
Abstract
Purpose
Ventricular arrhythmias (VAs) are a common cause of sudden death in acute myocardial infarction (MI), for which hypertension is a major risk factor. Nicorandil opens ATP-sensitive potassium (KATP) channels, which are expressed by nerve terminals and cardiomyocytes and regulate the release of norepinephrine (NE). However, the effects of nicorandil on ischemic NE release in cardiac tissue remain unclear. Therefore, we herein investigated whether nicorandil suppressed interstitial NE concentrations and VAs during acute MI in pressure overload–induced hypertrophic hearts.
Methods
Rats were divided into two groups: an abdominal aortic constriction (AAC) group and sham-operated (Sham) group. Four weeks after constriction, cardiac geometry and functions were examined using echocardiography and hemodynamic analyses. Myocardial ischemia was induced by coronary artery occlusion for 100 min with or without the administration of nicorandil. VAs were assessed by electrocardiography, and NE concentrations in the ischemic region were measured using a micro-dialysis method.
Results
AAC induced left ventricular hypertrophy with diastolic dysfunction. VAs markedly increased in the early phase (0–20 min) of ischemia in both groups and were more frequent in the AAC group. Cardiac interstitial NE concentrations were higher in the AAC group before ischemia and significantly increased during ischemia in both groups. Nicorandil significantly suppressed ischemia-induced VAs and NE increases in the AAC group.
Conclusion
Ischemia-induced VAs were more frequent in hypertrophic hearts and associated with high interstitial concentrations of NE. The attenuation of ischemia-induced increases in NE through neuronal KATP opening by nicorandil may suppress ischemia-induced VAs in hypertrophic hearts.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hypertension is a common lifestyle-related disease that is highly prevalent in the elderly. Left ventricular (LV) hypertrophy is a cardiac manifestation of hypertensive cardiac disorders and an important risk factor for sudden cardiac death and arrhythmias [1,2,3]. Hypertension is also a prevalent risk factor for ischemic heart diseases, such as angina pectoris and myocardial infarction, the incidence of which was estimated to be 2- to 3-fold higher than that in normotensive individuals in the Framingham study [3]. In acute myocardial infarction, severe ventricular arrhythmias (VAs), such as ventricular fibrillation (Vf) and sustained ventricular tachycardia (VT), are frequent causes of death because they occur during the first hour of the onset of ischemia before medical monitoring [4, 5]. In acute ischemia, VAs consist of 2 phases. The first phase of VAs, occurring in the first 2 to 30 min, is attributed to abnormal automaticity and reentry, while the latter phase, occurring 1.5 to 72 h after the onset of acute myocardial infarction, occurs due to abnormal automaticity in surviving Purkinje fibers and is associated with the reperfusion of ischemic areas [6]. The coexistence of acute ischemia and LV hypertrophy was previously shown to induce greater alterations in the electrophysiological state than in non-hypertrophic hearts, and increased the inducibility of VAs [7].
Adenosine triphosphate (ATP)-sensitive potassium (KATP) channels are expressed by various types of cells in cardiac tissue, such as cardiac myocytes, vascular smooth muscle cells, and autonomic neurons, and KATP channels are present in the sarcolemma and mitochondria of these cells [8,9,10]. The KATP channel almost closes under normoxic conditions; however, under ischemic conditions, the depletion of intracellular ATP concentrations and the accumulation of ischemia-associated metabolites, such as adenosine diphosphate and lactate, open the channel. Nicorandil is a hybrid compound of an ATP channel opener and nitrate and is typically used in the treatment of angina pectoris as a coronary vasodilator. In patients with acute myocardial infarction, the administration of nicorandil before coronary interventions reduces infarct sizes and ischemia–reperfusion-induced arrhythmia by attenuating the no-reflow phenomenon [11,12,13]. Furthermore, the administration of nicorandil prior to ischemia has been shown to ameliorate ischemia–reperfusion injury, including arrhythmia, as pharmacological preconditioning in several types of animals [14,15,16]. The suppression of cardiac inflammation and direct myocyte protection were previously identified as some of the protective effects of nicorandil [14,15,16]. In cardiac myocytes, the attenuation of Ca overload and mitochondrial protection were found to contribute to the beneficial effects of KATP channel openers in ischemia–reperfusion injury [17, 18]. KATP channels are also expressed by the presynaptic and postsynaptic membranes of sympathetic nerves, which regulate neurotransmitter release, including norepinephrine (NE), in cardiac tissue [9, 19, 20]. In acute neuron injury, the opening of KATP channels has been shown to protect neurons [21]. However, the effects of nicorandil on neuronal NE release and associated VAs under acute ischemia in hearts, particularly hypertrophic hearts, remain unclear. Therefore, the present study investigated whether nicorandil suppressed ischemia-induced neuronal NE release and VAs using chronic pressure overload–induced cardiac hypertrophic rats by abdominal aortic constriction (AAC).
Methods
All procedures conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. The experimental protocol was approved by the Bioethics Committee of Kyoto Pharmaceutical University and was conducted in accordance with the rules for Animal Experimentation of Kyoto Pharmaceutical University.
Experimental Animals and Protocols
Eight-week-old male Sprague Dawley rats were anesthetized with an intraperitoneal injection of sodium pentobarbital (35 mg/kg). Rats were randomly divided into two groups: (1) an AAC group and (2) sham-operated (Sham) group. AAC was performed by placing a ligature around the abdominal aorta and a 24-gauge needle and then removing the needle. Sham-operated animals were treated similarly except that the suture around the abdominal aorta was not tied. Four weeks after surgery, 6–7 rats in each group were subjected to echocardiography and hemodynamic analyses under anesthesia with urethane (1.2 mg/kg). Rats were then sacrificed, and the hearts and lungs were extracted and weighed. The LV was extracted, weighed, and stored for later analyses. The remaining rats in each group were further divided into the following groups: (1–1) an untreated AAC (AAC-cont, n = 5) group, (1–2) nicorandil (20 μg/kg/min)-treated AAC (AAC-nico, n = 5) group, (2–1) untreated sham (Sham-cont, n = 7) group, and (2–2) nicorandil (20 μg/kg/min)-treated sham (Sham-nico, n = 6) group. Under anesthesia with urethane (1.2 mg/kg) and controlled ventilation, electrocardiography was recorded from limb electrodes to assess VAs and a polyethylene tube was inserted into the femoral vein for the infusion of nicorandil. Another polyethylene tube was inserted into the femoral artery to measure blood pressure. Body temperature was maintained at 37 ℃ by a controlled warm bed. After thoracotomy, the heart was exposed and a microdialysis probe (EVAL, length 5 mm, OD 0.225 mm, and ID 0.175 mm; SB Kawasumi Co., Kawasaki, Japan) was implanted in the anterior wall of the LV (Fig. 1a, b). A 6–0 silk suture was inserted around the left anterior coronary artery and set to the occluder. After 120 min of stabilization, VAs were assessed for 20 min before ischemia and following acute myocardial ischemia by occlusion of the coronary artery for 100 min in the presence or absence of the infusion of nicorandil (20 μg/kg/min) (Fig. 1c).

Structure of the microdialysis probe (a), schema of interstitial fluid collection in the ischemic region of the left ventricle (b), and the experimental protocol (c). Five millimeters of a PAN dialysis membrane (a) was inserted between polyethylene capillaries and inserted in the anterior wall of the left ventricle (the area of left anterior coronary perfusion). Ringer’s solution was perfused from the inlet polyethylene capillary, and the dialysate was collected from the outlet polyethylene capillary (b). Time course of the experimental protocol (c). Sham, sham-operated group; AAC, abdominal aortic constriction group. Four weeks after surgery, Sham- and AAC-treated rats were further divided into the four groups: untreated sham group (Sham-cont), nicorandil-treated sham group (Sham-nico), untreated AAC group (AAC-cont), and nicorandil-treated AAC group (AAC-nico). Rats were subjected to ischemia by occlusion of the left anterior coronary artery for 100 min in the presence or absence of the infusion of nicorandil
LV Echocardiographic Studies
Four weeks after AAC surgery, LV function was assessed by transthoracic echocardiography with a 15-MHz sector scan probe (Sonos 5500, Phillips Medical Systems Japan, Tokyo, Japan). Diastolic interventricular septum wall thickness, diastolic posterior wall thickness, LV end-diastolic diameter (LVEDd) and end-systolic diameter (LVEDs), LV fractional shortening (LVFS), and the LV ejection fraction (LVEF) were measured using M-mode recordings at the level of the mitral tip.
Hemodynamic Analyses
After echocardiography, hemodynamic parameters were assessed. A 1.6-F catheter-tip pressure manometer (PRIMETECH Co., Tokyo, Japan) was inserted via the right carotid artery into the LV and connected to a transducer. LV systolic pressure (LVSP), LV end-diastolic pressure (LVEDP), and the maximal rates of LV pressure increases (+ dP/dtmax) and decreases (− dP/dtmax) were measured.
Histological Analysis
After hemodynamic analyses, hearts were extracted and washed in isotonic saline. After separation from the right ventricle, the LV was fixed with phosphate-buffered 4% paraformaldehyde solution and embedded with paraffin. Paraffin-embedded tissue samples were cut into 4-μm-thick sections. Sections were deparaffinized with xylene, stained with hematoxylin–eosin (HE) and Sirius Red, and examined using an optical microscope system (Olympus IX71). The sizes of cardiac myocytes were assessed by cross-sectional area measurements using HE-stained sections, and the percentage area of interstitial fibrosis was calculated using Sirius Red–stained sections. In the assessment of cross-sectional myocyte areas, a suitable area of myocytes was defined as that with myofiber shapes and circular capillary profiles (indicative of a true transverse section). The circumferences of more than 100 myocytes were traced, and the computerized pixels of each sample were counted. Cardiac interstitial fibrosis was quantified by counting the pink pixel content in a computer-assisted image analysis program using Sirius Red–stained sections and calculated as the sum of the area of fibrosis divided by the sum of the cardiac tissue area in the field. At least five independent fields of the myocardium in each sample were photographed and quantified.
Measurement of NE Levels in Cardiac Tissue
Myocardial interstitial NE concentrations were assessed using the microdialysis method [22]. The microdialysis probe implanted in the anterior wall of the LV was perfused with Ringer’s solution at a rate of 1.5 μL/min and stabilized for 120 min (Fig. 1a, b). The composition of Ringer’s solution was as follows (mmol/L): NaCl 147, KCl 4.02, and CaCl2 2.7. After stabilization, the dialysate was sampled for 20 min before and after ischemia in the presence or absence of the infusion of nicorandil during the ischemic period. Dialysate samples were frozen at − 80 °C for later analyses. NE contents in collected dialysates were measured using high-performance liquid chromatography and an electrochemical detection system [23].
Statistical Analysis
StatView software version 5.0 (SAS Institute, Inc., NC, USA) and EZR software were used to analyze the significance of differences. All values are expressed as the mean ± SEM. Data were examined by Student’s t test (Fig. 2b, c, d, e, f, Fig. 3a, b, Fig. 7b (before versus after ischemia), and Table 1), a one-way ANOVA for repeated measures followed by Fisher’s protected least significant difference test (Fig. 4a, Fig. 5a, Fig. 6a), or a one-way ANOVA for multiple comparisons followed by the protected least significant difference test (Fig. 4b, Fig. 5b, Fig. 6b, Fig. 7a). The proportion of rats exhibiting Vf was analyzed using a contingency table analysis. A p value < 0.05 was considered to be significant.

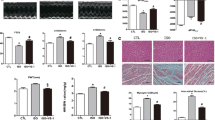
Echocardiographic assessment 4 weeks after surgery in Sham and AAC groups. Representative M-mode echocardiography (a). The bar graph indicates the left ventricular end-diastolic diameter (LVEDd) (b), left ventricular fractional shortening (LVFS) (c), left ventricular ejection fraction (LVEF) (d), the thickness of the intraventricular septum (IVS) (e), and the thickness of the posterior wall of the left ventricle (LVPW) (f) (n = 7). *p < 0.05 versus Sham

Histological assessment of cardiomyocyte hypertrophy and interstitial fibrosis in the left ventricle 4 weeks after surgery. Representative photomicrographs of hematoxylin–eosin-stained LV sections (a) and Sirius Red–stained LV sections (b) 4 weeks after surgery. Bars indicate 50 μm (a) or 100 μm (b). Cross-sectional area of myocytes (a, n = 5) and the percentage of interstitial fibrosis (b, n = 6) in the Sham and AAC groups. *p < 0.05 versus Sham

Effects of nicorandil on ischemia-induced premature ventricular contractions (PVCs). Ventricular arrhythmias were assessed by electrocardiography. The line graph shows the time course of PVCs in the Sham and AAC groups in the presence or absence of nicorandil (a), and the bar graph shows the number of PVCs during the first 20 min of ischemia (b). Sham-cont, untreated sham-operated group (n = 7); Sham-nico, nicorandil-treated sham-operated group (n = 5); AAC-cont, untreated AAC-operated group (n = 5); AAC-nico, nicorandil-treated AAC-operated group (n = 6). ¶p < 0.05 versus others, *p < 0.05 versus Sham-cont, #p < 0.05 versus AAC-cont

Effects of nicorandil on ischemia-induced ventricular tachycardia (VT). VT was assessed by electrocardiography. The line graph shows the time course of VT in the Sham and AAC groups in the presence or absence of nicorandil (a), and the bar graph shows the number of VT during the first 20 min of ischemia (b). Sham-cont, untreated sham-operated group (n = 7); Sham-nico, nicorandil-treated sham-operated group (n = 5); AAC-cont, untreated AAC-operated group (n = 5); AAC-nico, nicorandil-treated AAC-operated group (n = 6). *p < 0.05 versus Sham-cont, †p < 0.05 versus Sham-nico, #p < 0.05 versus AAC-cont

Effects of nicorandil on ischemia-induced premature ventricular fibrillation (Vf). Vf was assessed by electrocardiography. The line graph shows the time course of Vf in the Sham and AAC groups in the presence or absence of nicorandil (a), and the bar graph shows the number of Vf during the first 20 min of ischemia (b). Sham-cont, untreated sham-operated group (n = 7); Sham-nico, nicorandil-treated sham-operated group (n = 5); AAC-cont, untreated AAC-operated group (n = 5); AAC-nico, nicorandil-treated AAC-operated group (n = 6). *p < 0.05 versus Sham-cont

Effects of nicorandil on ischemia-induced interstitial norepinephrine (NE) concentrations. Interstitial NE concentrations in the ischemic region of the left ventricle were assessed using microdialysis methods. The bar graph shows interstitial NE concentrations before ischemia (a). The line graph shows the time course of NE concentrations in the Sham and AAC groups in the presence or absence of nicorandil before and after the first 20 min of ischemia. Sham-cont, untreated sham-operated group (n = 7); Sham-nico, nicorandil-treated sham-operated group (n = 5); AAC-cont, untreated AAC-operated group (n = 5); AAC-nico, nicorandil-treated AAC-operated group (n = 6). *p < 0.05 versus Sham-cont, #p < 0.05 versus AAC-cont, †p < 0.05 versus before ischemia
Results
Body and Organ Weights and Hemodynamic Analyses
Body and organ weights and hemodynamic data 4 weeks after surgery are shown in Table 1. Body weights were similar between the Sham and AAC groups. However, the ratio of heart weight-to-body weight and the ratio of LV weight-to-body weight significantly increased in the AAC group, suggesting that chronic pressure overload by AAC induced LV hypertrophy. AAC also significantly increased LVSP. In addition, heart rates (HRs) were lower in the AAC group than in the Sham group. Maximum increases in LV pressure (+ dP/dtmax), an indicator of LV systolic function, and LVEDP as well as the maximum decrease in LV pressure (− dP/dtmax), an indicator of LV diastolic function, were measured. + dP/dtmax was similar between the Sham and AAC groups. LVEDP was significantly higher (by fourfold), and − dP/dtmax was lower (by 35%) in the AAC group than in the Sham group, indicating that AAC induced LV diastolic dysfunction with preserved systolic function.
Echocardiographic Assessments
Echocardiographic assessments of LV geometry and function 4 weeks after AAC surgery are shown in Fig. 2. Four weeks after AAC surgery, the diastolic diameters of the LV were similar in the AAC and Sham groups. In addition, LVFS and LVEF, indicators of LV systolic function, were similar between the Sham and AAC groups. On the other hand, the interventricular septum and posterior wall of the LV were markedly thicker in the AAC group.
Histological Assessments
Myocyte hypertrophy and interstitial fibrosis in the LV are shown in Fig. 3. Figure 3a (left photos) and 3b (left photos) shows representative sections stained with HE and Sirius Red, respectively, 4 weeks after surgery. Quantitative measurements of the cross-sectional areas of myocytes, an indicator of myocyte hypertrophy, are shown in Fig. 3a (bar graph). The cross-sectional area was significantly increased in the AAC group, indicating myocyte hypertrophy. Furthermore, in Sirius Red–stained sections, the percentage of pink areas, which indicate interstitial fibrosis, was significantly higher in the AAC group than in the Sham group (Fig. 3b, bar graph).
Assessment of Ischemia-Induced VAs
Acute ischemia–induced VAs were assessed by electrocardiography. The time courses of premature ventricular contractions (PVCs), VT, and Vf are shown in Figs. 4a, 5a, and 6a, respectively. Before ischemia, VAs were not detected in the Sham or AAC group. During ischemia by left anterior coronary occlusion, the number of PVCs markedly increased within 20 min and then gradually decreased (Fig. 4a). The time course change in PVCs was greater in the AAC-cont group than in the other groups. The total number of PVCs in the first 20 min of ischemia is shown in Fig. 4b. The frequency of ischemia-induced PVCs was 2.7-fold higher in the AAC-cont group than in the Sham-cont group. The administration of nicorandil during ischemia did not affect blood pressure. On the other hand, the frequency of PVCs was slightly lower in the Sham-nico group than in the Sham-cont group, significantly lower by 80% in the AAC-nico group than in the AAC-cont group, and similar in the AAC-nico and Sham-cont groups. VT was observed during the early phase of ischemia, similar to PVCs (Fig. 5a). The time course change in VT was significantly greater in the AAC-cont group than in the Sham groups and slightly greater in the AAC-cont group than in the AAC-nico group (p = 0.07) (Fig. 5a). The total frequency of VT in the first 20 min of ischemia is shown in Fig. 5b. Similar to the results on PVCs, the frequency of VT was significantly higher in the AAC-cont group than in the Sham-cont group. The infusion of nicorandil significantly suppressed VT during the first 20 min of ischemia in AAC rats. Vf was also observed in the early phase of ischemia (Fig. 6a), with 60% and 14.2% of rats exhibiting Vf in the AAC-cont group and Sham-cont group, respectively. During the first 20 min of ischemia, the frequency of Vf was higher in the AAC-cont group than in the Sham-cont group. The infusion of nicorandil slightly reduced (p = 0.06) the proportion of rats exhibiting Vf in the AAC groups, while Vf did not occur in the Sham-nico or AAC-nico group.
NE Concentrations in the Ischemic Region of the LV
Increases in intracellular NE concentrations play critical roles in VAs in the early phase of ischemia. Therefore, interstitial NE concentrations in the ischemic region of the LV were assessed using the microdialysis method and are shown in Fig. 7. NE concentrations before ischemia were approximately 3-fold higher in the AAC-cont and AAC-nico groups than in the Sham-cont and Sham-nico groups (Fig. 7a). During the first 20 min of ischemia, NE concentrations in the ischemic region significantly increased in the Sham-cont, Sham-nico, and AAC-cont groups, and tissue NE concentrations were 2.6-fold higher in the early phase of ischemia in the AAC-cont group than in the Sham-cont group. Furthermore, nicorandil significantly suppressed ischemia-induced increases in NE in the AAC-nico group.
Discussion
The present results demonstrated that 4 weeks of pressure overload by AAC induced LV hypertrophy and associated histological LV remodeling, such as myocyte hypertrophy and interstitial fibrosis, leading to LV diastolic dysfunction with preserved systolic function. Acute ischemia by the occlusion of the coronary artery induced VAs more frequently in AAC-induced hypertrophic hearts than in non-hypertrophic Sham hearts. In addition, cardiac interstitial NE concentrations before ischemia were higher in the AAC group than in the Sham group and were significantly increased in both groups by ischemia. The infusion of nicorandil during ischemia suppressed ischemia-induced VAs and abolished Vf in association with the suppression of ischemia-induced increases in interstitial NE concentrations in AAC-induced hypertrophic hearts.
In the present study, + dP/dtmax, LVFS, and LVEF were similar between the AAC and Sham groups. On the other hand, LVEDP was significantly increased and − dP/dtmax was smaller in the AAC group, indicating that AAC induced LV diastolic dysfunction with preserved systolic function. Accumulating evidence suggests that pressure overload–induced hypertrophy impairs LV diastolic function, which is consistent with the present results [24]. Four weeks after surgery, HR was significantly lower in the AAC group than in the Sham group. HR is dependent on the duration of diastole [25, 26]. Pressure overload–induced LV hypertrophy caused cardiac diastolic dysfunction in the present study, while a clinical study reported a longer duration of diastole due to diastolic dysfunction in hypertensive patients [27]. Therefore, diastolic dysfunction due to pressure overload may prolong the duration of cardiac diastole, which resulted in reduced HR in the present study.
In hypertrophic hearts, cardiac fibrosis–induced anatomical uncoupling and reentry mechanisms contribute to arrhythmia. In the present model, interstitial fibrosis and myocyte hypertrophy were observed after AAC. In the AAC model used in the present study, the abdominal aorta between the left and right renal arteries was constricted, which reduced renal blood flow and enhanced renin production in addition to pressure overload by aortic constriction. A previous study reported increases in the level of the well-known fibrosis-promoting cytokine, transforming growth factor β, the production of which is enhanced by the renin-angiotensin pathway, and the content of collagen in the cardiac tissue of a similar AAC-induced LV hypertrophy model [28]. Therefore, not only constriction-induced pressure overload, but also the promotion of the renin-angiotensin system may have increased LV hypertrophy and interstitial fibrosis in the present model. Interstitial fibrosis produces autonomic uncoupling and the zig-zag propagation of electrical wave conduction, which activates the reentry mechanism [29]. VAs were not observed in the AAC groups (AAC-cont and AAC-nico groups) or Sham groups (Sham-cont and Sham-nico groups) under normoxic conditions in the present study. Our observation period under normoxic conditions before ischemia was only 20 min, which may have been too short to detect VAs. On the other hand, acute ischemia by coronary occlusion induced VAs more frequently in the AAC-cont group than in the Sham-cont group, which is consistent with previous findings [7]. Therefore, ischemia-induced arrhythmogenicity in addition to vulnerable reentry formation by histological changes in the hypertrophic heart may have exacerbated ventricular ischemia in the present study.
The frequency of VAs markedly increased under ischemia in both the Sham-cont and AAC-cont groups. Reentry and abnormal automaticity play critical roles in the acute phase of ischemia [6]. Automaticity is partly mediated by autonomic neuron activity, as indicated by interstitial NE concentrations. The addition of a catecholamine to the perfusate of an extracted perfusion heart model in mice increased ischemic arrhythmias, while local cardiac denervation during the acute ischemic period reduced the frequency of VAs [30, 31], suggesting the crucial role of interstitial NE concentrations in ischemia-induced arrhythmia. In the present study, the frequency of VAs was the highest from 0 to 20 min after the onset of ischemia, and interstitial NE concentrations were higher in this period than in the non-ischemic period (− 20 to 0 min) in both the Sham-cont and AAC-cont groups. Therefore, consistent with previous findings, increases in interstitial NE concentrations during the early phase of ischemia may contribute to ischemia-induced VAs in both non-hypertrophic and hypertrophic hearts.
NE concentrations before ischemia were higher in hypertrophic hearts than in non-hypertrophic hearts in the present study, and this phenomenon was consistent with previous findings [32]. NE concentrations before ischemia in the AAC-cont group were higher than those after ischemia in the Sham-cont group. In contrast, VAs were more frequent in the ischemic period in the Sham-cont group than in the non-ischemic period in the AAC-cont group. Therefore, sudden increases in interstitial NE concentrations during the early phase of ischemia may be critical for the induction of VAs along with absolute interstitial NE concentrations. In addition to increases in NE, other ischemia-induced changes, such as Ca overload and metabolic changes, may play a role in VAs during the early phase of ischemia [6]. Ischemia-induced VAs were more frequent in the AAC-cont group than in the Sham-cont group. On the other hand, increases in NE concentrations during the early phase of ischemia were similar between the Sham-cont and AAC-cont groups. A previous study reported that pressure-induced hypertrophic hearts were more sensitive to catecholamines than non-hypertrophic hearts [33]. Therefore, the greater sensitivity of NE in AAC-induced hypertrophic hearts may play a critical role in the frequency of VAs in the early phase of ischemia in the present study.
In the present study, nicorandil suppressed ischemia-induced VAs in association with the attenuation of interstitial NE increases in the ischemic cardiac tissue of AAC-induced hypertrophic hearts. KATP channels are expressed in nerve terminals and cardiomyocytes and mediate the release of NE from sympathetic nerve terminals [34]. Furthermore, the affinity of nicorandil to KATP channels was shown to be higher in nerve terminals than in cardiomyocytes [35]. Therefore, in the present study, nicorandil activated KATP channels in neurons, thereby inhibiting NE release during ischemia in AAC-induced hypertrophic hearts. On the other hand, the massive myocardial release of NE into interstitial cardiac tissue was noted during ischemia and the mechanism underlying this increase was reportedly via non-exocytotic release [36, 37]. In a preliminary examination of a similar acute ischemic model, longer (1-h) ischemia markedly increased interstitial NE concentrations. However, the most frequent duration of ischemia that induced VAs was within 20 min and NE increases in this period were significant, but not massive. Therefore, the increase in NE observed in the early phase of ischemia in the present study may be attributed to exocytotic release, and the activation of KATP channels by nicorandil suppressed its release. Moreover, the activation of KATP channels in neurons attenuated acute neuron injury [21]. Nicorandil-induced KATP opening may also protect neurons from ischemic injury, leading to the suppression of the non-exocytotic release of NE observed in the present study.
Nicorandil suppressed ischemia-induced VAs in the AAC group and slightly inhibited them in the Sham group, whereas ischemia-induced interstitial increases in NE were suppressed in the AAC group, but not in the Sham group. In the present hypertrophic model, pressure overload affects not only the heart, but also other upper parts of the body. A previous study reported that chronic hypertension in a systemic hypertension rat model impaired neurons through nerve microangiopathy [38]. Hypertension delayed the conduction velocity of neurons and was associated with histological impairments, such as axonal atrophy, endoneurial edema, microangiopathy, and myelin splitting [38]. Furthermore, morphological changes, such as endoneurial edema, may induce hypoxia in neurons. On the other hand, hypoxia-inducible factor 2α (HIF2α) increased the number of KATP channels in perinatal rat adrenal chromaffin cells [39]. Therefore, in the present model of chronic pressure overload by AAC, KATP channels in neurons may have been increased by HIF2α and, thus, the suppression of NE by nicorandil was enhanced in the AAC-nico group. In the AAC group, nicorandil significantly suppressed ischemia-induced increases in interstitial NE, whereas NE concentrations after ischemia in the AAC-nico group were still higher than those after ischemia in the Sham-cont group. On the other hand, the frequency of ischemia-induced VAs was similar between the AAC-nico and Sham-cont groups. Therefore, a sudden increase in interstitial NE may play critical roles on VAs along with absolute NE levels.
The suppression of interstitial NE enhancement in ischemic period by nicorandil through neuronal KATP opening is the attentional focus in the present study. On the other hand, KATP is also expressed in cardiac myocytes and Horinaka et al. [40] reported that nicorandil suppressed lethal ischemic VAs in association with upregulation of endothelial nitric oxide synthase and sulfonylurea receptor 2 expression in non-ischemic region. Therefore, we cannot exclude the possibility that nicorandil affects myocytes in non-ischemic region and might play a role in the reduction of VAs.
The present results indicate that nicorandil suppressed ischemia-induced VAs in association with the attenuation of ischemia-induced increases in NE in hypertrophic cardiac tissue. Nicorandil opens KATP channels in nerve terminals and cardiac myocytes, and the opening of KATP in nerve terminals by nicorandil may inhibit the release of NE, thereby suppressing ischemia-induced VAs. However, in the present study, the effects of KATP inhibitors were not examined because selective inhibitors of neuronal KATP were not available. Therefore, further examinations are needed to elucidate the role of KATP in nerve terminals on the nicorandil-induced suppression of ischemia-induced VAs.
In conclusion, ischemia-induced arrhythmias were more frequently observed in chronic pressure overload–induced LV hypertrophic hearts than in non-hypertrophic hearts. Moreover, interstitial NE concentrations in cardiac tissue were higher in hypertrophic hearts than in non-hypertrophic hearts, and ischemia further increased NE concentrations. Nicorandil attenuated ischemia-induced increases in NE through neuronal KATP opening, which suppressed ischemia-induced VAs in hypertrophic hearts.
Data Availability
The datasets analyzed in the present study are available from the corresponding author on reasonable request.
References
Haider AW, Larson MG, Benjamin EJ, Levy D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J Am Coll Cardiol. 1998;32:1454–9.
Levy D, Anderson KM, Savage DD, Balkus SA, Kannel WB, Castelli WP. Risk of ventricular arrhythmias in left ventricular hypertrophy: the Framingham Heart Study. Am J Cardiol. 1987;60:560–5.
Levy D, Wilson PW, Anderson KM, Castelli WP. Stratifying the patient at risk from coronary disease: new insights from the Framingham Heart Study. Am Heart J. 1990;119:712–7.
Gheeraert PJ, De Buyzere ML, Taeymans YM, et al. Risk factors for primary ventricular fibrillation during acute myocardial infarction: a systematic review and meta-analysis. Eur Heart J. 2006;27:2499–510.
Luqman N, Sung RJ, Wang C-L, Kuo C-T. Myocardial ischemia and ventricular fibrillation: pathophysiology and clinical implications. Int J Cardiol. 2007;119:283–90.
Di Diego JM, Antzelevitch C. Ischemic ventricular arrhythmias: experimental models and their clinical relevance. Heart Rhythm. 2011;8:1963–8.
Kohya T, Kimura S, Myerburg RJ, Bassett AL. Susceptibility of hypertrophied rat hearts to ventricular fibrillation during acute ischemia. J Mol Cell Cardiol. 1988;20:159–68.
Moreau C, Jacquet H, Prost AL, D’hahan N, Vivaudou M. The molecular basis of the specificity of action of K(ATP) channel openers. EMBO J. 2000;19:6644–51.
Kang CS, Chen CC, Lin CC, Chang NC, Lee TM. Effect of ATP-sensitive potassium channel agonists on sympathetic hyperinnervation in postinfarcted rat hearts. Am J Physiol Heart Circ Physiol. 2009;296:H1949–59.
Garlid KD, Paucek P, Yarov-Yarovoy V, Sun X, Schindler PA. The mitochondrial KATP channel as a receptor for potassium channel openers. J Biol Chem. 1996;271:8796–9.
Lee HC, An SG, Choi JH, et al. Effect of intra-coronary nicorandil administration prior to reperfusion in acute ST segment elevation myocardial infarction. Circ J. 2008;72:1425–9.
Zhou J, Xu J, Cheng A, Li P, Chen B, Sun S. Effect of nicorandil treatment adjunctive to percutaneous coronary intervention in patients with acute myocardial infarction: a systematic review and meta-analysis. J Int Med Res. 2020;48:300060520967856.
Wang ZD, Li H, Liu M, et al. Effect of intravenous application of nicorandil on area of myocardial infarction in patients with STEMI during the perioperative stage of PCI. Clin Hemorheol Microcirc. 2021;77:411–23.
Das B, Sarkar C. Mitochondrial K ATP channel activation is important in the antiarrhythmic and cardioprotective effects of non-hypotensive doses of nicorandil and cromakalim during ischemia/reperfusion: a study in an intact anesthetized rabbit model. Pharmacol Res. 2003;47:447–61.
Mizumura T, Nithipatikom K, Gross GJ. Effects of nicorandil and glyceryl trinitrate on infarct size, adenosine release, and neutrophil infiltration in the dog. Cardiovasc Res. 1995;29:482–9.
Das B, Sarkar C, Karanth KS. Selective mitochondrial K(ATP) channel activation results in antiarrhythmic effect during experimental myocardial ischemia/reperfusion in anesthetized rabbits. Eur J Pharmacol. 2002;437:165–71.
Eaton M, Hernandez LA, Schaefer S. Ischemic preconditioning and diazoxide limit mitochondrial Ca overload during ischemia/reperfusion: role of reactive oxygen species. Exp Clin Cardiol. 2005;10:96–103.
Chen M, Zhou JJ, Kam KW, Qi JS, Yan WY, Wu S, et al. Roles of KATP channels in delayed cardioprotection and intracellular Ca(2+) in the rat heart as revealed by kappa-opioid receptor stimulation with U50488H. Br J Pharmacol. 2003;140:750–8.
Miura T, Kawamura S, Tatsuno H, et al. Ischemic preconditioning attenuates cardiac sympathetic nerve injury via ATP-sensitive potassium channels during myocardial ischemia. Circulation. 2001;104:1053–8.
Ohe K, Sperlagh B, Santha E, et al. Modulation of norepinephrine release by ATP-dependent K+ channel activators and inhibitors in human isolated right atrium. Cardiovasc Res. 1999;43:125–34.
Yoshida M, Nakakimura K, Cui YJ, Matsumoto M, Sakabe T. Adenosine A(1) receptor antagonist and mitochondrial ATP-sensitive potassium channel blocker attenuate the tolerance to focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2004;24:771–9.
Akiyama T, Yamazaki T, Ninomiya I. In vivo monitoring of myocardial interstitial norepinephrine by dialysis technique. Am J Physiol. 1991;261:H1643–7.
Naseratun N, Kobara M, Watanabe Y, Toba H, Nakata T. Comparison of effects of L/N-type and L-type calcium channel blockers on post-infarct cardiac remodelling in spontaneously hypertensive rats. Clin Exp Pharmacol Physiol. 2020;47:1545–53.
Richards DA, Aronovitz MJ, Calamaras TD, et al. Distinct phenotypes induced by three degrees of transverse aortic constriction in mice. Sci Rep. 2019;9:5844.
Zhang W, Bogale S, Golriz F, Krishnamurthy R. Relationship between heart rate and quiescent interval of the cardiac cycle in children using MRI. Pediatr Radiol. 2017;47:1588–93.
Sarnari R, Kamal RY, Friedberg MK, Silverman NH. Doppler assessment of the ratio of the systolic to diastolic duration in normal children: relation to heart rate, age and body surface area. J Am Soc Echocardiogr. 2009;22:928–32.
Trevi G, Sheiban I, Gorni R. Myocardial hypertrophy and arterial hypertension. G Ital Cardiol. 1995;25:1331–8.
Zheng RH, Bai XJ, Zhang WW, et al. Liraglutide attenuates cardiac remodeling and improves heart function after abdominal aortic constriction through blocking angiotensin II type 1 receptor in rats. Drug Des Devel Ther. 2019;13:2745–57.
Assayag P, Carré F, Chevalier B, Delcayre C, Mansier P, Swynghedauw B. Compensated cardiac hypertrophy: arrhythmogenicity and the new myocardial phenotype. I Fibrosis Cardiovasc Res. 1997;34:439–44.
Stables CL, Curtis MJ. Development and characterization of a mouse in vitro model of ischaemia-induced ventricular fibrillation. Cardiovasc Res. 2009;83:397–404.
Schlaich MP, Kaye DM, Lambert E, et al. Angiotensin II and norepinephrine release: interaction and effects on the heart. J Hypertens. 2005;23:1077–82.
Bischoff KO, Meesmann W, Wild U. Contractility and elevated catecholamine sensitivity of pressure-produced hypertrophied heart. Z Kardiol. 1974;63:971–87.
Chen J, Li M, Yu Y, et al. Prevention of ventricular arrhythmia complicating acute myocardial infarction by local cardiac denervation. Int J Cardiol. 2015;184:667–73.
Burgdorf C, Dendorfer A, Kurz T, et al. Role of neuronal KATP channels and extraneuronal monoamine transporter on norepinephrine overflow in a model of myocardial low flow ischemia. J Pharmacol Exp Ther. 2004;309:42–8.
Liu GS, Thornton J, Van Winkle DM, Stanley AW, Olsson RA, Downey JM. Protection against infarction afforded by preconditioning is mediated by A1 adenosine receptors in rabbit heart. Circulation. 1991;84:350–6.
Schömig A, Fischer S, Kurz T, Richardt G, Schömig E. Nonexocytotic release of endogenous noradrenaline in the ischemic and anoxic rat heart: mechanism and metabolic requirements. Circ Res. 1987;60:194–205.
Schömig A, Dart AM, Dietz R, Mayer E, Kübler W. Release of endogenous catecholamines in the ischemic myocardium of the rat. Part A: locally mediated release. Circ Res. 1984;55:689–701.
Nukada H, Baba M, Ogasawara S, McMorran D, Yagihashi S. Neuropathy in the spontaneously hypertensive rat: an electrophysiological and histological study. Muscle Nerve. 2016;54:756–62.
Salman S, Brown ST, Nurse CA. Chronic nicotine induces hypoxia inducible factor-2α in perinatal rat adrenal chromaffin cells: role in transcriptional upregulation of KATP channel subunit Kir6.2. Am J Physiol Cell Physiol. 2012;302:C1531-8.
Horinaka S, Kobayashi N, Yabe A, et al. Nicorandil protects against lethal ischemic ventricular arrhythmias and up-regulates endothelial nitric oxide synthase expression and sulfonylurea receptor 2 mRNA in conscious rats with acute myocardial infarction. Cardiovasc Drugs Ther. 2004;18:13–22.
Acknowledgements
The authors gratefully thank Dr. T. Akiyama M.D., Ph.D. (Department of Cardiac Physiology, National Cerebral and Cardiovascular Center, Osaka, Japan) for the excellent technical assistance with the microdialysis method.
Funding
The present study was supported in part by the “The Open Research Program” of Kyoto Pharmaceutical University from the Ministry of Education, Science, and Culture of Japan.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Miyuki Kobara designed and performed the experiments and wrote the first draft of the manuscript. Toshihiro Amano performed the experiments and data analysis. Hiroe Toba performed the data analysis. Tetsuo Nakata contributed to the concept of the study and drafting the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval
All procedures conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. The protocol was approved by the Bioethics Committee of Kyoto Pharmaceutical University.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kobara, M., Amano, T., Toba, H. et al. Nicorandil Suppresses Ischemia-Induced Norepinephrine Release and Ventricular Arrhythmias in Hypertrophic Hearts. Cardiovasc Drugs Ther 37, 53–62 (2023). https://doi.org/10.1007/s10557-022-07369-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-022-07369-1