
Abstract
Purpose
Hepatotoxicity has emerged as a major cause of statin treatment interruption. Although organic anion-transporting polypeptide 1B1 (SLCO1B1), multidrug resistance protein 1 (ABCB1), and breast cancer resistance protein (ABCG2) have been identified as transporters of statins, knowledge of their role in statin-associated hepatotoxicity remains limited. Therefore, we aimed to conduct a comprehensive analysis to elucidate the association between hepatotoxicity and SLCO1B1, ABCB1, and ABCG2 polymorphisms.
Methods
This study retrospectively analyzed prospectively collected samples. We selected 10 single nucleotide polymorphisms (SNPs) of SLCO1B1, 9 SNPs of ABCB1, and 12 SNPs of ABCG2. We developed two models for multivariable analyses (Model I: clinical factors only; Model II: both clinical and genetic factors), and the attributable risk (%) of variables in Model II was determined.
Results
Among 851 patients, 66 (7.8%) developed hepatotoxicity. In Model I, lipophilic statins, atrial fibrillation (Afib), and diabetes mellitus showed a significant association with hepatotoxicity. In Model II, lipophilic statins and Afib, SLCO1B1 rs11045818 A allele, SLCO1B1 rs4149035 T allele, and ABCG2 rs2622629 TT genotype were associated with higher hepatotoxicity risk. Among them, the SLCO1B1 rs11045818 A allele exhibited the highest attributable risk (93.2%). The area under the receiver operating characteristic curve in Model I was 0.62 (95% CI: 0.55–0.69), and it was increased to 0.71 in Model II (95% CI: 0.64–0.77).
Conclusion
This study investigated the correlation between hepatotoxicity and polymorphisms of transporter genes in patients taking statins. The findings could help improve personalized treatments for patients receiving statin therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hydroxymethyl glutaryl coenzyme A reductase (HMG-CoA) inhibitors, commonly referred to as statins, are widely used in the treatment of dyslipidemia for the management of atherosclerotic cardiovascular diseases (ASCVD) [1]. The pharmacological effects of statins include a reduction in plasma concentrations of total cholesterol (TC) and low-density lipoprotein cholesterol (LDL-C) and an increase in the level of high-density lipoprotein cholesterol [2]. Their lipid-lowering activity has been associated with a significant decrease in mortality, nonfatal acute myocardial infarction, stroke, and coronary revascularization for patients with high cardiovascular risk [3].
Statins are well-tolerated by most people; however, adverse drug events (ADEs) such as muscle symptoms, new-onset diabetes mellitus (DM), and hepatotoxicity have been observed [4]. In particular, liver toxicity has emerged as one of the major causes contributing to the interruption of statin treatment [5]. The symptoms of hepatic ADEs are diverse; although the most common symptoms are asymptomatic and usually involve the temporary elevation of transaminases [6], severe hepatotoxicity leading to liver failure or death has been reported in post-marketing surveillance [7].
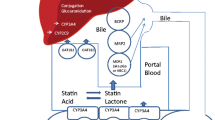
Statins are substrates for transporter proteins from the solute carrier (SLC) and ATP-binding cassette (ABC) superfamilies [8]. Organic anion-transporting polypeptide 1B1 (OATP1B1), encoded by the SLCO1B1 gene, is an influx transporter that facilitates the hepatic uptake of all statins, thereby regulating systemic exposure to statins. In particular, polymorphisms of SLCO1B1 have been identified as a genetic factor for statin-induced myopathy in a genome-wide association study (GWAS) and systematic reviews [9, 10]. Multidrug resistance protein 1 (encoded by ABCB1) and breast cancer resistance protein (encoded by ABCG2) are efflux transporters associated with the hepatobiliary excretion of statins [8].
Inter-individual differences in the response to statins suggest a genetic factor as a potential contributor to the variable response to statin therapy [11]. However, there is limited information on the correlation between gene polymorphisms linked to transporters and hepatotoxicity. Various studies have focused on hepatotoxicity for one specific type of statin [12, 13]. Additionally, some studies have defined the primary outcome as ADEs, with hepatotoxicity included as part of composite outcomes [14, 15]. Therefore, we aimed to conduct a comprehensive pharmacogenomic investigation by examining the association between hepatotoxicity and polymorphisms of transporter genes, including SLCO1B1, ABCB1, and ABCG2.
Methods
Study Participants and data Collection
This study was conducted at Ewha Womans University Seoul Hospital and Ewha Womans University Mokdong Hospital. We identified patients aged ≥ 18 years who started taking statins (atorvastatin, fluvastatin, lovastatin, pitavastatin, pravastatin, rosuvastatin, or simvastatin) between January 2000 and May 2021 for the primary or secondary prevention of ASCVD. Genomic DNA samples were prospectively collected during regularly scheduled clinic visits from February to May 2021. Patients were excluded if they (1) received statins less than 3 months (2) lacked liver function test results, (3) presented with elevated aspartate aminotransferase (AST), alanine aminotransferase (ALT), or alkaline phosphatase (ALP) at statin initiation, (4) had underlying chronic liver diseases, (5) had inappropriate follow-up data, or (6) had insufficient DNA samples for analysis.
The primary endpoint was hepatotoxicity, defined as grade II or higher according to the Common Terminology Criteria for Adverse Events (CTCAE) version 5.0 [16]. The CTCAE defines grade II toxicity levels of AST, ALT, and ALP as 3.0–5.0, 3.0–5.0, and 2.5–5.0 times the upper limit of normal (ULN), respectively. As transaminase levels exceeding three times the ULN require additional tests or the discontinuation of statins in clinical settings, we set the cutoff for hepatotoxicity at grade II in this analysis [17, 18]. We retrospectively collected demographic and clinical information from electronic medical records, including data on age, height, weight, estimated glomerular filtration rate (eGFR), liver function tests (AST, ALT, ALP), serum lipid levels, statin prescription data (type, dosage, duration), alcohol and smoking status, comorbidities and comedications.
This study was approved by the Institutional Review Boards (IRBs) of Ewha Womans University Seoul Hospital and Ewha Womans University Mokdong Hospital (IRB numbers: 2020-11-014 and 2021-02-026, respectively). We followed the ethical guidelines outlined in the 1964 Declaration of Helsinki and its subsequent amendments. Prior to participation, all patients provided written informed consent.
Single Nucleotide Polymorphism (SNP) and Haplotype Selection
We selected 10 SNPs of SLCO1B1 [19,20,21], 9 SNPs of ABCB1 [22, 23], and 12 SNPs of ABCG2 [24,25,26,27] based on previous findings. For SLCO1B1, further haplotype analysis was conducted using 4 haplotypes: SLCO1B1*1A (c.388 A-c.521T), *1B (c.388G-c.521T), *5 (c.388 A-c.521 C), and *15 (c.388G-c.521 C). HaploReg v4.2 was used to assess the linkage disequilibrium (LD) and minor allele frequency in Asian populations [28]. We obtained genetic information for these SNPs from the National Center for Biotechnology Information [29].
Genotyping Methods
Genomic DNA was extracted from the patients’ blood samples using QIAamp DNA Blood Mini Kit (QIAGEN GmbH, Hilden, Germany) or saliva samples using OraGene-600 (DNA Genotek, Ottawa, ON, Canada). The genotypes of 31 SNPs were analyzed by TaqMan SNP genotyping assay (Applied Biosystems, Foster City, CA, USA) or SNaPshot Multiplex Kit (Applied Biosystems, Foster City, CA, USA).
Statistical Analysis
Chi-squared test or Fisher’s exact test was used to analyze categorical variables and unpaired t-test was used to compare continuous variables. Crude odds ratios (ORs) and adjusted ORs (AORs) with 95% confidence intervals (CIs) were calculated by univariate and multivariable regression analyses, respectively. A multivariable logistic regression model was used to identify independent risk factors for hepatotoxicity. The model incorporated variables with p < 0.05 in univariate analysis along with strong confounders such as age and sex. Attributable risk (%) was calculated by the equation \((1-1/\text{A}\text{O}\text{R}) \times 100\).
The model fit of the prediction model was assessed by the Hosmer-Lemeshow goodness-of-fit test. The discrimination of the model was further evaluated by calculating the area under the receiver operating characteristic curve (AUROC). All statistical analyses were performed using SPSS v20.0 (IBM Corp., Armonk, NY, USA) and p < 0.05 was considered statistically significant.
Results
Among the 1,005 enrolled patients, 154 patients were excluded for the following reasons: received statins for less than 3 months (5 patients), did not have liver function test results (55 patients), had elevated liver enzyme levels before administration of statins (58 patients), had underlying liver diseases (29 patients), died during statin therapy (1 patient), had inappropriate follow-up data (2 patients), and had insufficient samples for DNA analysis (4 patients) (Fig. 1). Consequently, 851 patients were included, among whom 66 patients (7.8%) had CTCAE grade II or higher hepatotoxicity.

Flowchart of patient selection
Table 1 shows the baseline characteristics of the study population. Among the patients, 67.3% of them were male, and the median age was 63 years (interquartile range (IQR): 27–91 years). The median follow-up period was 2.0 years (IQR 1.2–3.2 years). In comparison with patients without comorbidities, patients with DM and atrial fibrillation (Afib) were more susceptible to hepatotoxicity (p = 0.013 and p = 0.037, respectively). Among the patients, 60.2% of them used lipophilic statins (including atorvastatin, fluvastatin, lovastatin, pitavastatin, and simvastatin), and 39.8% of them used hydrophilic statins (such as pravastatin and rosuvastatin). There was more hepatotoxicity among patients taking lipophilic statins than among those taking hydrophilic statins (9.4% vs. 5.3%, p = 0.030). The most commonly used comedication was antiplatelets (81.8%), followed by angiotensin-converting enzyme inhibitors (ACEIs)/angiotensin II receptor blockers (ARBs) (52.1%) and calcium channel blockers (38.3%). None of the patients used nicotinic acid.
The results of genotype analysis for SLCO1B1 are presented in Table 2. For SLCO1B1 rs11045818, A allele carriers had an increased risk of hepatotoxicity compared with that of GG genotype carriers (41.7% vs. 7.1%, p = 0.001). There was a higher incidence of hepatotoxicity among patients with the T allele of rs4149035 than among those with the CC genotype (11.3% vs. 6.4%, p = 0.019). None of the SLCO1B1 haplotypes demonstrated statistical significance for hepatotoxicity (Table 2). Within the ABCG2 gene, there was increased hepatotoxicity among individuals with the rs2622629 TT genotype compared with those with the C allele (12.6% vs. 7.0%, p = 0.041) (Table 3). In the case of rs4367138, hepatotoxicity was more common among patients with wild-type homozygotes (GG) than among those carrying the variant (A) allele (14.9% vs. 7.0%, p = 0.020). However, no significant differences were found between the two groups in ABCB1.
Two models were constructed for multivariable logistic regression analyses; Model I included clinical factors only, and Model II included both clinical and genetic factors (Table 4). In Model I, lipophilic statins, Afib, and DM showed a significant association with hepatotoxicity. In Model II, lipophilic statins increased the risk of hepatotoxicity by 2.1 times (95% CI: 1.2–3.9), and Afib increased the risk by 2.2 times (95% CI: 1.1–4.4). Regarding genetic factors, A allele of SLCO1B1 rs11045818 had the most significant impact on hepatotoxicity risk (AOR: 14.7, 95% CI: 4.1–53.1) compared to the GG genotype. Individuals carrying the SLCO1B1 T allele showed a 1.9-fold higher risk of liver toxicity (95% CI: 1.1–3.3) than those with the CC genotype. Furthermore, compared with individuals carrying the variant allele, ABCG2 rs2622629 TT genotype carriers exhibited a 2.4-fold (95% CI: 1.2–4.6) increased risk of hepatotoxicity.
The SLCO1B1 rs11045819 A allele was associated with a high attributable risk for hepatotoxicity at 93.2%, followed by ABCG2 rs2622629 TT genotype (58.1%) and SLCO1B1 rs4149035 T allele (46.3%) (Table 4). The Hosmer-Lemeshow test revealed a satisfactory fit for Model I and Model II (χ2 = 2.11, p = 0.72 and χ2 = 2.60, p = 0.86, respectively). The AUROC in Model I (0.62, 95% CI: 0.55–0.69), the AUROC was increased in Model II (0.71, 95% CI: 0.64–0.77) by adding genetic factors (Fig. 2).

The receiver operating characteristic (ROC) curve for hepatotoxicity using clinical and genetic factors. The blue line represents the predicted probability of Model I, while the green line represents that of Model II. The yellow line is the reference
Discussion
Identifying the clinical and genetic risk factors associated with hepatotoxicity is essential for preventing ADEs in patients receiving statin therapy [30]. This study indicated that SLCO1B1 rs11045818, SLCO1B1 rs4149035, ABCG2 rs2622629, lipophilic statins, and Afib were significantly associated with hepatotoxicity. The AUROC for predicting statin-associated hepatotoxicity, which included clinical and genetic factors, was satisfactory.
The SLCO1B1 gene, located on chromosome 12, is predominantly expressed in the liver and is responsible for the uptake of endogenous substances or drugs such as statins and anti-bacterial agents into hepatocytes [31]. As the active transportation of statins into hepatocytes is mediated mainly by OATP1B1, polymorphisms of the SLCO1B1 gene could affect statin disposition, potentially contributing to the development of hepatotoxicity [8, 32]. Further supporting this, Jin et al. demonstrated that SNPs and haplotypes of SLCO1B1 were critical predisposing factors for methimazole-induced hepatotoxicity [32].
SLCO1B1 rs11045818, a synonymous variant, exhibited the highest attributable risk in our study [33]. This finding is in agreement with Alhawari et al., which revealed that the rs11045818 A allele was associated with a 27% increase in ALT levels in type 2 DM patients taking atorvastatin (p < 0.05) [34]. Furthermore, rs11045818 is in complete LD (r2 = 1) with SLCO1B1 rs11045819 (c.463 C > A), a missense variant [28, 35]. Rs11045819 is part of the SLCO1B1*14 haplotype, which enhances the function of OATP1B1 [35]. Due to the increased influx of statins, patients with a variant allele of rs11045819 were found to have a decreased area under the plasma concentration-time curve for simvastatin acid. In addition, the efficacy of statins in lowering LDL-C and TC levels was observed to be better for these patients than for those with wild-type homozygotes [36]. Therefore, SLCO1B1 rs11045818 could potentially affect OATP1B1 function, resulting in the enhanced hepatic uptake of statins, thereby increasing the risk of hepatotoxicity.
Analysis of SLCO1B1 rs4149035, an intron variant, showed a correlation between the wild-type allele (T allele) and increased liver toxicity risk. A Spanish cohort study of 384 pediatric patients with acute lymphoblastic leukemia revealed that a variant-type homozygote (CC) of rs4149035 was associated with increased methotrexate plasma concentration and nephrotoxicity [37]. In addition, SLCO1B1 rs4149035 was found to be highly correlated with SLCO1B1 rs4149033 (r2 = 0.98) [28]. A variant allele of rs4149033 increased the risk of rhabdomyolysis by 1.4 times (95% CI: 1.06 − 1.87) in cerivastatin-treated patients [38]. Another study identified a variant allele of rs4149033 as one of the risk factors associated with sudden cardiac death caused by coronary artery disease (OR: 1.30, 95% CI: 1.03 − 1.64) [39]. These studies collectively suggest that these SNPs could reduce the hepatic uptake of statins, leading to an increased risks of systemic complications [31, 40].
ABCG2, expressed in the liver, small intestines, and kidneys, resides on chromosome 4 [41]. It plays a vital role in regulating intestinal absorption and the biliary excretion of drugs. The reduced activity of ABCG2 has been shown to increase the absorption of statins in the gastrointestinal tract and decrease drug efflux in the biliary ducts [8]. Therefore, we hypothesized that these dual effects may result in drug accumulation in hepatocytes, possibly leading to hepatotoxicity. ABCG2 rs2622629, located in the intronic region of ABCG2, was associated with hepatotoxicity risk in this study. Although the specific function of this SNP in hepatotoxicity has not been characterized in detail, an eQTL analysis performed by GTEx demonstrated that C allele of rs2622629 was associated with higher ABCG2 expression in the thyroid and esophageal mucosa (p = 7.9 × 10−7) [42]. However, as no significant difference was detected in the liver (p = 0.7) [42], further studies are necessary to elucidate the role of this SNP.
In comparison with hydrophilic statins, lipophilic statins were associated with greater susceptibility to hepatotoxicity in our study. This result was consistent with previous findings showing that lipophilic statins were associated with the majority of statin-induced liver injuries, whereas hydrophilic statins accounted for a small proportion [7]. Moreover, a meta-analysis demonstrated a higher risk of ALT elevation among patients taking lipophilic statins compared with those taking hydrophilic statins (OR: 2.69, 95% CI: 1.84–3.95) [43]. These lipophilicity-dependent effects highlight key differences in the pharmacokinetic properties of statins. For example, lipophilic statins can easily penetrate the hepatic and intestinal membrane by passive diffusion, whereas hydrophilic statins depend on specific transporters [8, 44]. In addition, lipophilic statins undergo hepatic metabolism via cytochrome P450 (CYP), whereas hydrophilic statins are mainly excreted unchanged [45]. CYP-dependent metabolism generates reactive oxygen species (ROS) and is involved in cell apoptosis [30]. Therefore, the use of lipophilic statins can promote ROS production and lipid peroxidation, decreasing the mitochondrial membrane potential and subsequently inducing cytotoxicity [30, 46]. Taken together, the findings explain the association between lipophilic statins and hepatotoxicity.
Patients with Afib showed approximately a 2-fold increase in hepatotoxicity in this study. Makar et al. revealed that 27.6% of study participants with Afib experienced ALT elevation above the ULN (40 IU/L), and 2.8% of them had ALT levels exceeding three times the ULN [47]. These findings suggest that Afib patients have an increased risk of elevated liver enzymes. Additionally, a recent study has identified persistent Afib as a significant factor associated with elevated liver fibrosis markers [48]. However, research examining the mechanism underlying the effects of Afib on hepatotoxicity is lacking, underscoring the need for further investigation.
This study is the first to comprehensively investigate various types of statins to evaluate the genetic polymorphisms of transporters as potential risk factors for statin-associated hepatotoxicity. However, the present study has a risk of bias due to the retrospective study design. Moreover, this study included only Asian participants with a relatively small sample size, which would limit the generalizability of the findings. Further prospective large cohort studies are needed to validate our findings.
Conclusion
Our study elucidated the relationship between SLCO1B1, ABCB1, and ABCG2 gene polymorphisms and hepatotoxicity. The findings could contribute to a better understanding of the causes of statin-induced hepatotoxicity and facilitate the development of personalized treatments for patients receiving statin therapy.
Data Availability
The datasets analyzed during the current study are presented as a supplementary file.
Code Availability
Not applicable.
References
Grundy SM, Stone NJ, Bailey AL, et al. AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;139(25):e1082–143. https://doi.org/10.1161/cir.0000000000000625.
Sizar O, Khare S, Jamil RT, Talati R. Statin Medications. https://www.ncbi.nlm.nih.gov/books/NBK430940/.
Efficacy. Safety of statin therapy in older people: a meta-analysis of individual participant data from 28 randomised controlled trials. Lancet. 2019;393(10170):407–15. https://doi.org/10.1016/s0140-6736(18)31942-1.
Thompson PD, Panza G, Zaleski A, Taylor B. Statin-Associated Side effects. J Am Coll Cardiol. 2016;67(20):2395–410. https://doi.org/10.1016/j.jacc.2016.02.071.
Licata A, Giammanco A, Minissale MG, et al. Liver and statins: a critical Appraisal of the evidence. Curr Med Chem. 2018;25(42):5835–46. https://doi.org/10.2174/0929867325666180327095441.
Bhardwaj SS, Chalasani N. Lipid-lowering agents that cause drug-induced hepatotoxicity. Clin Liver Dis. 2007;11(3):597–613, vii. https://doi.org/10.1016/j.cld.2007.06.010.
Björnsson E, Jacobsen EI, Kalaitzakis E. Hepatotoxicity associated with statins: reports of idiosyncratic liver injury post-marketing. J Hepatol. 2012;56(2):374–80. https://doi.org/10.1016/j.jhep.2011.07.023.
Rocha KCE, Pereira BMV, Rodrigues AC. An update on efflux and uptake transporters as determinants of statin response. Expert Opin Drug Metab Toxicol. 2018;14(6):613–24. https://doi.org/10.1080/17425255.2018.1482276.
Turongkaravee S, Jittikoon J, Lukkunaprasit T, et al. A systematic review and meta-analysis of genotype-based and individualized data analysis of SLCO1B1 gene and statin-induced myopathy. Pharmacogenomics J. 2021;21(3):296–307. https://doi.org/10.1038/s41397-021-00208-w.
Kee PS, Chin PKL, Kennedy MA, Maggo SDS. Pharmacogenetics of Statin-Induced myotoxicity. Front Genet. 2020;11:575678. https://doi.org/10.3389/fgene.2020.575678.
Streja L, Packard CJ, Shepherd J, Cobbe S, Ford I. Factors affecting low-density lipoprotein and high-density lipoprotein cholesterol response to pravastatin in the West of Scotland Coronary Prevention Study (WOSCOPS). Am J Cardiol. 2002;90(7):731–6. https://doi.org/10.1016/s0002-9149(02)02599-7.
Qu KK, Zhang CN, Dong LX, et al. Association of ABCB1 polymorphisms with lipid homeostasis and liver injury response to atorvastatin in the Chinese population. Can J Physiol Pharmacol. 2020;98(1):15–22. https://doi.org/10.1139/cjpp-2019-0339.
Mirošević Skvrce N, Macolić Šarinić V, Šimić I, et al. ABCG2 gene polymorphisms as risk factors for atorvastatin adverse reactions: a case-control study. Pharmacogenomics. 2015;16(8):803–15. https://doi.org/10.2217/pgs.15.47.
Merćep I, Radman I, Trkulja V, et al. Loss of function polymorphisms in SLCO1B1 (c.521T > C, rs4149056) and ABCG2 (c.421C > A, rs2231142) genes are associated with adverse events of rosuvastatin: a case-control study. Eur J Clin Pharmacol. 2022;78(2):227–36. https://doi.org/10.1007/s00228-021-03233-7.
Yow HY, Hamzah S, Abdul Rahim N, Suppiah V. Pharmacogenomics of response to statin treatment and susceptibility to statin-induced adverse drug reactions in asians: a scoping review. Asian Biomed (Res Rev News). 2023;17(3):95–114. https://doi.org/10.2478/abm-2023-0050.
Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0. U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES. National Institutes of Health. 2017. https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf. Accessed 03 Apr 2023.
Meurer L, Cohen SM. Drug-Induced Liver Injury from statins. Clin Liver Dis. 2020;24(1):107–19. https://doi.org/10.1016/j.cld.2019.09.007.
McKenney JM, Davidson MH, Jacobson TA, Guyton JR. Final conclusions and recommendations of the National Lipid Association Statin Safety Assessment Task Force. Am J Cardiol. 2006;97(8a):c89–94. https://doi.org/10.1016/j.amjcard.2006.02.030.
Boivin AA, Cardinal H, Barama A, et al. Organic anion transporting polypeptide 1B1 (OATP1B1) and OATP1B3: genetic variability and haplotype analysis in white canadians. Drug Metab Pharmacokinet. 2010;25(5):508–15. https://doi.org/10.2133/dmpk.dmpk-10-sh-046.
Nguyen HH, Nguyen CTT, Mai TNP, Huong PT. Associations between four polymorphisms of the SLCO1B1 and effectiveness of the statins: a meta-analysis. Pharmacogenet Genomics. 2023;33(4):65–78. https://doi.org/10.1097/fpc.0000000000000490.
Peters BJ, Rodin AS, Klungel OH, et al. Pharmacogenetic interactions between ABCB1 and SLCO1B1 tagging SNPs and the effectiveness of statins in the prevention of myocardial infarction. Pharmacogenomics. 2010;11(8):1065–76. https://doi.org/10.2217/pgs.10.81.
Yoon HY, Song TJ, Yee J, Park J, Gwak HS. Association between Genetic Polymorphisms and bleeding in patients on direct oral anticoagulants. Pharmaceutics. 2022;14(9). https://doi.org/10.3390/pharmaceutics14091889.
Levran O, O’Hara K, Peles E, et al. ABCB1 (MDR1) genetic variants are associated with methadone doses required for effective treatment of heroin dependence. Hum Mol Genet. 2008;17(14):2219–27. https://doi.org/10.1093/hmg/ddn122.
Kim H, Song TJ, Yee J, et al. ABCG2 gene polymorphisms may affect the bleeding risk in patients on Apixaban and Rivaroxaban. Drug Des Devel Ther. 2023;17:2513–22. https://doi.org/10.2147/dddt.S417096.
Boocock J, Leask M, Okada Y, et al. Genomic dissection of 43 serum urate-associated loci provides multiple insights into molecular mechanisms of urate control. Hum Mol Genet. 2020;29(6):923–43. https://doi.org/10.1093/hmg/ddaa013.
Ye J, Zeng Z, Chen Y, et al. Examining an Association of Single Nucleotide Polymorphisms with Hyperuricemia in Chinese Flight attendants. Pharmgenomics Pers Med. 2022;15:589–602. https://doi.org/10.2147/pgpm.S364206.
Kiyotani K, Mushiroda T, Imamura CK, et al. Significant effect of polymorphisms in CYP2D6 and ABCC2 on clinical outcomes of adjuvant tamoxifen therapy for breast cancer patients. J Clin Oncol. 2010;28(8):1287–93. https://doi.org/10.1200/jco.2009.25.7246.
Ward LD, Kellis M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 2016;44(D1):D877–81. https://doi.org/10.1093/nar/gkv1340.
Sayers EW, Bolton EE, Brister JR, et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2022;50(D1):D20–6. https://doi.org/10.1093/nar/gkab1112.
Averbukh LD, Turshudzhyan A, Wu DC, Wu GY. Statin-induced Liver Injury patterns: a clinical review. J Clin Transl Hepatol. 2022;10(3):543–52. https://doi.org/10.14218/jcth.2021.00271.
Niemi M, Pasanen MK, Neuvonen PJ. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev. 2011;63(1):157–81. https://doi.org/10.1124/pr.110.002857.
Jin S, Li X, Fan Y, et al. Association between genetic polymorphisms of SLCO1B1 and susceptibility to methimazole-induced liver injury. Basic Clin Pharmacol Toxicol. 2019;125(6):508–17. https://doi.org/10.1111/bcpt.13284.
Alhawari H, Jarrar Y, AlKhatib MA, et al. The Association of 3-Hydroxy-3-Methylglutaryl-CoA Reductase, apolipoprotein E, and Solute Carrier Organic Anion Genetic Variants with Atorvastatin Response among Jordanian patients with type 2 diabetes. Life (Basel). 2020;10(10). https://doi.org/10.3390/life10100232.
Giannakopoulou E, Ragia G, Kolovou V, et al. No impact of SLCO1B1 521T > C, 388A > G and 411G > A polymorphisms on response to statin therapy in the Greek population. Mol Biol Rep. 2014;41(7):4631–8. https://doi.org/10.1007/s11033-014-3334-z.
Ramsey LB, Gong L, Lee SB, et al. PharmVar GeneFocus: SLCO1B1. Clin Pharmacol Ther. 2023;113(4):782–93. https://doi.org/10.1002/cpt.2705.
Mykkänen AJH, Taskinen S, Neuvonen M, et al. Genomewide Association Study of Simvastatin Pharmacokinetics. Clin Pharmacol Ther. 2022;112(3):676–86. https://doi.org/10.1002/cpt.2674.
Lopez-Lopez E, Ballesteros J, Piñan MA, et al. Polymorphisms in the methotrexate transport pathway: a new tool for MTX plasma level prediction in pediatric acute lymphoblastic leukemia. Pharmacogenet Genomics. 2013;23(2):53–61. https://doi.org/10.1097/FPC.0b013e32835c3b24.
Marciante KD, Durda JP, Heckbert SR, et al. Cerivastatin, genetic variants, and the risk of rhabdomyolysis. Pharmacogenet Genomics. 2011;21(5):280–8. https://doi.org/10.1097/FPC.0b013e328343dd7d.
Hernesniemi JA, Lyytikäinen LP, Oksala N, et al. Predicting sudden cardiac death using common genetic risk variants for coronary artery disease. Eur Heart J. 2015;36(26):1669–75. https://doi.org/10.1093/eurheartj/ehv106.
Kiander W, Sjöstedt N, Manninen R, et al. Functional in vitro characterization of SLCO1B1 variants and simulation of the clinical pharmacokinetic impact of impaired OATP1B1 function. Eur J Pharm Sci. 2022;176:106246. https://doi.org/10.1016/j.ejps.2022.106246.
Krishnamurthy P, Schuetz JD. Role of ABCG2/BCRP in biology and medicine. Annu Rev Pharmacol Toxicol. 2006;46:381–410. https://doi.org/10.1146/annurev.pharmtox.46.120604.141238.
Battle A, Brown CD, Engelhardt BE, Montgomery SB. Genetic effects on gene expression across human tissues. Nature. 2017;550(7675):204–13. https://doi.org/10.1038/nature24277.
Bytyçi I, Bajraktari G, Bhatt DL, et al. Hydrophilic vs lipophilic statins in coronary artery disease: a meta-analysis of randomized controlled trials. J Clin Lipidol. 2017;11(3):624–37. https://doi.org/10.1016/j.jacl.2017.03.003.
Schachter M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundam Clin Pharmacol. 2005;19(1):117–25. https://doi.org/10.1111/j.1472-8206.2004.00299.x.
Fong CW. Statins in therapy: understanding their hydrophilicity, lipophilicity, binding to 3-hydroxy-3-methylglutaryl-CoA reductase, ability to cross the blood brain barrier and metabolic stability based on electrostatic molecular orbital studies. Eur J Med Chem. 2014;85:661–74. https://doi.org/10.1016/j.ejmech.2014.08.037.
Karahalil B, Hare E, Koç G, et al. Hepatotoxicity associated with statins. Arh Hig Rada Toksikol. 2017;68(4):254–60. https://doi.org/10.1515/aiht-2017-68-2994.
Makar GA, Weiner MG, Kimmel SE, et al. Incidence and prevalence of abnormal liver associated enzymes in patients with atrial fibrillation in a routine clinical care population. Pharmacoepidemiol Drug Saf. 2008;17(1):43–51. https://doi.org/10.1002/pds.1514.
Miyamoto R, Nagao K, Matsuto K, et al. Relationship between atrial fibrillation and a liver fibrogenesis marker in patients with acute heart failure. Int J Cardiol. 2023;374:51–7. https://doi.org/10.1016/j.ijcard.2023.01.001.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (grant number: NRF-2023R1A2C1007463).
Author information
Authors and Affiliations
Contributions
All the authors have made substantial contributions to the conception of the study. SAC, JSK, TJS, and HSG contributed to designing the study. YC and TJS contributed to material preparation and YAP, DHL, MP and JY contributed to data collection. SAC and JSK performed data analysis and interpretation. SAC and JSK contributed to the drafting of the manuscript. TJS and HSG contributed to the critical revision of the manuscript. All authors approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics Approval
The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of Ewha Womans University Seoul Hospital and Ewha Womans University Mokdong Hospital (IRB number: 2020-11-014 and 2021-02-026, respectively).
Consent for Publication
All authors approved the final manuscript and the submission to this journal.
Consent to Participate
Written informed consent was obtained from all patients.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Choi, SA., Kim, J.S., Park, YA. et al. Transporter Genes and statin-induced Hepatotoxicity. Cardiovasc Drugs Ther (2024). https://doi.org/10.1007/s10557-024-07580-2
Accepted:
Published:
DOI: https://doi.org/10.1007/s10557-024-07580-2