTS and NIT represent the PL, TS and IT recorded from the nonoperated foot, EPL, ETS and EIT represent the PL, IT and TS recorded from the operated, experimental foot. An SFI value of approximately 0 indicates better recovery, whereas an SFI value of approximately 100 represents total dysfunction.
Histological assessment of regenerated nerve and gastrocnemius muscle
After conventional feeding for 12 weeks, the rats were anesthetized prior to intracardial perfusion with 0.1 M PBS followed by 4% paraformaldehyde in PBS (pH 7.4). Then the nerve samples and gastrocnemius muscle were harvested, postfixed in buffered 4% paraformaldehyde for 4 h, dehydrated by increasing concentrations of ethanol, embedded in paraffin, cut into 3-μm thickness for hematoxylin–eosin (HE) staining (Department of Pathology, Zhenjiang First People's Hospital) and Masson staining (wanleibio, China). The sections were observed by light microscopy (DP73, Olympus, Japan).
Wet weight analysis of gastrocnemius muscle
12 weeks after surgery, the nerve samples were removed, and the gastrocnemius muscle of the bilateral lower extremity was rapidly and completely resected and weighed using a precision electronic balance. The rat gastrocnemius wet weight recovery rate was calculated according to the following formula: (Wet weight of the experimental side gastrocnemius/wet weight of the healthy side gastrocnemius) × 100%.
Statistical analysis
A one-way ANOVA with multiple comparisons was adopted to evaluate the difference among three or more groups. The experiment was independently carried out at least three times. Data are shown as the mean ± standard deviation (SD), and a p value ≤ 0.05 was statistically significant.
Results
The characteristics of neural stem cells differentiated from human embryonic stem cells
The hESCs cultured under a feeder-free condition grew into large compact colonies with clear and smooth edges (Fig. 2a). Approximately 18 days after culture in neural induction medium, the cells began to become oval or partly triangular, resulting in bipolar or tripolar shapes. Upon reaching more than 75% confluence, cells were digested and seeded on new Matrigel-coated wells. The cells could be extensively passaged to 50 generations while retaining their characteristic morphological characteristics and could be frozen and thawed (Fig. 2b–d). The derived cells were characterized by PCR and immunostaining analysis for the expression of NSC markers. Compared with hESCs, several NSC genes, including Pax6 and Sox1, were significantly upregulated in the derived cells, whereas the pluripotency gene Oct4 was downregulated (Fig. 2e). Immunofluorescence staining revealed that the hESC-NSCs expressed Nestin and Pax6 (Fig. 3a, b). Meanwhile, flow cytometry analysis revealed that human embryonic stem cell-derived neural stem cells expressed CD44 and CD133 and weakly expressed O1 on the surface, while Nestin, β-III tubulin and GFAP were expressed in the cytoplasm of hESC-NSCs (Fig. 3c, d). Since hESC-NSCs could be extensively passaged to 50 generations, and the shapes of these cells remained unchanged in this process, to further characterize in vitro-cultured hESC-NSCs, we used P2, P8 and P18 hESC-NSCs to observe the expression of Nestin, Pax6, Sox1, and Sox2 (Fig. 4a). Flow cytometry analysis showed the expression of Nestin, β-III tubulin, GFAP, O1, and CD133, in P2, P8 and P18 hESC-NSCs (Fig. 4b).

Human embryonic stem cell and hESC-derived neural stem cells. a hESCs were cultured on Matrigel. b–d hESC-NSCs at passages 8, 13 and 35. e PCR for gene expression in hESCs and P8 hESC-NSCs (a Scale bars 200 µm; b–d Scale bars 100 µm; e NC, negative control)

Cellular localization of P8 hESC-NSCs markers. a, b hESC-NSCs immunofluorescence staining for Nestin and Pax6. c Cell membrane analysis of Nestin, β-III tubulin, GFAP, O1, CD133 and CD44 in P8 hESC-NSCs. d Intracellular analysis of Nestin, β-III tubulin, and GFAP in P8 hESC-NSCs. (a, b Scale bars 100 µm)

Expression of neural markers in different passages of hESC-NSCs. a qPCR analysis was performed for Nestin, Pax6, Sox1 and Sox2, and the expression levels of P8 hESC-NSCs and P18 hESC-NSCs were compared with those of P2 hESC-NSCs (n = 3, *p < 0.05; **p < 0.01; ****p < 0.0001. N.S., not significant). b Flow cytometry analysis of Nestin, β-III tubulin, GFAP, O1, and CD133 in different passages of hESC-NSCs. (Black entity: isotype control; solid line: P2 hESC-NSCs; thick broken line: P8 hESC-NSCs; thin broken line: P18 hESC-NSCs)
hESC-NSC-MVs promote axonal growth of DRG
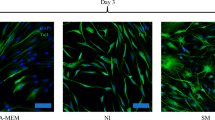
The diameter distribution of hESC-NSC-MVs was found to be 50–1000 nm by a NanoSight nanoparticle analysis system (Fig. 5a, b). Dorsal root ganglion explants were used to test the effects of the supplementation on neurite outgrowth. In vitro experiment, hESC-NSC-MVs and PBS (as a control) were cultured with DRG and then the DRG were stained with β-III tubulin. The neurite length of DRG in the hESC-NSC-MVs group was much longer than that in the PBS group, representative photomicrographs of dorsal root ganglia with β-III tubulin markers are shown in Fig. 6a–b. The difference was statistically significant (Fig. 6c).

Nanoparticle Tracking Analysis (NTA) by NanoSight of hESC-NSC-MVs. a The shape of hESC-NSC-MVs. b The average size of hESC-NSC-MVs

hESC-NSC-MVs cocultured with DRG for 36 h. a, b β-III tubulin immunofluorescence staining of DRG after co-culture with PBS (a) and hESC-NSC-MVs (b). c Quantitative comparison of the neurite length of DRG between the hESC-NSC-MVs group and the PBS (control) group (Scale bars 100 µm; n = 3, **p < 0.01)
General observations after the operation
A week after the operation, the rats were dragging their hind limbs, and their toes were closed together. On the third day, the hind limbs in the rats from all groups had become red and swollen, but no autophagy had occurred. Subsequently, redness and swelling became more aggravated, and autophagy occurred in the PBS and hESC-NSCs groups. Symptoms were mild in the hESC-NSC-MVs group. Four weeks after the operation, the toes were slightly separated, and there were withdrawal and escape reactions after plantar acupuncture. The recovery of the hESC-NSC-MVs group was better than that of the PBS group and the hESC-NSCs group. Twelve weeks after the operation, the redness and swelling had subsided in the hESC-NSC-MV group, and the dragging of the hind limbs was much improved. In the hESC-NSC-MVs group, the appearance of the affected hindlimb (Fig. 7a) was closer to that of the healthy side than it was in the PBS group or the hESC-NSCs group, but in the hESC-NSCs group, the toes closed together in the hESC-NSCs group (Fig. 7b) and the valgus deformity of the foot could be seen in the static state in PBS group (Fig. 7c). Rat walking status as shown in the animation (Online Resource 1 https://v.qq.com/x/page/q086765kvnw.html; Online Resource 2: https://v.qq.com/x/page/l0867cjh8hd.html; Online Resource 3: https://v.qq.com/x/page/y0867v4vqjx.html).

Macroscopic view of rats after operation. Hindlimb status in the hESC-NSC-MVs group (a), hESC-NSCs group (b), PBS group (c) 12 weeks after surgery
Sciatic nerve index and recovery rate of the wet weight of the gastrocnemius muscle
The SFI was measured 12 weeks after the operation. The results showed that the hESC-NSC-MVs group was significantly better than the hESC-NSCs and PBS groups (Fig. 8a). Meanwhile, the gastrocnemius muscles on the experimental side of each group were atrophied to varying degrees compared with the contralateral side (Fig. 8b–d). The recovery rate of the wet weight of the gastrocnemius muscle was higher in the hESC-NSC-MVs group than in the hESC-NSCs and PBS groups, and the difference was statistically significant (Fig. 8e).

Sciatic nerve index and gastrocnemius wet weight 12 weeks after surgery. a The sciatic functional index (SFI) in each group. b–d Macroscopic view of the gastrocnemius muscle from the operated limb 12 weeks after surgery in the PBS (b), hESC-NSCs (c), and hESC-NSC-MVs group (d), e wet weight analysis of the gastrocnemius muscle 12 weeks after the surgery (n = 9, **p < 0.01; ***p < 0.001; N.S., not significant)
Histological analysis of the regenerated nerve
Twelve weeks after the operation, the sciatic nerves of each of the rats were dissected, and the silicone tubes were opened carefully. It was found that the nerve conduits slightly adhered to the surrounding tissues and were easily peeled. The newly regenerative nerve fibers in the silicone conduits could be directly and clearly seen by naked eyes (Fig. 9a–b). As shown in Fig. 9b, in the middle of the nerve conduits, the new axons generated and completely connected the two ends of the sciatic nerve. After Masson staining, the myelin sheath was stained as a pale red wheel, the axons were dark stained centrally, and the epineurium and surrounding connective tissue were stained dark blue. In the PBS group, regenerated nerve fibers were observed, but the arrangement was loose, disordered and incomplete (Fig. 9c–e). There were significantly more regenerated nerve fibers in the hESC-NSCs group than in the PBS group, which showed loose nerve fibers and few axons (Fig. 9f–h). There were significantly more regenerated nerve fibers in the hESC-NSC-MVs group than in either of the other two groups, and the regenerated nerve fibers were orderly and densely arranged, with more axons and a thicker myelin sheath (Fig. 9i–k). As could be seen in the figure, the regenerated nerve fibers in the hESC-NSC-MVs group were closer to the normal nerve fibers (Fig. 9i–n).

Regenerated nerve after repair of a sciatic nerve defect at 12 weeks. a macroscopic view of nerve conduit with regenerative nerve fiber in rat. b The harvested regenerative nerve fiber 12 weeks after surgery. Masson staining of transverse sections from the middle segment of regenerated nerves 12 weeks after surgery, c–e PBS group, f–h hESC-NSCs group, i–k hESC-NSC-MVs group, l–n normal group. (Scale bars; c, f, i, l: 200 µm; d, g, j, m: 100 µm; e, h, k, n: 50 μm)
Histological analysis of the gastrocnemius muscle
To evaluate the atrophy of rat gastrocnemius muscle resulted from dysfunction of the sciatic nerves, gastrocnemius muscle of the three groups were stained with HE staining. The atrophy is stopped accompanied with gradual functional recovery of the sciatic nerves. Twelve weeks after the operation, the gastrocnemius muscle tissues of the PBS, hESC-NSCs and hESC-NSC-MVs groups were stained with HE. The results showed that the gastrocnemius muscle fibers of the PBS group were atrophic and fused, and the gap between cells was large (Fig. 10a). The gastrocnemius muscle fibers of the hESC-NSCs group were atrophic to a more moderate degree (Fig. 10b). The gastrocnemius muscle fibers of the hESC-NSC-MVs group were mildly atrophic, and the cell gap was small (Fig. 10c). The percentage of muscle fiber area was in the similar range between the hESC-NSC-MVs group and normal group (Fig. 10d), which were significantly higher than that in the PBS at 12 weeks after surgery (Fig. 10e). This finding indicates that hESC-NSC-MVs is capable of synergistically reversing muscle atrophy.

Histological analysis of gastrocnemius muscle in each group. Representative light micrographs of the transverse-sectioned gastrocnemius muscle following HE staining for the operated limb in the a PBS group, b hESC-NSCs group, c hESC-NSC-MVs group, d normal group at 12 weeks postoperatively. The average percentage of muscle fiber in each groupwas shown in (e). (Scale bars a–d 100 µm. *p < 0.05; ***p < 0.001)
Discussion
Nerves cannot regenerate on their own, and the current “golden standard” autografts provide the best outcomes but require suitable donor material. The lack of engraftable human nerves has been a major obstacle in cell-based therapies for regenerating damaged nerves (Sayad and Zaminy 2017). NSCs are promising transplants for diseases associated with the nervous system, so it is extremely important to obtain them in great quantities (Das et al. 2016; Lee et al. 2017). hESC- NSCs are attracting growing attention from researchers, several strategies have been employed to achieve neural stem cells from hESCs in vitro (Woo et al. 2009; Roubal et al. 2016; Salimi et al. 2014; Yasui et al. 2017). Among these strategies, this is primarily accomplished by embryoid body (EB) formation in neural induction medium to achieve neural stem cells. However, since hESCs are pluripotent, cells on the surface of EB and those inside the aggregates will encounter a varied gradient of morphogens. To overcome the limitations of EB protocol, another way to achieve high efficiency of neural stem cells is based on monolayer differentiation system of hESCs. In our monolayer culture protocol, after neural induction, hESCs changed from large compact colonies with clear and smooth edges to oval or partly triangular monolayers. Compared with hESCs, Pax6 and Sox1 were significantly upregulated in hESC-NSCs, whereas the pluripotency gene Oct4 was downregulated. Sox2 and Nestin showed no obvious differences, similar to previous reports (Oh et al. 2018). Analyses of microarray data revealed that brain-derived NSCs and hESC-NSCs were shown to express distinct groups of genes but, nonetheless, shared many properties involving NSC markers (Shin et al. 2007). Immunofluorescence staining indicated that hESC-NSCs expressed Nestin and Pax6 and had a high nucleocytoplasmic ratio. Flow cytometry analysis showed that our hESC-NSCs were homogeneous groups and expressed CD44, CD133, and weakly O1 on the membrane surface, while Nestin, β-III tubulin and GFAP were only expressed intracellularly. We succeeded in purifying and proliferating neural stem cells from hESCs as in other laboratories (Zhao et al. 2017).
The expression of Nestin in neural stem cells gradually decreases with longer culture times and greater passage numbers, and neural stem cells gradually lose the capabilities of early neural stem cells in long-term subcultures and became a kind of fate-determining progenitor cell (Banda and Grabel 2016; Li et al. 2017; Xu et al. 2009). In our study, comparison of different passages of hESC-NSCs revealed that the expression of Nestin, Pax6, Sox1, and Sox2 in P8 hESC-NSCs was highest, but according to the results of flow cytometry analysis, the expression of Nestin, β-III tubulin, GFAP, and CD133 showed no significant differences in P2, P8, and P18 hESC-NSCs. P2 and P8 hESC-NSCs weakly expressed O1, while P18 hESC-NSCs had almost no expression of O1. The expression of Nestin was contradictory between the transcriptional and antigen levels, possibly because of the sensitivity of the methods. qPCR results indicated that most of the stemness characteristics had arisen around the eighth generation of hESC-NSCs and were reduced gradually in subsequent passages. Therefore, P8 hESC-NSCs were chosen for the subsequent animal experiments.
NSCs play a vital role in the development of the nervous system and the repair of injured nerves (Wang et al. 2017a, b). However, how this role would be affected by other factors, such as their own proliferation, migration and differentiation, is currently unknown. In addition, NSCs are nuclear cells, so even if their immunogenicity is low, there is still a risk of tumorigenesis. NSC-MVs were derived from NSCs and possessed the same biological activity as the NSCs. It had been reported that MSC-MVs played the same therapeutic role as MSCs (Wei et al. 2016), and we tried to explore whether NSC-MVs had the same effect as NSCs. The particle size of MVs we extracted from NSCs was in the range of 50–1000 nm. The culture of neural tissues in vitro has become a very useful technical in neurobiology research. Through the study of nerve tissues in vitro, the pathogenesis of some neurological diseases was discussed at the cellular and molecular levels, which provided an experimental model and theoretical basis for clinical treatment. Dorsal root ganglia (DRG) are rich in sensory neurons in the peripheral nervous system. They have been widely used in neuroscience research for applications such as axon guidance and regeneration, myelination of the central and peripheral systems, and tissue engineering (Motti et al. 2017; Ying et al. 2016; Santos et al. 2017). In this study, hESC-NSC-MVs were cocultured with DRG, and hESC-NSC-MVs were shown to promote axonal regeneration of DRG. This suggested that hESC-NSC-MVs could promote the regeneration of nerves in vitro.
Severe peripheral nerve injury might lead to local hemorrhage and edema accompanied by severe inflammation. Fibroblasts proliferate and secrete a large number of collagen fibers at the injury site to form scars, inhibit the protrusion of proximal axons, and affect nerve regeneration and functional recovery (Cinteza et al. 2015; Gaudet et al. 2011). Excessive inflammation inhibits nerve regeneration, so immunosuppressive agents have been used to improve the microenvironment of and promote nerve regeneration (Li et al. 2010). This kind of inflammation reaction is reflected that a localized physical condition in which part of limbs becomes reddened, swollen, hot and painful. One week after the operation, most of the rats with sciatic nerve injury showed redness and swelling, and some even presented with ulcers and autophagy. As time passed, it was found that hESC-NSC-MVs could effectively inhibit redness and ulcer formation, and while there was no autophagy in the hESC-NSC-MV group, the PBS and hESC-NSCs groups did display it. Autophagy is caused by spontaneous neuralgia after nerve injury. The mechanism is still unknown, but we speculated that hESC-NSC-MVs possessed a substance that inhibited inflammation.
In 1982, the sciatic nerve function index was first used to evaluate functional recovery after sciatic nerve injury, and now it has become one of the most indispensable indexes to assess sciatic nerve injury (De et al. 1982). To detect the functional recovery of the injured sciatic nerve, the SFI was measured at 12 weeks. The results showed that the SFI of the hESC-NSC-MVs group was greater to that of the PBS group and hESC-NSCs group. However, the SFI index never reached healthy levels, presumably because the regenerated nerve fibers did not establish good connections with the target organs or because the muscles had atrophied due to prolonged denervation.
It has been shown that NSCs could promote the regeneration of nerve fibers after sciatic nerve injury (Fu et al. 2011; Huang et al. 2017). By Masson staining, hESC-NSC-MVs were shown to significantly improve the prognosis of injured nerves and promote the regeneration of nerve fibers and axon growth in rats with sciatic nerve injury. Compared with hESC-NSCs, hESC-NSC-MVs had more obvious effects on improving the injured nerve, so it was inferred that NSCs mainly depended on paracrine MVs in the repair of the injured nerve.
Because muscles are supported and nourished by nerves, nerve injuries could lead to dysfunction and atrophy of the muscles they control (Sando and Cederna 2016; Tuffaha et al. 2016; Wu et al. 2016). The morphological observation of the gastrocnemius muscles 12 weeks after the operation showed that the gastrocnemius muscles atrophied most severely in the PBS group, and the atrophy was less grave but still severe in the NSCs group. Gastrocnemius muscle atrophy was inhibited in the hESC-NSC-MVs group.
In summary, hESC-NSC-MVs could effectively promote the regeneration of the sciatic nerve after a defect injury. This effect was stronger in the hESC-NSC-MVs group than in the hESC-NSCs group, suggesting that MVs are an important substance secreted by NSCs to promote nerve regeneration. MVs are secreted by NSCs, but in this experiment, the treatment of hESC-NSC-MVs was superior to that of hESC-NSCs. These results suggested that hESC-NSCs were foreign bodies in the rats, which could activate the immune response of the body to reject hESC-NSCs, making them unable to effectively repair nerve injury. In addition, the survival time of hESC-NSCs in vivo, and whether it played a role in NSC function, is unknown. Therefore, NSCs are the most important seed cells in the process of nerve injury repair, and MVs are a new and important intercellular regulatory molecule. Compared with hESC-NSCs, hESC-NSC-MVs possess low immunogenicity, conferring great clinical significance in the treatment of nervous system diseases. It is expected that hESC-NSC-MVs will play a very important role as a new treatment for nervous system diseases in the future.
Conclusion
In this study, we successfully differentiated human embryonic stem cells into neural stem cells and isolated MVs from NSCs. Furthermore, the effect of hESC-NSC-MVs on nerve repair was preliminarily studied, and their value in sciatic nerve defect repair was evaluated. However, the MV substances that played a role in this repair and the mechanisms used still needed further exploration. In conclusion, hESC-NSC-MVs could be used as a potential tissue engineering repair material in the clinic, providing a new method for regenerative repair of peripheral nerve defects.
References
Banda E, Grabel L (2016) Directed differentiation of human embryonic stem cells into neural progenitors. Methods Mol Biol 1307:289–298
Bruno S, Collino F, Deregibus MC, Grange C, Tetta C, Camussi G (2013) Microvesicles derived from human bone marrow mesenchymal stem cells inhibit tumor growth. Stem Cells Dev 22:758–771
Cai C, Grabel L (2007) Directing the differentiation of embryonic stem cells to neural stem cells. Dev Dyn 236:3255–3266
Chen J, Liu Z, Hong MM et al (2014) Proangiogenic compositions of microvesicles derived from human umbilical cord mesenchymal stem cells. PLoS ONE 9(12):e115316
Cinteza D, Persinaru I, Maciuceanu ZB et al (2015) Peripheral nerve regeneration—an appraisal of the current treatment options. Maedica (Buchar) 10(1):65–68
Das MM, Avalos P, Suezaki P et al (2016) Human neural progenitors differentiate into astrocytes and protect motor neurons in aging rats. Exp Neurol 280:41–49
De ML, Freed WJ, Wyatt RJ (1982) An index of the functional condition of rat sciatic nerve based on measurements made from walking tracks. Exp Neurol 77(3):634–643
Fournier BP, Loison-Robert LS, Ferré FC et al (2016) Characterisation of human gingival neural crest-derived stem cells in monolayer and neurosphere cultures. Eur Cell Mater 31:40–58
Fu KY, Dai LG, Chiu IM et al (2011) Sciatic nerve regeneration by microporous nerve conduits seeded with glial cell line-derived neurotrophic factor or brain-derived neurotrophic factor gene transfected neural stem cells. Artif Organs 35(4):363–372
Fu X, Rong Z, Zhu S et al (2014) Genetic approach to track neural cell fate decisions using human embryonic stem cells. Protein Cell 5:69–79
Gadkari R, Zhao L, Teklemariam T, Hantash BM (2014) Human embryonic stem cell derived-mesenchymal stem cells: An alternative mesenchymal stem cell source for regenerative medicine therapy. Regen Med 9:453–465
Gaudet AD, Popovich PG, Ramer MS et al (2011) Wallerian degeneration: gaining perspective on inflammatory events after peripheral nerve injury. J Neuroinflam 8(1):110
Gonzalez-Perez F, Hernández J, Heimann C et al (2017) Schwann cells and mesenchymal stem cells in laminin- or fibronectin-aligned matrices and regeneration across a critical size defect of 15 mm in the rat sciatic nerve. J Neurosurg Spine 28(1):109–118
Hess JR, Brenner MJ, Fox IK et al (2007) Use of cold-preserved allografts seeded with autologous Schwann cells in the treatment of a long-gap peripheral nerve injury. Plast Reconstr Surg 119(1):246–259
Houschyar KS, Momeni A, Pyles MN et al (2016) The role of current techniques and concepts in peripheral nerve repair. Plast Surg Int 2016:1–8
Huang CW, Huang WC, Qiu X et al (2017) The differentiation stage of transplanted stem cells modulates nerve regeneration. Sci Rep 7(1):17401
Jie C, Li C, Chen L (2015) The role of microvesicles derived from mesenchymal stem cells in lung diseases. Biomed Res Int 2015:985814
Lee DC, Chen JH, Hsu TY et al (2017) Neural stem cells promote nerve regeneration through IL12-induced Schwann cell differentiation. Mol Cell Neurosci 79:1–11
Li B, Chen HW, Hu ZX (2007) Differentiation of human embryonic stem cells to neural progenitors. Zool Res 28:311–319
Li X, Wang W, Wei G et al (2010) Immunophilin FK506 loaded inchitosan guide promotes peripheral nerve regeneration. Biotechnol Lett 32(9):1333–1337
Li YC, Tsai LK, Wang JH et al (2014) A neural stem/precursor cell monolayer for neural tissue engineering. Biomaterials 35:1192–1204
Motti D, Lerch JK, Danzi MC et al (2017) Identification of miRNAs involved in DRG neurite outgrowth and their putative targets. Febs Lett 591(14):2091–2105
Oh JH, Jung CR, Lee MO et al (2018) Comparative analysis of human embryonic stem cell derived neural stem cells as an in vitro human model. Int J Mol Med 41:783–790
Pollard SM (2013) In vitro expansion of fetal neural progenitors as adherent cell lines. Methods Mol Biol 1059:13–24
Prajumwongs P, Weeranantanapan O, Jaroonwitchawan T et al (2016) Human embryonic stem cells: a model for the study of neural development and neurological diseases. Stem Cells Int 10:1–9
Roubal I, Park SJ, Kim Y (2016) Derivation of neural precursor cells from human embryonic stem cells for DNA methylomic analysis. Methods Mol Biol 1341:345–357
Salimi A, Nadri S, Ghollasi M et al (2014) Comparison of different protocols for neural differentiation of human induced pluripotent stem cells. Mol Biol Rep 41:1713–1721
Sando IC, Cederna PS (2016) Discussion: Growth hormone therapy accelerates axonal regeneration, promotes motor reinnervation, and reduces muscle atrophy following peripheral nerve injury. Plastic Reconstr Surg 137(6):1781–1783
Santos D, Wieringa P, Moroni L et al (2017) PEOT/PBT guides enhance nerve regeneration in long gap defects. Adv Healthc Mater. https://doi.org/10.1002/adhm.201600298
Sayad SF, Zaminy A (2017) Stem cell therapy for nerve injury. World J Stem Cells 9:144–151
Shin S, Sun Y, Liu Y et al (2007) Whole genome analysis of human neural stem cells derived from embryonic stem cells and stem and progenitor cells isolated from fetal tissue. Stem Cells 25:1298–1306
Shroff G, Dhanda Titus J, Shroff R (2017) A review of the emerging potential therapy for neurological disorders: human embryonic stem cell therapy. Am J Stem Cells 6:1–12
Sun Y, Campisi J, Higano C et al (2012) Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat Med 18(9):1359–1368
Svendsen CN, Caldwell MA, Ostenfeld T (1999) Human neural stem cells: isolation, expansion and transplantation. Brain Pathol 9:499–513
Tsai MJ, Pan HA, Liou DY et al (2010) Adenoviral gene transfer of bone morphogenetic protein-7 enhances functional recovery after sciatic nerve injury in rats. Gene Ther 17(10):1214–1224
Tuffaha SH, Budihardjo JD, Sarhane KA et al (2016) Growth hormone therapy accelerates axonal regeneration, promotes motor reinnervation, and reduces muscle atrophy following peripheral nerve injury. Plast Reconstr Surg 137(6):1771–1780
Varejao AS, Meek MF, Ferreira AJ et al (2001) Functional evaluation of peripheral nerve regeneration in the rat: walking track analysis. J Neurosci Methods 108(1):1–9
Wang Y, Li WY, Jia H et al (2016) KLF7-transfected Schwann cell graft transplantation promotes sciatic nerve regeneration. Neuroscience 340:319–332
Wang C, Lu CF, Peng J et al (2017a) Roles of neural stem cells in the repair of peripheral nerve injury. Neural Regen Res 12(12):2106–2112
Wang S, Qiu J, Guo W et al (2017b) A nanostructured molybdenum disulfide film for promoting neural stem cell neuronal differentiation: toward a nerve tissue-engineered 3D scaffold. Adv Biosyst 1(5):1600042
Wei JJ, Chen YF, Xue CL et al (2016) Protection of nerve injury with exosome extracted from mesenchymal stem cell. Zhongguo Yi Xue Ke Xue Yuan Xue Bao Acta Academiae Medicinae Sinicae 38(1):33–36
Woo SM, Kim J, Han HW et al (2009) Notch signaling is required for maintaining stem-cell features of neuroprogenitor cells derived from human embryonic stem cells. BMC Neurosci 10:97
Wu R, Li W, Chen F et al (2016) Evaluation of artificial nerve conduit and autografts in peripheral nerve repair in the rat model of sciatic nerve injury. Neurol Res 38(5):461–466
Xu HF (2009) Optimized culture of human embryonic stem cells and differentiation into neural stem cells. Huazhong Univer Sci Technol 1–84
Xu L, Zhou S, Feng GY et al (2012) Neural stem cells enhance nerve regeneration after sciatic nerve injury in rats. Mol Neurobiol 46(2):265–274
Yasui T, Uezono N, Nakashima H et al (2017) Hypoxia epigenetically confers astrocytic differentiation potential on human pluripotent cell-derived neural precursor cells. Stem Cell Rep 8:1743–1756
Ying W, Li WY, Hua J et al (2016) KLF7-transfected Schwann cell graft transplantation promotes sciatic nerve regeneration. Neuroscience 340:319–332
Zhang Q, Nguyen P, Xu Q et al (2017) Neural progenitor-like cells induced from human gingiva-derived mesenchymal stem cells regulate myelination of schwann cells in rat sciatic nerve regeneration. Stem Cells Transl Med 6:458–470
Zhao ZR, Yu WD, Shi C et al (2017) Correlation between receptor-interacting protein 140 expression and directed differentiation of human embryonic stem cells into neural stem cells. Neural Regen Res 12:118–124
Zheng J, Jian S, Lu X et al (2016) BDNF promotes the axonal regrowth after sciatic nerve crush through intrinsic neuronal capability upregulation and distal portion protection. Neurosci Lett 621:1–8
Zuchero JB (2014) Purification and culture of dorsal root ganglion neurons. Cold Spring Harb Protoc 2014(8):813–814
Acknowledgments
We appreciate the stem cell bank of the Chinese Academy of Sciences under a Materials Transfer Agreement for providing us hESC lines (SHhES2). This work was supported by the National Natural Science Foundation of China (Grant No. 81571221), Natural Science Foundation of Jiangsu Province (Grant No. BK20151346) and Qing Lan Project of Jiangsu Province.
Author information
Authors and Affiliations
Contributions
JH designed the research study, and XC and KY performed the research. JY and HW created the animal model. JH, XS and HX supplied study materials, instrumentation tools, computing resources, and other analysis tools. JG, LZ, XJ, TC, YD and BT analyzed the data. XC and KY wrote the article.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that there are no competing interests.
Ethical approval
All experimental procedures were conducted in accordance with Chinese legislation on animal protection and were approved by the Ethics Committee of Jiangsu University.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rats walked freely in the PBS group (MP4 755 kb)
Rats walked freely in the hESC-NSCs group (MP4 1254 kb)
Rats walked freely in the hESC-NSC-MVs group (MP4 1331 kb)
Rights and permissions
About this article

Cite this article
Chen, X., Ye, K., Yu, J. et al. Regeneration of sciatic nerves by transplanted microvesicles of human neural stem cells derived from embryonic stem cells. Cell Tissue Bank 21, 233–248 (2020). https://doi.org/10.1007/s10561-020-09816-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10561-020-09816-5