
Abstract
Purpose
Small and big conductance Ca2+-sensitive potassium (KCa) channels are involved in cardioprotective measures aiming at reducing myocardial reperfusion injury. For levosimendan, infarct size–reducing effects were shown. Whether activation of these channels is involved in levosimendan-induced postconditioning is unknown. We hypothesized that levosimendan exerts a concentration-dependent cardioprotective effect and that both types of Ca2+-sensitive potassium channels are involved.
Methods
In a prospective blinded experimental laboratory investigation, hearts of male Wistar rats were randomized and placed on a Langendorff system, perfused with Krebs-Henseleit buffer at a constant pressure of 80 mmHg. All hearts were subjected to 33 min of global ischemia and 60 min of reperfusion. At the onset of reperfusion, hearts were perfused with various concentrations of levosimendan (0.03–1 μM) in order to determine a concentration-response relationship. To elucidate the involvement of KCa-channels for the observed cardioprotection, in the second set of experiments, 0.3 μM levosimendan was administered in combination with the subtype-specific KCa-channel inhibitors paxilline (1 μM, big KCa-channel) and NS8593 (0.1 μM, small KCa-channel) respectively. Infarct size was determined by tetrazolium chloride (TTC) staining.
Results
Infarct size in controls was 60 ± 7% and 59 ± 6% respectively. Levosimendan at a concentration of 0.3 μM reduced infarct size to 30 ± 5% (P < 0.0001 vs. control). Higher concentrations of levosimendan did not induce a stronger effect. Paxilline but not NS8593 completely abolished levosimendan-induced cardioprotection while both substances alone had no effect on infarct size.
Conclusions
Cardioprotection by levosimendan-induced postconditioning shows a binary phenomenon, either ineffective or with maximal effect. The cardioprotective effect requires activation of big but not small KCa channels.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
After myocardial infarction, the restoration of coronary blood flow is crucial for the survival of the patient. Myocardial ischemia is mostly not predictable making cardioprotective strategies for these situations necessary. Cardioprotection against ischemia and reperfusion injury can be achieved by postconditioning, an intervention that occurs at the end of an ischemic period and the onset of reperfusion. Zhao et al. [1] described the phenomenon of ischemic postconditioning where short cycles of ischemia and reperfusion at the onset of reperfusion-induced significant infarct size reduction. The effect of ischemic postconditioning can be mimicked pharmacologically, e.g., with volatile anesthetics [2] or opioids [3]. Also for calcium (Ca2+) sensitizer, levosimendan cardioprotective properties were shown [4]. A cardioprotective effect by levosimendan postconditioning was described by du Toit et al. [5]. The authors showed in isolated Guinea pigs hearts an involvement of the reperfusion injury salvage kinase (RISK) pathway and ATP-sensitive potassium (KATP)-channels. Matsumoto et al. demonstrated an involvement of the mitochondrial permeability transition pore (mPTP) and a decrease of the cardioprotective effect of levosimendan-induced conditioning by hyperglycemia [6]. Levosimendan is a positive inotropic drug without any effect on diastolic function [7]. An interaction of levosimendan with cardiac troponin C causes an increased sensitivity for Ca2+ in cardiomyocytes. Clinically, levosimendan is indicated for the treatment of heart failure. Whether cardioprotection by levosimendan-induced postconditioning is concentration-dependent is unknown.
Activation of Ca2+-sensitive potassium channels was shown to be involved in cardioprotection by postconditioning induced by the volatile anesthetic isoflurane [8]. One can distinguish between big (BKCa) and small (SKCa) conductance calcium-sensitive potassium channels. Opening of BKCa channels is related to increased cytosolic Ca2+ and a voltage gradient [9]. SKCa channels are voltage-independent and activation of these channels triggers an interaction of calmodulin in the C-terminus region [10]. Stowe et al. demonstrated that BKCa and SKCa channels activated by respective agonists independently from each other induced significant infarct size reduction [11]. Whether both types of Ca2+-sensitive potassium channels play a role in levosimendan-induced postconditioning has not been investigated so far.
In the present study, we set out to determine the lowest cardioprotective concentration of levosimendan and the underlying mechanism involved in levosimendan-induced cardioprotection against ischemia and reperfusion injury. We hypothesize that the infarct size–reducing effect of levosimendan is concentration-dependent and that activation of small and/or big conductance Ca2+-sensitive potassium channels is required for its cardioprotective property.
Methods
The study was performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (publication number 85-23, revised 1996) and was approved by the Animal Ethics Committee of the University of Duesseldorf (O 27/12), Germany. Experiments were done and results were reported in accordance with the ARRIVE guidelines.
Surgical Preparation
Surgical preparation was performed as described previously [3]. Male Wistar rats (mean body weight part one, 287 ± 16 g; part two, 317 ± 23 g) were anesthetized by intraperitoneal injection of pentobarbital (90 mg/kg). Hereafter, animals were thoracotomized for the removal of the hearts. The hearts were mounted on a Langendorff system and perfused with Krebs-Henseleit solution, enriched with 95% O2 and 5% CO2. The solution contains (in mM) 118 NaCl, 4.7 KCl, 1.2 MgSO4, 1.17 KH2PO4, 24.9 NaHCO3, 2.52 CaCl2, 0.5 EDTA, 11 glucose, and 1 lactate at 37 °C. For the duration of the experiments, a constant pressure (80 mmHg) and temperature (37 °C) was maintained [12]. We inserted a fluid-filled balloon into the left ventricle and kept an end-diastolic pressure of 2–8 mmHg. The hearts underwent an equilibration period of 20 min. We measured heart rate, maximal left ventricular pressure (LVP max), coronary flow and minimal left ventricular pressure (LVP min) continuously and digitized it at a sampling rate of 500 Hz by use of an analogue to digital converter system (PowerLab/8SP, ADInstrument Pty Ltd., Castle Hill, Australia). Left ventricular–developed pressure (LVDP) was calculated as maximal LVP–minimal LVP. Data were continuously recorded on a personal computer using Chart for Windows v5.0 (ADInstruments Pty Ltd., Castle Hill, Australia).
Experimental Protocol
After surgical preparation, all hearts underwent a baseline period of 20 min and 33 min of global ischemia followed by 60 min of reperfusion. Global ischemia of 33 min was achieved by stopping coronary perfusion (no-flow ischemia) of the heart with Krebs-Henseleit buffer for 33 min. Furthermore, the heart was surrounded by oxygen-free buffer solution to prevent diffusion of oxygen from the outside into the myocardium. Start of reperfusion was induced by removing the surrounding buffer and restoring the coronary perfusion of the heart.
Global ischemia caused a sufficient infarct size, representing the primary endpoint of this study, to show relevant cardioprotective effects. Additionally, this degree of myocardial damage does not completely impair myocardial function during reperfusion.
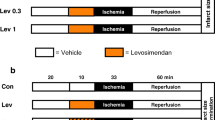
The present study was divided into two parts. In the first part, we determined the lowest cardioprotective concentration of levosimendan needed for infarct size reduction. Hearts were randomly assigned to five groups (n = 6 per group, Fig. 1a):
Control (Con): Hearts received no further treatment.
Levosimendan (Lev): Hearts were perfused with 0.03, 0.1, 0.3, and 1 μM Lev for 10 min at the onset of reperfusion.

a, b Experimental protocol. a Part 1 of the study: Con = control, Lev = levosimendan; b Part 2 of the study: Con = control, Lev = levosimendan, Pax = paxilline; NS8593 = N-[(1R)-1,2,3,4-tetrahydro-1-naphthalenyl]-1H-benzimidazol-2-amine hydrochloride
Levosimendan in a concentration of 0.3 μM showed the strongest infarct size–reducing effect, thus this concentration was chosen for part two of the study assessing the underlying mechanism of levosimendan-induced postconditioning. Hearts were randomly assigned to six groups (n = 5–6 per group, Fig. 1b):
Control (Con, n = 6): Hearts received no further treatment.
Levosimendan (Lev, n = 6): Hearts were perfused with 0.3 μM Lev for 10 min at the onset of reperfusion.
Paxilline + Levosimendan (Pax + Lev, n = 6): Hearts were perfused with the BKCa-channel inhibitor paxilline in a concentration of 1 μM [13] combined with 0.3 μM Lev for 10 min at the onset of reperfusion.
Paxilline (Pax, n = 6): Hearts were perfused with 1 μM Pax for 10 min only, to rule out an intrinsic effect of Pax on infarct size.
NS8593 + Levosimendan (NS8593 + Lev, n = 6): Hearts were perfused with the SKCa-channel inhibitor NS8593 (N-[(1R)-1,2,3,4-Tetrahydro-1-naphthalenyl]-1H-Benzimidazol-2-amine hydrochloride) in a concentration of 0.1 μM [14] combined with 0.3 μM Lev for 10 min at the onset of reperfusion.
NS8593 (NS8593, n = 5): Hearts were perfused with 0.1 μM NS8593 for 10 min only, to rule out an intrinsic effect of NS8593 on infarct size.
At the end of reperfusion, hearts were cut into transverse slices, starting from the cardiac apex to just before the cardiac valvular plane. The slices were stained with 0.75% TTC solution. The size of the infarcted area was determined by planimetry using SigmaScan Pro 5 computer software (SPSS Science Software, Chicago, IL) by two blinded investigators. For this purpose, the individual TTC stained slices were scanned and infarction area was manually determined on the computer by selecting the infarcted and non-infarcted area. The non-vital infarction area appears colorless and the vital non-infarcted area of the myocardium red. Infarct size was calculated as percentage of infarcted area to non-infarcted area from the whole heart.
Statistical Analysis
Calculation of sample size was done by using GraphPad StatMate™ (GraphPad Software, San Diego, CA, USA) and resulted in a group size of n = 6 for detecting a 25% difference in infarct size with a power of 80% (α < 0.05 (two-tailed)). Hemodynamic variables were measured continuously and detected during baseline, ischemia, and reperfusion. To compare hemodynamic variables between groups or between different time points within groups, we used a two-way analysis of variance (ANOVA) and a Tukey post hoc test (GraphPad Software, San Diego, CA, USA). An investigator blinded to the experimental groups determined the infarct sizes. A one-way ANOVA was chosen, followed by a Tukey post hoc test to analyze infarct size. Data are presented as mean ± SD. Differences were regarded as statistically significant when P < 0.05.
Results
Infarct Size—Concentration-Related Effect of Levosimendan Postconditioning
Figure 2a illustrates infarct sizes for various concentrations of levosimendan. Control hearts showed an infarct size of 60 ± 7%. The lowest cardioprotective concentration of levosimendan was 0.3 μM with an infarct size of 30 ± 5% (P < 0.0001 vs. Con). Increasing the concentration to 1 μM did not lead to a further infarct size reduction (Lev 1: 32 ± 4%; p = 0.9871 vs. Lev 0.3). Lower concentrations than 0.3 μM did not induce cardioprotection (Lev 0.1: 50 ± 8%; p = 0.1111 vs. Con and Lev 0.03: 56 ± 7%; p = 0.8197 vs. Con).

a, b Infarct size measurement. a The infarct size of controls (Con) and levosimendan (Lev); b the infarct size of controls (Con), levosimendan (Lev) with or without the KCa-channel inhibitors paxilline (Pax) and NS8593 (NS8593). Data are mean ± SD. *P < 0.05 vs. Con and #P < 0.05 vs. Lev respectively
Infarct Size—Mechanism of Levosimendan Postconditioning
Figure 2b represents infarct sizes from part two of the study assessing the involvement of BKCa and/or SKCa channels in levosimendan-induced postconditioning. Infarct size of the control group was 59 ± 6%. Administration of 0.3 μM levosimendan reduced infarct size to 31 ± 8% (P < 0.0001 vs. Con). The cardioprotective effect of levosimendan was completely abrogated by the BKCa-channel inhibitor paxilline (Pax + Lev: 62 ± 6%, P < 0.0001 vs. Lev) but not by the SKCa-channel inhibitor NS8593 (NS8593 + Lev: 35 ± 7%, P = 0.9021 vs. Lev). Both inhibitors alone had no effect on infarct size (Pax: 62 ± 4%; P = 0.9021 vs. Con and NS8593: 60 ± 4%; P > 0.9999 vs. Con).
Cardiac Function
There were no differences in heart rate, LVDP, and coronary flow between the groups (Tables 1 and 2). After ischemia and during reperfusion, LVDP and coronary flow were statistically different from baseline in all groups in both parts of the study (Tables 1 and 2). In part two of the study, heart rate was statistically different compared to baseline after ischemia in the Pax + Lev group and in the Pax group (Table 2). LVP min, dP/dt max, and dP/dt min were statistically different during the reperfusion period compared to baseline (Supplement Table S1 and S2).
Discussion
The results of the current study demonstrate that postconditioning with the calcium-sensitizer levosimendan shows a binary phenomenon, either ineffective or with maximal effect. The lowest cardioprotective concentration of levosimendan in our study model was 0.3 μM. We are the first to demonstrate that this cardioprotective effect requires activation of BKCa but not SKCa channels.
Cardioprotective interventions by postconditioning are promising because the majority of myocardial ischemic events cannot be predicted. Although short cycles of ischemia and reperfusion are a very strong stimulus to induce infarct size reduction—with the exception of cardiac surgery and cardiological interventions—such measure is not feasible for the clinical scenario. Fortunately, cardioprotection achieved by ischemic stimuli can be mimicked pharmacologically thereby inducing a comparable infarct size reduction.
Levosimendan is clinically used for patients with heart failure and leads, when administered preoperatively, to lower mortality [15]. It increases the calcium sensitivity by binding to troponin C and has a positive inotropic effect, without influencing relaxation in the diastole. In addition, it opens ATP-dependent potassium channels (KATP) in the smooth vascular muscles, lowering pre- and afterload by vasodilation. Hönisch et al. demonstrated postconditioning properties of levosimendan in the rat heart in vivo [4]. The infarct size reduction was in the same range as with ischemic postconditioning but the combination of both interventions did not lead to a more pronounced cardioprotective effect. In addition, this group showed that cardioprotection was mediated by PI3K (phosphatidylinositol 3 kinase) pathway and activation of KATP-channels.
The current results show that the lowest cardioprotective concentration of levosimendan in our model is 0.3 μM and an increase in concentration to 1 μM had no additional effect. Employing the lowest effective concentration of levosimendan allows for maximal cardioprotection by minimizing possible drug-related side effects. Concentration-dependent effects of levosimendan postconditioning have not been studied so far. Based on our results, the effect of levosimendan-induced cardioprotection is more concentration-related than dependent as we detected an on-off phenomenon, either ineffective or with maximal effect.
Involvement of KCa-channel activation in cardioprotection was described by us [3, 13] and others [8, 16]. Stowe et al. demonstrated in the isolated heart model using global ischemia that activation of BKCa and SKCa channels independently from each other confers cardioprotection [11]. In the present study, also global ischemia was used. In the Langendorff model, both forms, regional and global, of ischemia are used [17,18,19]. With a direct comparison of both ischemia forms, Kim et al. demonstrated that global ischemia induced larger infarct sizes than regional ischemia [20]. Due to this larger infarct sizes, an excellent discrimination between viable and infarcted area is possible. A further advantage of global ischemia is the high reproducibility due to equal severity and extent of ischemia. In particular, induction of regional ischemia is often inconsistent in localization and severity. Interestingly we detected a strong effect of levosimendan on infarct size but no hemodynamic improvement during the reperfusion period. Consistent with current literature [21], for isolated perfused hearts, infarct size seems the most sensitive marker to assess cardioprotection. The exact reason for this is unclear, but the occurrence of myocardial stunning is often discussed, especially after global ischemia.
The study by Stowe et al. showed that cardioprotection induced by the BKCa-channel activator NS1619 was not affected by the SKCa-channel inhibitor NS8593 and vice versa [11]. Furthermore, simultaneous activation of BKCa and SKCa channels did not further enhance the infarct size reducing effect [11]. Our results emphasize that activation of KCa channels play a pivotal role in pharmacological postconditioning induced by levosimendan. Interestingly, the results further demonstrate that only BKCa channels are crucial for the cardioprotective effect as the SKCa-channel inhibitor NS8593 had no influence on the infarct size reduction induced by levosimendan. Only the blocker of BKCa-channels paxilline completely abrogated the effect of levosimendan. Paxilline is a mycotoxin produced by the fungus Penicillium paxilli blocking all subunits of BKCa channels [22] selectively [23,24,25]. In contrast to the results by Stowe et al. [11], where activation by selective agonists of both channels led to infarct size reduction, levosimendan’s cardioprotective effect might be mainly conferred via activation of BKCa channels and inhibition of SKCa channels would have very little impact. Another explanation could be that inhibition of the signaling pathway downstream of SKCa-channel activation with subsequent loss of cardioprotection would elucidate an involvement of these channels in levosimendan conditioning. It is important to distinguish between both channels due to their differential activation depending on the actual status of the cell. BKCa channels are voltage-gated and directly regulated by Ca2+. In contrast, SKCa channels are voltage-independent and are indirectly regulated by Ca2+ via calmodulin. Both KCa channels regulate mitochondrial bioenergetics but are activated during different conditions of the cell [11]. Thus, time point and mode of activation seem to be important. The importance to distinguish both channels is also visible by their specific inhibitors and activators. The study by Stowe et al. demonstrated that the cardioprotective effect of BKCa- and SKCa-channel activation was completely abolished by the administration of the O2·- dismutator TBAP (Mn (III) tetrakis (4-benzoic acid) porphyrin). TBAP hereby interferes with the signaling pathway downstream of KCa-channel activation demonstrating that O2·- are released by activation of BKCa and SKCa channels suggesting that both channels share a common final step in cardioprotection. However, the exact involvement of SKCa channels in levosimendan postconditioning has to be investigated in a future study.
Activation of BKCa-channels seems to represent a crucial step in the signaling cascade of pharmacological postconditioning. Previously, we could show that these channels are also involved in morphine-induced postconditioning [3].
As aforementioned, ischemic postconditioning is not feasible for the clinical setting making pharmacological postconditioning an interesting and promising option for myocardial protection in the clinic. Unfortunately, these promising interventions are not implemented in the clinic yet. Results from a controlled experimental setup, focusing on one specific aspect such as infarct size reduction and excluding confounding factors such as concomitant diseases or effect-modulating medications, are difficult to extrapolate to the clinical setting. Drugs like beta-blockers or the antidiabetic glibenclamide were shown to abrogate infarct size reduction by the volatile anesthetic desflurane [26, 27]. Furthermore, age was shown to have an impact on cardioprotective properties of conditioning interventions [28]. Many cardioprotective interventions, ischemically or pharmacologically, failed to reduce infarct size in the aged heart (for review see [29]). Aging might interfere with cardioprotective signaling pathways suggesting that downstream activation within the signaling cascades might be promising for myocardial protection of the aged heart. Previously, we demonstrated that activation of BKCa channels with NS1619 reduced infarct size in the aged rat heart in vivo and that the extent of infarct size reduction, employing the same concentration of the BKCa-channel activator, was comparable to young rats [30]. Particularly, these results emphasize BKCa channels as an important and promising target for myocardial protection making future research in this field indispensable.
Diabetes and hyperglycemia were also shown to abolish cardioprotection by conditioning interventions [31, 32]. Matsumoto et al. demonstrated that hyperglycemia raised the threshold for postconditioning with levosimendan [6]. Only a ten times higher concentration of levosimendan was able to restore the hyperglycemia-induced abrogated cardioprotective effect [6]. Whether BKCa-channel activation of levosimendan or other effects of this drug were responsible for cardioprotection under hyperglycemia can only be speculated.
Limitations
An animal in vitro study has several limitations. Considering the hemodynamic data, we measure the isovolumetric work of the heart, thus only the intrinsic properties of the rat heart, without neurohumoral and peripheral vascular influence. Thus, it is difficult to transfer this isovolumetric working Langendorff heart to the clinical situation. However, and that is, in turn, the advantage of the model, as we detected no differences in hemodynamic variables between the groups, we can conclude that the infarct size reduction is due to the effect of levosimendan.
Levosimendan is a clinically used inotropic drug for the treatment of heart failure. Against this background, next to its inotropic activity, cardioprotective properties of levosimendan would be particularly attractive. In this context, one has to consider that we have examined the hearts of young and healthy rats. Patients who would benefit from such an intervention are usually old, multimorbid, and take several different medications representing possible confounders for cardioprotection. Therefore, our results cannot be related to the clinic immediately, but suggest a possible direction and open up new possibilities for levosimendan, which should be considered.
Conclusion
The current results show that the calcium-sensitizer levosimendan induces cardioprotection by postconditioning in a more concentration-related than a concentration-dependent manner. The lowest cardioprotective concentration inducing the strongest cardioprotective effect was 0.3 μM. Furthermore, it seems that activation of BKCa channels but not SKCa channels is an elementary step in the signaling cascade of myocardial protection induced by levosimendan.
References
Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, et al. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol. 2003;285(2):H579–88.
Steurer MP, Steurer MA, Baulig W, Piegeler T, Schlapfer M, Spahn DR, et al. Late pharmacologic conditioning with volatile anesthetics after cardiac surgery. Crit Care. 2012;16(5):R191.
Huhn R, Heinen A, Weber NC, Schlack W, Preckel B, Hollmann MW. Ischaemic and morphine-induced post-conditioning: impact of mK (Ca) channels. Br J Anaesth. 2010;105(5):589–95.
Honisch A, Theuring N, Ebner B, Wagner C, Strasser RH, Weinbrenner C. Postconditioning with levosimendan reduces the infarct size involving the PI3K pathway and KATP-channel activation but is independent of PDE-III inhibition. Basic Res Cardiol. 2010;105(2):155–67.
du Toit EF, Genis A, Opie LH, Pollesello P, Lochner A. A role for the RISK pathway and K (ATP) channels in pre- and post-conditioning induced by levosimendan in the isolated guinea pig heart. Br J Pharmacol. 2008;154(1):41–50.
Matsumoto S, Cho S, Tosaka S, Higashijima U, Maekawa T, Hara T, et al. Hyperglycemia raises the threshold of levosimendan- but not milrinone-induced postconditioning in rat hearts. Cardiovasc Diabetol. 2012;11:4.
Papp Z, Edes I, Fruhwald S, De Hert SG, Salmenpera M, Leppikangas H, et al. Levosimendan: molecular mechanisms and clinical implications: consensus of experts on the mechanisms of action of levosimendan. Int J Cardiol. 2012;159(2):82–7.
Kinoshita M, Tsutsumi YM, Fukuta K, Kasai A, Tanaka K. Isoflurane-induced postconditioning via mitochondrial calcium-activated potassium channels. J Med Investig. 2016;63(1–2):80–4.
Stefani E, Ottolia M, Noceti F, Olcese R, Wallner M, Latorre R, et al. Voltage-controlled gating in a large conductance Ca2+−sensitive K+channel (hslo). Proc Natl Acad Sci U S A. 1997;94(10):5427–31.
Jenkins DP, Strobaek D, Hougaard C, Jensen ML, Hummel R, Sorensen US, et al. Negative gating modulation by (R)-N-(benzimidazol-2-yl)-1,2,3,4-tetrahydro-1-naphthylamine (NS8593) depends on residues in the inner pore vestibule: pharmacological evidence of deep-pore gating of K (Ca)2 channels. Mol Pharmacol. 2011;79(6):899–909.
Stowe DF, Yang M, Heisner JS, Camara AKS. Endogenous and agonist-induced opening of mitochondrial big versus small Ca2+−sensitive K+ channels on cardiac cell and mitochondrial protection. J Cardiovasc Pharmacol. 2017;70(5):314–28.
Behmenburg F, Dorsch M, Huhn R, Mally D, Heinen A, Hollmann MW, et al. Impact of mitochondrial Ca2+-sensitive potassium (mBKCa) channels in sildenafil-induced cardioprotection in rats. PLoS One. 2015;10(12):e0144737.
Behmenburg F, Trefz L, Dorsch M, Strothoff M, Mathes A, Raupach A, et al. Milrinone-induced postconditioning requires activation of mitochondrial Ca(2+)-sensitive potassium (mBKCa) channels. J Cardiothorac Vasc Anesth. 2018;32(5):2142–8.
Pasdois P, Quinlan CL, Rissa A, Tariosse L, Vinassa B, Costa AD, et al. Ouabain protects rat hearts against ischemia-reperfusion injury via pathway involving src kinase, mitoKATP, and ROS. Am J Physiol Heart Circ Physiol. 2007;292(3):H1470–8.
Levin R, Degrange M, Del Mazo C, Tanus E, Porcile R. Preoperative levosimendan decreases mortality and the development of low cardiac output in high-risk patients with severe left ventricular dysfunction undergoing coronary artery bypass grafting with cardiopulmonary bypass. Exp Clin Cardiol. 2012;17(3):125–30.
Shintani Y, Node K, Asanuma H, Sanada S, Takashima S, Asano Y, et al. Opening of Ca2+−activated K+ channels is involved in ischemic preconditioning in canine hearts. J Mol Cell Cardiol. 2004;37(6):1213–8.
Bell RM, Mocanu MM, Yellon DM. Retrograde heart perfusion: the Langendorff technique of isolated heart perfusion. J Mol Cell Cardiol. 2011;50(6):940–50.
Najafi M, Noroozi E, Javadi A, Badalzadeh R. Anti-arrhythmogenic and anti-inflammatory effects of troxerutin in ischemia/reperfusion injury of diabetic myocardium. Biomed Pharmacother. 2018;102:385–91.
Leistner M, Sommer S, Kanofsky P, Leyh R, Sommer SP. Ischemia time impacts on respiratory chain functions and Ca(2+)-handling of cardiac subsarcolemmal mitochondria subjected to ischemia reperfusion injury. J Cardiothorac Surg. 2019;14(1):92.
Kim JH, Kim J, Park YH, Chun KJ, Kim JS, Jang YH, et al. Cardiodynamics and infarct size in regional and global ischemic isolated heart model: comparison of 1 hour and 2 hours reperfusion. Korean Circ J. 2012;42(9):600–5.
Lindsey ML, Bolli R, Canty JM Jr, Du XJ, Frangogiannis NG, Frantz S, et al. Guidelines for experimental models of myocardial ischemia and infarction. Am J Physiol Heart Circ Physiol. 2018;314(4):H812–H38.
Hu H, Shao LR, Chavoshy S, Gu N, Trieb M, Behrens R, et al. Presynaptic Ca2+−activated K+ channels in glutamatergic hippocampal terminals and their role in spike repolarization and regulation of transmitter release. J Neurosci. 2001;21(24):9585–97.
Sanchez M, McManus OB. Paxilline inhibition of the alpha-subunit of the high-conductance calcium-activated potassium channel. Neuropharmacology. 1996;35(7):963–8.
Wang X, Fisher PW, Xi L, Kukreja RC. Essential role of mitochondrial Ca2+−activated and ATP-sensitive K+ channels in sildenafil-induced late cardioprotection. J Mol Cell Cardiol. 2008;44(1):105–13.
Zhou Y, Lingle CJ. Paxilline inhibits BK channels by an almost exclusively closed-channel block mechanism. J Gen Physiol. 2014;144(5):415–40.
Lange M, Redel A, Smul TM, Lotz C, Nefzger T, Stumpner J, et al. Desflurane-induced preconditioning has a threshold that is lowered by repetitive application and is mediated by beta 2-adrenergic receptors. J Cardiothorac Vasc Anesth. 2009;23(5):607–13.
Thornton JD, Thornton CS, Sterling DL, Downey JM. Blockade of ATP-sensitive potassium channels increases infarct size but does not prevent preconditioning in rabbit hearts. Circ Res. 1993;72(1):44–9.
Schulman D, Latchman DS, Yellon DM. Effect of aging on the ability of preconditioning to protect rat hearts from ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2001;281(4):H1630–6.
Wojtovich AP, Nadtochiy SM, Brookes PS, Nehrke K. Ischemic preconditioning: the role of mitochondria and aging. Exp Gerontol. 2012;47(1):1–7.
Huhn R, Weber NC, Preckel B, Schlack W, Bauer I, Hollmann MW, et al. Age-related loss of cardiac preconditioning: impact of protein kinase A. Exp Gerontol. 2012;47(1):116–21.
Huhn R, Heinen A, Hollmann MW, Schlack W, Preckel B, Weber NC. Cyclosporine A administered during reperfusion fails to restore cardioprotection in prediabetic Zucker obese rats in vivo. Nutr Metab Cardiovasc Dis. 2010;20(10):706–12.
Huhn R, Heinen A, Weber NC, Hollmann MW, Schlack W, Preckel B. Hyperglycaemia blocks sevoflurane-induced postconditioning in the rat heart in vivo: cardioprotection can be restored by blocking the mitochondrial permeability transition pore. Br J Anaesth. 2008;100(4):465–71.
Acknowledgments
In partial fulfillment of the requirements for an MD thesis (M. van de Snepscheut). We thank J. Reinle and L. Goetze for their practical support.
Funding
This study was supported by institutional and departmental sources.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical Approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Stroethoff, M., Bunte, S., Raupach, A. et al. Impact of Ca2+-Sensitive Potassium Channels in Levosimendan-Induced Postconditioning. Cardiovasc Drugs Ther 33, 581–588 (2019). https://doi.org/10.1007/s10557-019-06908-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-019-06908-7