
Abstract
Colorectal cancer (CRC) patients frequently develop liver metastases, which are the major cause of cancer-related mortality. The molecular basis and management of colorectal liver metastases (CRLMs) remain a challenging clinical issue. Recent genomic evidence has demonstrated the liver tropism of CRC and the presence of a stricter evolutionary bottleneck in the liver as a target organ compared to lymph nodes. This bottleneck challenging CRC cells in the liver is organ-specific and requires adaptation not only at the genetic level, but also at the phenotypic level to crosstalk with the hepatic microenvironment. Here, we highlight the emerging evidence on the clonal evolution of CRLM and review recent insights into the molecular mechanisms orchestrating the bidirectional interactions between metastatic CRC cells and the unique liver microenvironment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Colorectal cancer (CRC) is the third most deadly cancer worldwide [1] and accounts for up to 10% of all cancer-related deaths [2]. Colorectal liver metastases (CRLM), the most common type of CRC metastasis, are the leading cause of CRC mortality [3]. It is estimated that 30–50% of CRC patients will suffer from CRLM throughout the course of their disease [3]. In fact, the incidence of metastatic liver cancer is 18–40 times higher than that of primary liver malignancies [4], with the colorectum being the most common site of origin [3, 5].
In the late nineteenth century, Paget proposed that organ-specific metastatic colonization occurs only when seeds (cancer cells) fall on congenial soil (organ microenvironment), known as the seed and soil theory [6]. As the largest parenchymal organ in the human body, the liver is endowed with unique anatomical features, making it the most common site for CRC metastasis [7]. On the one hand, the liver is highly vascularized and has a rich blood supply. With a unique dual vascular organization, it receives blood from both the hepatic artery (25%) and the portal vein (75%) [8]. As the venous drainage of the colon and rectum primarily flows into the portal vein, the liver serves as the first stop for circulating CRC cells. On the other hand, blood from the hepatic artery and the portal vein converge in the liver sinusoids, wherein the flow rate is exceptionally low [9]. Moreover, the sinusoidal vasculature is highly permeable, as liver sinusoidal endothelial cells (LSECs) are fenestrated and have an incomplete basement membrane [10]. Physiologically, these unique characteristics allow a sufficient exchange of materials between plasma and hepatocytes. Nonetheless, in the setting of cancer metastasis, the same traits make the liver intrinsically susceptible to trapping disseminated tumor cells in the microvasculature.
There are several possible reasons for the difficulty in treating CRLMs. The therapeutic challenge with surgery lies in the fact that curative resection is achieved in most patients. First, a large proportion of patients are diagnosed with unresectable diseases due to the lack of effective diagnostic methods for early detection. In addition, insufficient knowledge of the route of spread (lymphatic or hematogenous) and chronology (early or late event of tumorigenesis) results in ambiguity in the extent of surgery. In cases where CRLMs develop rapidly after primary malignant transformation and spread via a hematogenous route, the benefit of a more radical surgical strategy may be limited [11,12,13]. Moreover, since the liver is the primary site of drug metabolism, chemotherapeutic efficacy is severely compromised. In addition, therapeutic responses to targeted therapies, such as EGFR, BRAF, and HER2 blockade, may be heterogeneous due to the genomic heterogeneity of metastases [14,15,16].
As biological research techniques continue to evolve, novel mechanisms underlying the development of CRLM are gradually being uncovered. For instance, genomic evolutionary analyses have reconstructed the phylogenetic trees of CRLM, enabling the exploration of driver gene heterogeneity and the chronology of metastatic seeding [17,18,19,20]. This provided us with a new perspective to determine which tumor clones are genetically selected by the liver, namely, those with driver alterations such as KRAS, NRAS, SMAD4, and BRAF. Nevertheless, therapeutic targeting of these oncogenes has achieved only moderate efficacy. This may be the result of gene expression plasticity influenced by the liver tumor microenvironment (TME). Single-cell RNA sequencing and multi-omics studies have recently provided a more comprehensive view of the unique metabolic and immune landscape of the hepatic niche, which exerts selective pressure on CRC cells both genetically and phenotypically [21,22,23]. Overall, the molecular basis and management of CRLM is a challenging clinical question that remains to be addressed. In this review, we discuss recent advances in the mechanisms of CRLM—how tumor cells evolve and crosstalk with the liver TME. In addition, we also highlight the corresponding clinical therapeutic implications of these findings.
2 The clonal evolution of CRLM
2.1 The temporal and spatial patterns of CRLM
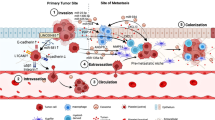
Comparative genetic studies of primary CRC and CRLMs have shed light on the timing and route of metastasis. Ancestral tree reconstructions have shown that subclones harboring driver gene mutations competent for metastasis can arise both early and late in the evolutionary process of CRC [17]. This was further supported by a recent genomic study showing that metastatic seeding occurs as early as approximately 2–4 years prior to the clinical diagnosis of primary CRC [18]. For many years, researchers have theorized that CRC spreads in a stepwise manner [24], with primary cancer cells first metastasizing to regional lymph nodes and then seeding distant organs. Nonetheless, recent studies have yielded conflicting results [19, 25,26,27]. As illuminated by genomic analyses, in most patients, subclones within the primary tumor directly seed liver metastases without colonizing the lymphatic system, supporting a hematogenous dissemination pattern [19, 25,26,27]. Given that venous drainage from the intestine reaches the liver via the portal vein, this would explain why the liver is the most common distant site. Collectively, evolutionary analyses suggest that in some cases, CRC cells may have hematogenously colonized the liver at an early stage of primary tumorigenesis, which raises some questions about surgical management, including the timing of surgery, the value of lymph node resection, and the extent of resection (Fig. 1).

The clonal evolution of CRLM. Metastatic spread of CRC can arise through distinct routes and exhibit different intra- and inter-lesion genomic heterogeneity. Top Panel: the possibility that CRC cells may bypass the lymph nodes and hematogenously colonize the liver raises some questions about the value of lymph node dissection and the extent of surgical resection; middle panel: CRLM may be derived from a single clone (monoclonal seeding) or from multiple distinct clones (polyclonal seeding) within the primary tumor, thus showing varying degrees of intratumoral heterogeneity. In polyclonal cases, a biopsy of a single region may underestimate genetic diversity; bottom panel: Within individual patients, untreated CRLMs follow monophyletic seeding, where separate lesions share the same original clones and are genetically similar to each other, suggesting a specific selective pressure in the liver. In this case, a biopsy of a single lesion is sufficient to guide treatment decisions. In contrast, CRC systemic metastases adhere to polyphyletic seeding, where inter-metastatic driver gene heterogeneity has been observed between lymph nodes, liver, and lung lesions, implying different levels of evolutionary bottlenecks. It should be noted that the clonal evolution of the treated CRLMs is more complex, as detailed in the text
2.2 The intratumor heterogeneity of CRLM
It is well established that primary CRC has a substantial level of genetic intratumor heterogeneity (ITH). For CRLM, the extent of ITH is largely influenced by the seeding pattern of tumor cells. CRC cells spreading to the liver were initially thought to follow a single-cell seeding mechanism, in which metastases arise from single clones within the primary tumor (monoclonal seeding) [28, 29]. This model indicates a competitive relationship between different clones in primary CRC [30]. However, there is growing evidence that CRLMs can also be seeded by multiple distinct clones (polyclonal seeding) [25, 26], suggesting that intercellular cooperation may be required [31, 32]. This notion is consistent with previous studies showing that circulating tumor cell (CTC) clusters that originate as oligoclonal groups from the primary tumor exhibit a higher ability to seed metastasis than single CTCs [33, 34]. Polyclonal metastasis may also occur through multiple rounds of seeding, in which case the first-arriving clone reshapes the liver microenvironment, making it favorable for later clones to colonize. In practical terms, polyclonality means that a biopsy at a single region of CRLM may underestimate its genetic diversity. A number of investigations of the cancer genome have shown that in most cases, the ITH within individual liver metastases was lower than that within their primary counterparts and lymph node metastases (relatively monoclonal) (Fig. 1) [18, 26, 35,36,37]. This is logically expected, given that cells arriving at the same distant sites tend to be single or in small clusters.
2.3 The intertumor heterogeneity of CRLM
In addition to intratumor heterogeneity, different levels of genetic diversity between metastases have also been observed within individual patients. When different metastases in distant organs have low inter-lesion heterogeneity and share the same parental clone, this pattern is referred to as monophyly. Within individual patients, untreated CRLMs resemble each other genetically and exhibit minimal functional driver gene heterogeneity, suggesting monophyletic seeding [35, 38]. In other words, for patients with multiple CRLMs, a biopsy of a single lesion is sufficient to depict the actionable genome and guide treatment decisions [36]. Polyphyletic seeding, on the other hand, describes another pattern in which different subclones in the primary site give rise to genetically distinct metastases [30, 35]. This is the case in CRC systemic metastases, where intermetastatic driver gene heterogeneity has been observed between liver and lung lesions [39] (Fig. 1).
The implications of genetic divergence among CRLMs extend beyond individual liver metastases. On the one hand, the interlesion diversity between the liver and lung strongly argues that specific selective pressures for a particular subpopulation of cells exist in the liver microenvironment that differ from those in the lung. In fact, this coincides with previous studies showing that metastasis-private mutations in CRLM were enriched in pathways postulated to foster colonization in the liver microenvironment, such as cell adhesion, extracellular matrix (ECM) remodeling, and hepatic stellate activation [40]. On the other hand, while CRLMs exhibit a monophyletic pattern, lymph node metastases of CRC show a high degree of interlesion diversity, suggesting a stricter evolutionary bottleneck in the liver [35]. It has been proposed for more than 100 years that certain cancers can exhibit organ-specific patterns of metastatic colonization; however, genomic evidence has remained scarce. Although no certain genetic traits have been defined as exclusive to CRLM, the observation that they are genetically homogeneous further corroborated the theory of metastatic tropism to the liver from an evolutionary point of view.
2.4 Genomic evolution of CRLM under therapeutic pressure
The clonal evolution of CRLM is stimulated not only by selective pressure from the liver microenvironment but also by therapeutic strategies. Phylogenetic analysis revealed that CRLM subclones can migrate within the liver, evade surgical resection, continue to evolve under chemotherapeutic pressure, and expand again to develop recurrence [41]. Of note, the evolutionary bottleneck imposed by surgery, chemotherapy, and targeted therapy varies. Although an increased degree of genetic divergence can always be observed in treated CRLMs, in most cases, no additional clinically actionable mutations are obtained after surgery and chemotherapy [42]. This suggests that a single biopsy and sequencing of metastases is sufficient for therapeutic decision-making over the conventional treatment course [38]. Conversely, when specific genes are targeted with small molecule inhibitors (in CRLM, EGFR, HER2, MEK, BRAF, KRAS blockade, etc.), acquired genetic alterations can arise that converge into reactivation of the targeted pathways [14, 15, 42,43,44]. Furthermore, secondary mutations elicited by targeted therapy can occur heterogeneously and independently in separate CRLMs within the same patient, leading to differences in treatment response between lesions [15]. Apparently, genomic evolution in this setting can lead to changes in therapeutic indications, making sequencing of a single-lesion biopsy insufficient and genomic follow-up after administration of targeted agents necessary. Serial ctDNA monitoring combined with tissue biopsies represents an effective tool to detect the clinically relevant oncogenic alterations associated with acquired resistance and to guide the next-line strategy [14, 15].
2.5 Genomic evolution of CRLM by tumor sidedness
It is well known that right-sided colon cancer, left-sided colon cancer, and rectal cancer exhibit significant differences in terms of embryonic origin, histological type, lymphovascular drainage, genomic profile, and gut flora, and metastatic patterns [45]. Clinical observations have shown that cancers originating from the left-sided colon and rectum are more likely to metastasize to the liver compared to those arising from the right-sided colon [46,47,48]. However, right-sided colon cancer is associated with a higher number of liver metastases, involvement of a broader range of segments, and a poorer prognosis [49]. The factors contributing to these differences are multifaceted, but recent genomic-based phylogenetic analyses have started to shed light on some of the underlying mechanisms.
Regarding the routes of metastatic spread, blood from the proximal part of the colon and rectum drains directly to the liver via the portal vein. In contrast, the distal part of the rectum may drain through the internal iliac vein to the inferior vena cava, bypassing the liver and entering the systemic circulation to reach the lung, as recently evidenced by genomic studies [39]. Alternatively, cancer cells from any site in the colorectum can migrate from the lymph nodes into the systemic circulation through the subclavian vein. In a study by Naxerova et al., which analyzed the clonal origin of lymph nodes and distant metastases, the majority (6/7) of liver metastases from right-sided colon cancer spread hematogenously from the primary tumor without lymph node involvement. In contrast, for almost half (4/9) of the patients with left-sided colon and rectal cancer, liver metastases might have originated sequentially from lymph node metastases [19]. Although the sample size is limited, this finding provides molecular evidence supporting the hypothesis that cancer cells from different primary sites may exhibit preferential patterns of dissemination routes to the liver, with some traveling directly through the portal vein and others via the lymph nodes and systemic circulation before reaching the liver. Adding to the complexity, the evolutionary bottlenecks in the circulation, lymph nodes, liver, and lung differ significantly from one another [35, 39]. As a result, tumor cells disseminated from various anatomical locations are subject to distinct selection pressures before seeding the liver, potentially contributing to the diverse evolutionary trajectories of liver metastases.
In addition to the anatomical difference, tumor sidedness has been demonstrated to influence liver metastasis due to inherent disparities in mutational profiles and clonal population structures. Notably, right-sided tumors possess a higher prevalent of oncogenic mutations (e.g., KRAS, BRAF, PIK3CA, AKT1, RNF43, and SMAD4) within the ancestral clone, while concurrently presenting fewer DNA copy-number alterations compared to left-sided primaries [50, 51]. These oncogenic events are all associated with worse oncologic outcomes in CRLM, which could partially explain the inferior prognosis observed in patients with right-sided tumors as opposed to those with left-sided tumors [50]. Conversely, left-sided tumors are characterized by a greater number of subclones, suggesting a more polyclonal and divergent evolutionarily nature [52, 53]. A high number of subclones may give rise to clones with enhanced metastatic potential and has been associated with an increased likelihood of liver metastasis development [54], corroborating the clinical observation that left-sided tumors exhibit a higher propensity for liver metastasis. Furthermore, primary tumors with a low mutational burden are more inclined to develop liver metastases, attributable to their reduced immunogenicity. In contrast, CRCs with a high mutational load exhibit a more complex TME that mitigates metastatic potential [53]. It is important to considered that the elevated frequency of microsatellite-instability-high (MSI-H)/deficient mismatch repair (dMMR) subtypes in right-sided colon cancer, as this genotype/phenotype displays a diminished inclination for liver metastasis development [2, 55]. Thus, the findings necessitate cautious interpretation, particularly when comparing genomic evolution between primary sites, taking into account the number of MSI-H/dMMR cases in the cohort. Collectively, clonal evolution and the development of liver metastases differ based on the primary site of CRC. However, due to the scarcity of current studies, much of the above discussion relies on indirect evidence. Furthermore, more conclusive analyses are needed to elucidate the finer details.
Overall, phylogenetic studies have highlighted the genomic impact on CRLM. However, emerging studies are highlighting the role of nongenetic mechanisms underpinning CRLM. It has recently been determined by paired DNA–RNA sequencing that only a small proportion of phenotypic changes in untreated primary CRC are accounted for by genetic variation [20], emphasizing the need for analysis beyond the genome, including the crosstalk with the unique metabolic and immune microenvironment.
3 The crosstalk of CRLM with the liver microenvironment
3.1 Metabolic adaptation of CRLM to the liver microenvironment
Branches of the hepatic artery, portal vein, hepatic vein, and bile duct along with the connective tissue stretch into the liver parenchyma, dividing the parenchyma into many small functional units called hepatic lobules, which are the basic structural elements of the liver. In the hepatic lobule, the blood flows concentrically through portal spaces toward the central vein [56]. Accordingly, the microenvironments of the liver are spatially altered, with a higher concentration of oxygen as well as high energy-consumption metabolic reactions in the periportal region and a low oxygen supply and less ATP-demanding metabolic processes in the central vein area [9, 57]. Metastatic CRC cells encounter distinct metabolic microenvironments and progressively increase oxidative stress along the porto–central axis during initial dissemination to the liver [56]. Therefore, to successfully transit through changing microenvironments, migrating CRC cells selectively and dynamically adapt their metabolism [58]. Alterations in glucose, fructose, lipid, amino acid, and nucleotide metabolism have been revealed in CRLM (Fig. 2).

Crosstalk between CRLMs and the liver TME. The microenvironment of CRLM undergoes remarkable spatiotemporal remodeling. The nutrient and oxygen content of the liver microenvironment is spatially altered along the porto–central axis; accordingly, migrating CRC cells dynamically adapt their metabolism for metastatic survival. Meanwhile, metastatic outgrowth is accompanied by a progressive weakening of antitumor immunity. Initially, the immature TME of CRLM is infiltrated by cytotoxic T cells and attacked by the immune system. Subsequently, as the metastatic cells expand, the immunosuppressive cells are recruited to cultivate an immunologically “cold” desert. Moreover, other components of the liver TME that crosstalk with CRLMs are shown, including translocated gut bacteria, hepatocytes, fibroblasts, and tissue stiffness
For overt colonization in the liver, metastatic CRC cells reprogram metabolic pathways to fulfill the bioenergetic, biosynthetic, and antioxidant requirements. In addition to glucose, fructose also acts as a carbon source to support CRC cell proliferation in the liver. ALDOB, an enzyme that catalyzes the reversible conversion of fructose-1,6-bisphosphate to glyceraldehyde 3-phosphate to enhance fructose metabolism and fuel glycolysis, gluconeogenesis, and the pentose phosphate pathway, is upregulated in disseminated CRC cells [59]. Creatine, a metabolite that is enriched in the liver, also contributes to malignant energetic catalysis. During intrahepatic hypoxia, creatine kinase brain-type (CKB) is upregulated by CRC cells and secreted into the extracellular microenvironment, where it catalyzes the generation of phosphocreatine from exogenous ATP and liver-derived creatine. Phosphocreatine is then taken up by CRC cells through the SLC6A8 transporter, and its high-energy phosphate is utilized to produce endogenous ATP, thereby meeting the high bioenergetic demand for metastatic survival [60].
Hypoxia represents a metabolic barrier to CRC outgrowth in the liver because metabolic precursors necessary for biosynthesis become restricted under such circumstances. In this case, metabolites accumulated from other pathways can be directed to fuel macromolecule biosynthesis [61]. In the low oxygen environment of the liver, the highly expressed gluconeogenic enzyme phosphoenolpyruvate carboxykinase 1 (PCK1) promotes CRC growth by enhancing nucleotide synthesis. Mechanistically, PCK1 drives reductive carboxylation to generate the pyrimidine precursor aspartate, thus maintaining the pyrimidine nucleotide pools, which are the building blocks for DNA and RNA [62].
Alterations in the metabolism of CRLMs are also involved in protecting against oxidative stress in the hypoxic hepatic microenvironment. To better respond to the changing redox status, metastatic CRC cells are equipped with a variety of antioxidant systems, with glutathione (GSH) being one of the most important defenses [63]. The enhanced ability of CD110 + CRC cells to maintain redox homeostasis during liver colonization has been linked to activated lysine catabolism [64]. This CRC subpopulation is attracted by hepatic thrombopoietin (TPO), a glycoprotein hormone generated by the liver. TPO-induced c-myc specifically upregulates genes involved in lysine degradation via epigenetic modifications, resulting in increased glutamate, a byproduct of lysine catabolism, to fuel GSH synthesis [65]. Furthermore, reprogrammed glucose metabolism also contributes to antioxidant responses. Pyruvate kinase M2 (PKM2), the most prevalent pyruvate kinase expressed in highly glycolytic cancer cells, is inhibited in CRLM by upregulating glucose pyruvate kinase liver and red blood cells (PKLR) [66]. Consequently, the glycolytic flux is diverted into the pentose phosphate pathway, thereby producing nicotinamide adenine dinucleotide phosphate (NADPH) to promote the intracellular reducing power [67]. This allows for the maintenance of reduced glutathione under hypoxic conditions during colorectal cancer liver colonization [66].
3.2 Cellular interactions between CRC cells and components of the liver microenvironment
In addition to metabolic rewiring, liver metastasis is further favored by the interaction between CRC cells and specific components of the hepatic microenvironment. In this section, we focus on the dynamic interplay between CRC cells and immune cells, hepatocytes, fibroblasts, and the extracellular matrix (ECM) of the hepatic microenvironment that sustains metastatic spread in the liver parenchyma.
As a physiological barrier to the systemic circulation, the liver is constantly targeted by a variety of pathogens and antigens from the gut. Thus, a large resident population of cells, including liver sinusoidal endothelial cells (LSECs), Kupffer cells (KCs), hepatic stellate cells (HSCs) and other immune cells, provides an enhanced innate immune defense [68]. Meanwhile, these cells are also programmed to induce immunosuppressive polarization to achieve an immunological balance [56]. Under pathological conditions such as infections and transplantation, a tolerogenic state is maintained characterized by immunosuppressive signals and impaired T-cell-mediated antigen responses to limit the magnitude of tissue injury, allowing the liver to recover [69, 70]. However, this state of immune tolerance is hijacked by disseminated CRC cells to facilitate a microenvironment that favors metastasis and may underlie the poor response to immunotherapy in patients with CRLM (Fig. 2).
3.2.1 Immune cells
After colonization in the liver, CRC cells progressively cultivate an immune-tolerant microenvironment. Initially, the TME is immature, and the early lesions of CRLM are infiltrated by cytotoxic T cells and attacked by the immune system [71]. Subsequently, the metastatic cells expand, which is accompanied by the recruitment of cancer-associated fibroblasts (CAFs) and macrophage and T-cell exclusion. During this process, immunosuppressive M2-like macrophages are induced, while the inflammatory M1-like phenotype is suppressed in the liver milieu [72]. Certain cell populations, such as MRC1+ CCL18+ M2-like macrophages with high metabolic activation and SPP1+ macrophages, are gradually being identified by single-cell studies [21, 23]. Tumor-associated neutrophils and liver-resident natural killer cells also contribute to the remodeling of the immune microenvironment [23, 73,74,75].
As the TME is reshaped, T cells that initially infiltrate the micrometastasis migrate to its margin, while PD-L1 expression by CRC cells gradually decreases [76]. Meanwhile, the function of T cells is inhibited. TGF-β, a well-known immunosuppressive cytokine, suppresses T-cell proliferation and activity [77]. TGF-activated CAFs are recruited in the liver microenvironment by CRC cells, resulting in T-cell exclusion and TH1-effector phenotype inhibition [78]. In addition, immunosuppressive CD11b+F4/80+ macrophages are attracted, causing a systemic deletion of T cells through FAS–FASL apoptotic signaling [72].
Given that the liver receives 70% of its blood supply from the intestine via the portal system, it is constantly exposed to the gut-derived microbiome and its products. Emerging evidence has suggested the intimate crosstalk between the liver immune microenvironment and the gut microbiome. Gut commensal bacteria have been shown to exploit bile acid metabolism to impair antitumor immunity in the liver. Mechanistically, Clostridium species (e.g., C. scindens) affect the conversion of primary to secondary bile acids to inhibit the expression of chemokine CXCL16 by sinusoidal endothelial cells, thereby suppressing NKT cell recruitment and compromising immunosurveillance of tumors growing in the liver. In multiple mouse models of primary and metastatic liver tumors, depleting commensal bacteria using an antibiotic cocktail treatment is sufficient to induce tumor regression [79].
Additionally, gut microorganisms can migrate and colonize the liver to shape its immune microenvironment. In patient samples, identical resident bacteria have been observed between primary CRC and paired liver metastases, including Escherichia coli, Fusobacterium, Selenomonas, Bacteroides, and Prevotella [80, 81]. In primary CRC, a disrupted gut vascular barrier (GVB) permits the penetration of harmful intestinal bacteria into the systemic circulation [80, 82]. As a result, tumor-resident Escherichia coli spread to the liver, where they recruit macrophages, neutrophils, and inflammatory monocytes, thus promoting an inflammatory microenvironment conducive to the seeding of CRC [80].
It is also worth noting that the immune microenvironment of CRLM varies depending on the site of origin. MSI-H/dMMR CRLM, which mostly originates from the right colon, has a higher level of immune infiltration compared to its MSS/pMMR counterpart [83]. However, only 4% of metastatic CRCs exhibit this genotype/phenotype, a frequency significantly lower than that of primary CRCs, which is consistent with the observation that MSI-H/dMMR is less likely to metastasize [84]. Furthermore, MSS/pMMR CRLMs from different primary sites also display heterogeneous immune profiles. A recent study using single-cell RNA sequencing and spatial transcriptomics revealed that CRLMs from patients with ascending colon, transverse colon, and sigmoid colon cancer were differentially enriched for immunosuppressive cells, including varying proportions of macrophages, CTLA4+ CD8+ T cells, and FOXP3+ Treg cells [21]. In another cohort from a multi-omics study, most rectal cancer liver metastases exhibited increased infiltration of effector HLA-DR+ CD8+ T cells, while colon cancer liver metastases demonstrated a higher enrichment of naïve HLA-DR- CD8+ T cells [85]. However, the molecular mechanisms underlying the interaction between CRC cells of different anatomical origins and distinct liver immune microenvironments remain largely elusive and warrant further investigation.
In summary, CRLM and its resident bacteria progressively reshape the immune microenvironment to cultivate an immunologically “cold” desert in the liver and even more, potentially compromise systemic antitumor immunity. Consistent with this, a comprehensive single-cell analysis of CRLM traced the immune subsets of CRLM and confirmed that the immune composition within the TME is shaped by both liver tissue-specific immunity and migrating CRC cells [23]. Dynamic changes in the immune TME can be exploited to prevent metastatic outgrowth. Indeed, in preclinical models, neoadjuvant immunotherapy before surgery can eradicate minor recurrence of liver foci at its onset [76], and TGF-β inhibitors and liver-directed radiotherapy can abrogate the immunosuppressive state of the liver and confer susceptibility to anti-PD-1 therapy [72, 78].
3.2.2 Hepatocytes
Cell competition, first identified in Drosophila, is a cell fitness-sensing mechanism in which cells with higher fitness induce the elimination of their neighbors [86]. Recently, it has been discovered that similar modes of competition exist between cancer cells and adjacent normal cells [87, 88]. In human samples of CRLM, YAP, and TAZ, the two downstream effectors of Hippo signaling, are observed to be activated in peritumoral hepatocytes. Another experimental study in mouse models of intrahepatic cholangiocarcinoma (CCA) and melanoma-derived liver metastases showed that overactivation of YAP in hepatocytes adjacent to cancer cells is sufficient to exert a tumor-suppressive function [89]. These findings suggest a possible competitive mechanism between the normal liver microenvironment and disseminated CRC cells, in which the relative intensity of oncogenic signaling, rather than cell-autonomous function, determines metastasis survival.
Hepatocytes can also promote an inflammatory microenvironment to support metastatic outgrowth. Once hepatocytes are educated by primary tumor stroma-derived IL-6, they produce excessive amounts of myeloid chemoattractants such as serum amyloid A (SAA), resulting in myeloid cell accumulation and liver fibrosis, which is favorable for tumor seeding [90]. Remarkably, patients with CRLM showed increased serum SAA, and genetic ablation of Saa in a mouse model resulted in blockade of the prometastatic niche in the liver [90]. Of note, elevated expression of liver SAA was also observed in CRLMs induced by gut bacterial translocation [80]. In mouse models, depleting commensal bacteria using an antibiotic cocktail significantly reduces liver SAA [80] and induces tumor regression [79, 91].
3.2.3 Fibroblasts
Another crucial cellular component of the CRLM stroma is CAFs. A recent analysis of human specimens identified hepatic stellate cells (HSCs) as the primary origin of CAFs by genetic tracing and single-cell RNA sequencing [92]. In general, CAFs exert their functions by secreting growth factors, remodeling the ECM, and promoting an immunosuppressive environment [93, 94].
In CRLM, direct interactions between tumor cells and inflammatory CAFs lead to the secretion of hepatocyte growth factor (HGF) by CAFs, thus promoting the growth of tumors [92]. As the major source of CAFs in liver metastases, HSCs contribute to the establishment of an immunosuppressive hepatic microenvironment by promoting the recruitment of MDSCs. This was achieved through a paracrine interaction between CRLM and HSCs [95]. Bevacizumab-resistant CRLM upregulated fibroblast growth factor-binding protein 1 (FGFBP1), which promoted FAPα expression in HSCs through paracrine stimulation. FAPα induces the release of CXCL5 from HSCs, thereby increasing the infiltration of MDSCs in the microenvironment.
Another major role of CAFs is to dysregulate the deposition of ECM, resulting in increased tissue stiffness and mechano-signaling activation. Tumors are pseudoorgans that are characterized not only by unique biological dysfunctions but also by distinct physical abnormalities. Emerging evidence has suggested that increased tissue stiffness, one of the altered mechanical properties of solid tumors, is a critical factor that affects both cancer cells and their microenvironment [96]. Indeed, matrix stiffening is a feature common to multiple solid tumors, such as breast cancer, pancreatic cancer, and CRC [97]. In CRLM, previous analysis of human surgical specimens observed increased deposition of type IV collagen [98], and circulating type IV collagen was identified as a novel potential tumor marker [99]. Hyaluronan generated by myofibroblastic CAFs was also shown to enhance CRLM, whereas type I collagen produced by the same cell population inhibited metastatic growth by limiting tumor expansion mechanically [92]. Recently, a study measuring patient-derived samples with atomic force microscopy revealed enhanced tissue stiffness of liver metastasis compared to primary CRC [100]. Consistent with previous notions, this is attributed to highly activated metastasis-associated fibroblasts (MAFs) with significant matrix remodeling and cell contraction signatures via the angiotensin-ARHGEF1-RhoA axis. Intriguingly, this mechanical change in the liver microenvironment influences endothelial cell behavior, thus facilitating metastatic angiogenesis and antiangiogenic therapy resistance. Going forward, renin-angiotensin system (anti-RAS) inhibitors significantly inhibited MAF activity, attenuated tissue stiffness, and conferred susceptibility to the antiangiogenic effect of bevacizumab in CRLM patients [100]
4 Clinical implications
Systemic therapies for CRLM have revolutionized disease treatment options since the early 2000s, owing largely to the introduction of targeted therapies and immunotherapeutic agents [3]. According to the molecular subtypes, EGFR antibodies, BRAF inhibitors, and anti-ERBB2 therapy have been incorporated into the systemic management of CRLM; however, they have only achieved moderate efficacy. Therapeutic targeting of these oncogenes often fails to distinguish the crosstalk between CRLM and the liver TME. Thus, a better understanding of the sophisticated metabolic adaptation and cellular interaction underlying CRLM has yielded potential strategies for the development of novel liver-exclusive interventions (Table 1).
In preclinical models, targeting specific metabolites or metabolic enzymes presents a valid option for CRLM. For example, dietary restriction of fructose and creatine has shown antitumor effects [59, 103]. Another case in point is targeting several metabolic enzymes, including CKB and GATM in creatine metabolism, alpha-aminoadipic semialdehyde synthase (AASS) in lysine catabolism, and PCK1, PKM2, and PKLR in glucose metabolism [60, 62, 65, 66, 103]. Mechano-based therapies such as anti-RAS drugs that modulate the tissue stiffness of CRLMs are also emerging. As shown by retrospective analysis, combination administration of bevacizumab with anti-RAS treatment can lead to significantly prolonged survival compared with antiangiogenic therapy alone [100]. Furthermore, analysis of epigenetic features at single-cell resolution has revealed relatively stable genome-wide DNA methylation patterns within a single genetic sublineage of primary CRC and paired CRLM, making epigenetic drugs a possible therapeutic strategy [37].
In microsatellite-instable (MSI) CRC, immunotherapy has shown promising efficacy that profoundly prolongs survival more than conventional chemotherapy and presents curative potential [104]. Unfortunately, immunotherapy is generally ineffective in patients without this molecular profile, which comprises 95% of metastatic CRC. Inspiringly, the recent identification of dynamic changes in the immune TME suggests a temporal window of susceptibility of CRLM to immunotherapy and the effectiveness of immunotherapy at the early stage of metastatic colonization in mice [76]. Additionally, a combination of immune checkpoint-based therapies with other interventions that intercept the components of the immunosuppressive hepatic microenvironment may offer an approach to abolish this resistance in microsatellite-stable (MSS) CRLM. Galunisertib, a TGFBR1-specific inhibitor, can enable a pronounced immune infiltration with activated T cells that exhibit marked surface expression of programmed cell-death protein 1 (PD-1) in the liver milieu, thereby inducing a robust antitumor immune response when combined with anti-PD-1–PD-L1 therapy in MSS CRLM transgenic mouse models [78]. Alternatively, in mouse models, liver-directed radiotherapy reduced the number of immunosuppressive hepatic macrophages to prevent antigen-specific T-cell loss, leading to increased hepatic T-cell infiltration and restoration of the efficacy of immune checkpoint blockade [72]. Furthermore, MEK inhibition, chemotherapy, and antiangiogenic agents have been shown to potentiate the immune response and efficacy when combined with anti-PD-L1 treatment in preclinical models and are being evaluated in clinical trials [101, 102, 105, 106]. Overall, further laboratory investigations are needed to better understand the role of hepatic immune tolerance in therapeutic resistance to immunotherapy, and clinical trials are required to determine the optimal combinatorial treatment strategy in MSS CRLM.
5 Concluding remarks
Over the past few years, extensive genomic data and trajectory analysis have increased our understanding of cancer as an evolutionary process. Although there are currently no specific genetic features that can be attributed exclusively to CRLM, molecular profiling for KRAS/NRAS/BRAF V600E, microsatellite instability, and ERBB2 has enabled treatment selection tailored to specific patient subsets and yielded improved overall survival. Nonetheless, the clonal selection that confers fitness to CRC cells is not only limited to genetic alterations, but occurs on multiple levels. To gain a comprehensive understanding of the molecular mechanisms behind CRLM, researchers need to interrogate genetic mutations with unique characteristics of the liver TME and link all aspects that may contribute to the adaptive phenotype. In addition, the interactions of CRC cells with distinct elements of the liver microenvironment change continuously throughout the process of colonization, highlighting the need for further investigation of their heterogeneous contribution. In this context, the advent of single-cell analysis, spatial transcriptomics, and high-throughput multi-omics can facilitate deconvoluting the delicate molecular landscape of CRLM [20, 21, 23]. In conclusion, only a thorough understanding of the molecular basis will open new avenues for scientists and clinicians to improve the prognosis of CRLM patients.
References
Biller, L. H., & Schrag, D. (2021). Diagnosis and treatment of metastatic colorectal cancer: A review. JAMA, 325(7), 669–685. https://doi.org/10.1001/jama.2021.0106
Dekker, E., Tanis, P. J., Vleugels, J. L. A., Kasi, P. M., & Wallace, M. B. (2019). Colorectal cancer. Lancet, 394(10207), 1467–1480. https://doi.org/10.1016/S0140-6736(19)32319-0
Tsilimigras, D. I., Brodt, P., Clavien, P. A., Muschel, R. J., D'Angelica, M. I., Endo, I., et al. (2021). Liver metastases. Nature Reviews Disease Primers, 7(1), 27. https://doi.org/10.1038/s41572-021-00261-6
Milette, S., Sicklick, J. K., Lowy, A. M., & Brodt, P. (2017). Molecular pathways: Targeting the Microenvironment of liver metastases. Clinical Cancer Research, 23(21), 6390–6399. https://doi.org/10.1158/1078-0432.CCR-15-1636
Hess, K. R., Varadhachary, G. R., Taylor, S. H., Wei, W., Raber, M. N., Lenzi, R., et al. (2006). Metastatic patterns in adenocarcinoma. Cancer, 106(7), 1624–1633. https://doi.org/10.1002/cncr.21778
Paget, S. (1989). The distribution of secondary growths in cancer of the breast. The Lancet, 8(2), 98–101.
Chow, F. C., & Chok, K. S. (2019). Colorectal liver metastases: An update on multidisciplinary approach. World Journal of Hepatology, 11(2), 150–172. https://doi.org/10.4254/wjh.v11.i2.150
Vidal-Vanaclocha, F. (2008). The prometastatic microenvironment of the liver. Cancer Microenviron, 1(1), 113–129. https://doi.org/10.1007/s12307-008-0011-6
Mielgo, A., & Schmid, M. C. (2020). Liver tropism in cancer: The hepatic metastatic niche. Cold Spring Harbor Perspectives in Medicine, 10(3), a037259. https://doi.org/10.1101/cshperspect.a037259
Poisson, J., Lemoinne, S., Boulanger, C., Durand, F., Moreau, R., Valla, D., et al. (2017). Liver sinusoidal endothelial cells: Physiology and role in liver diseases. Journal of Hepatology, 66(1), 212–227. https://doi.org/10.1016/j.jhep.2016.07.009
Miskovic, D., & Mirnezami, R. (2021). Is complete mesocolic excision superior to conventional colectomy for colon cancer? Lancet Oncology, 22(7), 917–918. https://doi.org/10.1016/S1470-2045(21)00256-4
Fujita, S., Mizusawa, J., Kanemitsu, Y., Ito, M., Kinugasa, Y., Komori, K., et al. (2017). Mesorectal excision with or without lateral lymph node dissection for clinical stage II/III lower rectal cancer (JCOG0212): A multicenter, randomized controlled, noninferiority trial. Annals of Surgery, 266(2), 201–207. https://doi.org/10.1097/SLA.0000000000002212
Markowitz, S. D. (2017). Cancer bypasses the lymph nodes. Science, 357(6346), 35–36. https://doi.org/10.1126/science.aan8299
Siravegna, G., Lazzari, L., Crisafulli, G., Sartore-Bianchi, A., Mussolin, B., Cassingena, A., et al. (2018). Radiologic and genomic evolution of individual metastases during HER2 blockade in colorectal cancer. Cancer Cell, 34(1), 148–162. https://doi.org/10.1016/j.ccell.2018.06.004
Russo, M., Siravegna, G., Blaszkowsky, L. S., Corti, G., Crisafulli, G., Ahronian, L. G., et al. (2016). Tumor heterogeneity and lesion-specific response to targeted therapy in colorectal cancer. Cancer Discovery, 6(2), 147–153. https://doi.org/10.1158/2159-8290.CD-15-1283
Pietrantonio, F., Oddo, D., Gloghini, A., Valtorta, E., Berenato, R., Barault, L., et al. (2016). MET-driven resistance to dual EGFR and BRAF blockade may be overcome by switching from EGFR to MET inhibition in BRAF-mutated colorectal cancer. Cancer Discovery, 6(9), 963–971. https://doi.org/10.1158/2159-8290.CD-16-0297
Ryser, M. D., Mallo, D., Hall, A., Hardman, T., King, L. M., Tatishchev, S., et al. (2020). Minimal barriers to invasion during human colorectal tumor growth. Nature Communications, 11(1), 1280. https://doi.org/10.1038/s41467-020-14908-7
Hu, Z., Li, Z., Ma, Z., & Curtis, C. (2020). Multi-cancer analysis of clonality and the timing of systemic spread in paired primary tumors and metastases. Nature Genetics, 52(7), 701–708. https://doi.org/10.1038/s41588-020-0628-z
Naxerova, K., Reiter, J. G., Brachtel, E., Lennerz, J. K., van de Wetering, M., Rowan, A., et al. (2017). Origins of lymphatic and distant metastases in human colorectal cancer. Science, 357(6346), 55–60. https://doi.org/10.1126/science.aai8515
Househam, J., Heide, T., Cresswell, G. D., Spiteri, I., Kimberley, C., Zapata, L., et al. (2022). Phenotypic plasticity and genetic control in colorectal cancer evolution. Nature, 611(7937), 744–753. https://doi.org/10.1038/s41586-022-05311-x
Wu, Y., Yang, S., Ma, J., Chen, Z., Song, G., Rao, D., et al. (2021). Spatiotemporal immune landscape of colorectal cancer liver metastasis at single-cell level. Cancer Discovery, 12(1), 134–153. https://doi.org/10.1158/2159-8290.CD-21-0316
Li, C., Sun, Y. D., Yu, G. Y., Cui, J. R., Lou, Z., Zhang, H., et al. (2020). Integrated omics of metastatic colorectal cancer. Cancer Cell, 38(5), 734–747 e739. https://doi.org/10.1016/j.ccell.2020.08.002
Liu, Y., Zhang, Q., Xing, B., Luo, N., Gao, R., Yu, K., et al. (2022). Immune phenotypic linkage between colorectal cancer and liver metastasis. Cancer Cell, 40(4), 424–437. https://doi.org/10.1016/j.ccell.2022.02.013
Obenauf, A. C., & Massague, J. (2015). Surviving at a distance: Organ-specific metastasis. Trends in Cancer, 1(1), 76–91. https://doi.org/10.1016/j.trecan.2015.07.009
Wei, Q., Ye, Z., Zhong, X., Li, L., Wang, C., Myers, R. E., et al. (2017). Multiregion whole-exome sequencing of matched primary and metastatic tumors revealed genomic heterogeneity and suggested polyclonal seeding in colorectal cancer metastasis. Annals of Oncology, 28(9), 2135–2141. https://doi.org/10.1093/annonc/mdx278
Dang, H. X., Krasnick, B. A., White, B. S., Grossman, J. G., Strand, M. S., Zhang, J., et al. (2020). The clonal evolution of metastatic colorectal cancer. Science Advances, 6(24), eaay9691. https://doi.org/10.1126/sciadv.aay9691
Alves, J. M., Prado-Lopez, S., Cameselle-Teijeiro, J. M., & Posada, D. (2019). Rapid evolution and biogeographic spread in a colorectal cancer. Nature Communications, 10(1), 5139. https://doi.org/10.1038/s41467-019-12926-8
Yachida, S., Jones, S., Bozic, I., Antal, T., Leary, R., Fu, B., et al. (2010). Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature, 467(7319), 1114–1117. https://doi.org/10.1038/nature09515
Campbell, P. J., Yachida, S., Mudie, L. J., Stephens, P. J., Pleasance, E. D., Stebbings, L. A., et al. (2010). The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature, 467(7319), 1109–1113. https://doi.org/10.1038/nature09460
Turajlic, S., & Swanton, C. (2016). Metastasis as an evolutionary process. Science, 352(6282), 169–175. https://doi.org/10.1126/science.aaf2784
Turajlic, S., Furney, S. J., Lambros, M. B., Mitsopoulos, C., Kozarewa, I., Geyer, F. C., et al. (2012). Whole genome sequencing of matched primary and metastatic acral melanomas. Genome Research, 22(2), 196–207. https://doi.org/10.1101/gr.125591.111
Gundem, G., Van Loo, P., Kremeyer, B., Alexandrov, L. B., Tubio, J. M. C., Papaemmanuil, E., et al. (2015). The evolutionary history of lethal metastatic prostate cancer. Nature, 520(7547), 353–357. https://doi.org/10.1038/nature14347
Gkountela, S., Castro-Giner, F., Szczerba, B. M., Vetter, M., Landin, J., Scherrer, R., et al. (2019). Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell, 176(1-2), 98–112. https://doi.org/10.1016/j.cell.2018.11.046
Aceto, N., Bardia, A., Miyamoto, D. T., Donaldson, M. C., Wittner, B. S., Spencer, J. A., et al. (2014). Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell, 158(5), 1110–1122. https://doi.org/10.1016/j.cell.2014.07.013
Reiter, J. G., Hung, W. T., Lee, I. H., Nagpal, S., Giunta, P., Degner, S., et al. (2020). Lymph node metastases develop through a wider evolutionary bottleneck than distant metastases. Nature Genetics, 52(7), 692–700. https://doi.org/10.1038/s41588-020-0633-2
Priestley, P., Baber, J., Lolkema, M. P., Steeghs, N., de Bruijn, E., Shale, C., et al. (2019). Pan-cancer whole-genome analyses of metastatic solid tumours. Nature, 575(7781), 210–216. https://doi.org/10.1038/s41586-019-1689-y
Bian, S., Hou, Y., Zhou, X., Li, X., Yong, J., Wang, Y., et al. (2018). Single-cell multiomics sequencing and analyses of human colorectal cancer. Science, 362(6418), 1060–1063. https://doi.org/10.1126/science.aao3791
Reiter, J. G., Makohon-Moore, A. P., Gerold, J. M., Heyde, A., Attiyeh, M. A., Kohutek, Z. A., et al. (2018). Minimal functional driver gene heterogeneity among untreated metastases. Science, 361(6406), 1033–1037. https://doi.org/10.1126/science.aat7171
Chen, H. N., Shu, Y., Liao, F., Liao, X., Zhang, H., Qin, Y., et al. (2021). Genomic evolution and diverse models of systemic metastases in colorectal cancer. Gut, 71(2), 322–332. https://doi.org/10.1136/gutjnl-2020-323703
Ishaque, N., Abba, M. L., Hauser, C., Patil, N., Paramasivam, N., Huebschmann, D., et al. (2018). Whole genome sequencing puts forward hypotheses on metastasis evolution and therapy in colorectal cancer. Nature Communications, 9(1), 4782. https://doi.org/10.1038/s41467-018-07041-z
Alves, J. M., Prado-Lopez, S., Tomas, L., Valecha, M., Estevez-Gomez, N., Alvarino, P., et al. (2022). Clonality and timing of relapsing colorectal cancer metastasis revealed through whole-genome single-cell sequencing. Cancer Letters, 543, 215767. https://doi.org/10.1016/j.canlet.2022.215767
van de Haar, J., Hoes, L. R., Roepman, P., Lolkema, M. P., Verheul, H. M. W., Gelderblom, H., et al. (2021). Limited evolution of the actionable metastatic cancer genome under therapeutic pressure. Nature Medicine, 27(9), 1553–1563. https://doi.org/10.1038/s41591-021-01448-w
Ahronian, L. G., Sennott, E. M., Van Allen, E. M., Wagle, N., Kwak, E. L., Faris, J. E., et al. (2015). Clinical acquired resistance to RAF inhibitor combinations in BRAF-mutant colorectal cancer through MAPK pathway alterations. Cancer Discovery, 5(4), 358–367. https://doi.org/10.1158/2159-8290.CD-14-1518
Bertotti, A., Papp, E., Jones, S., Adleff, V., Anagnostou, V., Lupo, B., et al. (2015). The genomic landscape of response to EGFR blockade in colorectal cancer. Nature, 526(7572), 263–267. https://doi.org/10.1038/nature14969
Missiaglia, E., Jacobs, B., D'Ario, G., Di Narzo, A. F., Soneson, C., Budinska, E., et al. (2014). Distal and proximal colon cancers differ in terms of molecular, pathological, and clinical features. Annals of Oncology, 25(10), 1995–2001. https://doi.org/10.1093/annonc/mdu275
Amri, R., Bordeianou, L. G., Sylla, P., & Berger, D. L. (2015). Variations in metastasis site by primary location in colon cancer. Journal of Gastrointestinal Surgery, 19(8), 1522–1527. https://doi.org/10.1007/s11605-015-2837-9
Riihimaki, M., Hemminki, A., Sundquist, J., & Hemminki, K. (2016). Patterns of metastasis in colon and rectal cancer. Science Reporter, 6, 29765. https://doi.org/10.1038/srep29765
Russolillo, N., Sperti, E., Langella, S., Menonna, F., Allieta, A., Di Maio, M., et al. (2020). Impact of primary tumor location on patterns of recurrence and survival of patients undergoing resection of liver metastases from colon cancer. HPB (Oxford), 22(1), 116–123. https://doi.org/10.1016/j.hpb.2019.05.014
Engstrand, J., Nilsson, H., Stromberg, C., Jonas, E., & Freedman, J. (2018). Colorectal cancer liver metastases - a population-based study on incidence, management and survival. BMC Cancer, 18(1), 78. https://doi.org/10.1186/s12885-017-3925-x
Yaeger, R., Chatila, W. K., Lipsyc, M. D., Hechtman, J. F., Cercek, A., Sanchez-Vega, F., et al. (2018). Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell, 33(1), 125–136. https://doi.org/10.1016/j.ccell.2017.12.004
Kim, S. C., Park, J. W., Seo, H. Y., Kim, M., Park, J. H., Kim, G. H., et al. (2022). Multifocal organoid capturing of colon cancer reveals pervasive intratumoral heterogenous drug responses. Advanced Sciencev (Weinh), 9(5), e2103360. https://doi.org/10.1002/advs.202103360
Banerjee, S., Zhang, X., Kuang, S., Wang, J., Li, L., Fan, G., et al. (2021). Comparative analysis of clonal evolution among patients with right- and left-sided colon and rectal cancer. Iscience, 24(7), 102718. https://doi.org/10.1016/j.isci.2021.102718
Sobral, D., Martins, M., Kaplan, S., Golkaram, M., Salmans, M., Khan, N., et al. (2022). Genetic and microenvironmental intra-tumor heterogeneity impacts colorectal cancer evolution and metastatic development. Communications Biology, 5(1), 937. https://doi.org/10.1038/s42003-022-03884-x
Joung, J. G., Oh, B. Y., Hong, H. K., Al-Khalidi, H., Al-Alem, F., Lee, H. O., et al. (2017). Tumor heterogeneity predicts metastatic potential in colorectal cancer. Clinical Cancer Research, 23(23), 7209–7216. https://doi.org/10.1158/1078-0432.CCR-17-0306
Malesci, A., Laghi, L., Bianchi, P., Delconte, G., Randolph, A., Torri, V., et al. (2007). Reduced likelihood of metastases in patients with microsatellite-unstable colorectal cancer. Clinical Cancer Research, 13(13), 3831–3839. https://doi.org/10.1158/1078-0432.CCR-07-0366
Li, X., Ramadori, P., Pfister, D., Seehawer, M., Zender, L., & Heikenwalder, M. (2021). The immunological and metabolic landscape in primary and metastatic liver cancer. Nature Reviews Cancer, 21(9), 541–557. https://doi.org/10.1038/s41568-021-00383-9
Berndt, N., Kolbe, E., Gajowski, R., Eckstein, J., Ott, F., Meierhofer, D., et al. (2021). Functional consequences of metabolic zonation in murine livers: Insights for an old Story. Hepatology, 73(2), 795–810. https://doi.org/10.1002/hep.31274
Bergers, G., & Fendt, S. M. (2021). The metabolism of cancer cells during metastasis. Nature Reviews Cancer, 21(3), 162–180. https://doi.org/10.1038/s41568-020-00320-2
Bu, P., Chen, K. Y., Xiang, K., Johnson, C., Crown, S. B., Rakhilin, N., et al. (2018). Aldolase B-mediated fructose metabolism drives metabolic reprogramming of colon cancer liver metastasis. Cell Metabolism, 27(6), 1249–1262. https://doi.org/10.1016/j.cmet.2018.04.003
Loo, J. M., Scherl, A., Nguyen, A., Man, F. Y., Weinberg, E., Zeng, Z., et al. (2015). Extracellular metabolic energetics can promote cancer progression. Cell, 160(3), 393–406. https://doi.org/10.1016/j.cell.2014.12.018
Garcia-Bermudez, J., Baudrier, L., La, K., Zhu, X. G., Fidelin, J., Sviderskiy, V. O., et al. (2018). Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nature Cell Biology, 20(7), 775–781. https://doi.org/10.1038/s41556-018-0118-z
Yamaguchi, N., Weinberg, E. M., Nguyen, A., Liberti, M. V., Goodarzi, H., Janjigian, Y. Y., et al. (2019). PCK1 and DHODH drive colorectal cancer liver metastatic colonization and hypoxic growth by promoting nucleotide synthesis. Elife, 8, e52135. https://doi.org/10.7554/eLife.52135
Wang, K., Jiang, J., Lei, Y., Zhou, S., Wei, Y., & Huang, C. (2019). Targeting metabolic-redox circuits for cancer therapy. Trends in Biochemical Sciences, 44(5), 401–414. https://doi.org/10.1016/j.tibs.2019.01.001
Gao, W., Chen, L., Ma, Z., Du, Z., Zhao, Z., Hu, Z., et al. (2013). Isolation and phenotypic characterization of colorectal cancer stem cells with organ-specific metastatic potential. Gastroenterology, 145(3), 636–646. https://doi.org/10.1053/j.gastro.2013.05.049
Wu, Z., Wei, D., Gao, W., Xu, Y., Hu, Z., Ma, Z., et al. (2015). TPO-induced metabolic reprogramming drives liver metastasis of colorectal cancer CD110+ tumor-initiating Cells. Cell Stem Cell, 17(1), 47–59. https://doi.org/10.1016/j.stem.2015.05.016
Nguyen, A., Loo, J. M., Mital, R., Weinberg, E. M., Man, F. Y., Zeng, Z., et al. (2016). PKLR promotes colorectal cancer liver colonization through induction of glutathione synthesis. The Journal of Clinical Investigation, 126(2), 681–694. https://doi.org/10.1172/JCI83587
Anastasiou, D., Poulogiannis, G., Asara, J. M., Boxer, M. B., Jiang, J. K., Shen, M., et al. (2011). Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science, 334(6060), 1278–1283. https://doi.org/10.1126/science.1211485
Heymann, F., & Tacke, F. (2016). Immunology in the liver--from homeostasis to disease. Nature Reviews. Gastroenterology & Hepatology, 13(2), 88–110. https://doi.org/10.1038/nrgastro.2015.200
Crispe, I. N. (2014). Immune tolerance in liver disease. Hepatology, 60(6), 2109–2117. https://doi.org/10.1002/hep.27254
O'Leary, K. (2021). Liver metastases cultivate an immune desert. Nature Reviews. Cancer, 21(3), 143. https://doi.org/10.1038/s41568-021-00338-0
Canellas-Socias, A., Cortina, C., Hernando-Momblona, X., Palomo-Ponce, S., Mulholland, E. J., Turon, G., et al. (2022). Metastatic recurrence in colorectal cancer arises from residual EMP1(+) cells. Nature, 611(7936), 603–613. https://doi.org/10.1038/s41586-022-05402-9
Yu, J., Green, M. D., Li, S., Sun, Y., Journey, S. N., Choi, J. E., et al. (2021). Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nature Medicine, 27(1), 152–164. https://doi.org/10.1038/s41591-020-1131-x
Song, J., Song, H., Wei, H., Sun, R., Tian, Z., & Peng, H. (2022). Requirement of RORalpha for maintenance and antitumor immunity of liver-resident natural killer cells/ILC1s. Hepatology, 75(5), 1181–1193. https://doi.org/10.1002/hep.32147
Tian, S., Chu, Y., Hu, J., Ding, X., Liu, Z., Fu, D., et al. (2022). Tumour-associated neutrophils secrete AGR2 to promote colorectal cancer metastasis via its receptor CD98hc-xCT. Gut, 71(12), 2489–2501. https://doi.org/10.1136/gutjnl-2021-325137
Wang, H., Zhang, B., Li, R., Chen, J., Xu, G., Zhu, Y., et al. (2022). KIAA1199 drives immune suppression to promote colorectal cancer liver metastasis by modulating neutrophil infiltration. Hepatology, 76(4), 967–981. https://doi.org/10.1002/hep.32383
Canellas-Socias, A., Cortina, C., Hernando-Momblona, X., Palomo-Ponce, S., Mulholland, E. J., Turon, G., et al. (2022). Metastatic recurrence in colorectal cancer arises from residual EMP1<sup>+</sup> cells. Nature, 611(7936), 603–613. https://doi.org/10.1038/s41586-022-05402-9
Gorelik, L., & Flavell, R. A. (2000). Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity, 12(2), 171–181. https://doi.org/10.1016/s1074-7613(00)80170-3
Tauriello, D. V. F., Palomo-Ponce, S., Stork, D., Berenguer-Llergo, A., Badia-Ramentol, J., Iglesias, M., et al. (2018). TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature, 554(7693), 538–543. https://doi.org/10.1038/nature25492
Ma, C., Han, M., Heinrich, B., Fu, Q., Zhang, Q., Sandhu, M., et al. (2018). Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science, 360(6391). https://doi.org/10.1126/science.aan5931
Bertocchi, A., Carloni, S., Ravenda, P. S., Bertalot, G., Spadoni, I., Lo Cascio, A., et al. (2021). Gut vascular barrier impairment leads to intestinal bacteria dissemination and colorectal cancer metastasis to liver. Cancer Cell, 39(5), 708–724. https://doi.org/10.1016/j.ccell.2021.03.004
Bullman, S., Pedamallu, C. S., Sicinska, E., Clancy, T. E., Zhang, X., Cai, D., et al. (2017). Analysis of fusobacterium persistence and antibiotic response in colorectal cancer. Science, 358(6369), 1443–1448. https://doi.org/10.1126/science.aal5240
Spadoni, I., Zagato, E., Bertocchi, A., Paolinelli, R., Hot, E., Di Sabatino, A., et al. (2015). A gut-vascular barrier controls the systemic dissemination of bacteria. Science, 350(6262), 830–834. https://doi.org/10.1126/science.aad0135
Taieb, J., Svrcek, M., Cohen, R., Basile, D., Tougeron, D., & Phelip, J. M. (2022). Deficient mismatch repair/microsatellite unstable colorectal cancer: Diagnosis, prognosis and treatment. European Journal of Cancer, 175, 136–157. https://doi.org/10.1016/j.ejca.2022.07.020
Testa, U., Castelli, G., & Pelosi, E. (2020). Genetic alterations of metastatic colorectal cancer. Biomedicines, 8(10), 414. https://doi.org/10.3390/biomedicines8100414
Mouradov, D., Greenfield, P., Li, S., In, E. J., Storey, C., Sakthianandeswaren, A., et al. (2023). Onco-microbial community profiling identifies clinico-molecular and prognostic subtypes of colorectal cancer. Gastroenterology. https://doi.org/10.1053/j.gastro.2023.03.205
Moreno, E., & Basler, K. (2004). dMyc transforms cells into super-competitors. Cell, 117(1), 117–129. https://doi.org/10.1016/s0092-8674(04)00262-4
Flanagan, D. J., Pentinmikko, N., Luopajarvi, K., Willis, N. J., Gilroy, K., Raven, A. P., et al. (2021). NOTUM from Apc-mutant cells biases clonal competition to initiate cancer. Nature, 594(7863), 430–435. https://doi.org/10.1038/s41586-021-03525-z
Kanada, M., Bachmann, M. H., & Contag, C. H. (2016). Signaling by extracellular vesicles advances cancer hallmarks. Trends in Cancer, 2(2), 84–94. https://doi.org/10.1016/j.trecan.2015.12.005
Moya, I. M., Castaldo, S. A., Van den Mooter, L., Soheily, S., Sansores-Garcia, L., Jacobs, J., et al. (2019). Peritumoral activation of the hippo pathway effectors YAP and TAZ suppresses liver cancer in mice. Science, 366(6468), 1029–1034. https://doi.org/10.1126/science.aaw9886
Lee, J. W., Stone, M. L., Porrett, P. M., Thomas, S. K., Komar, C. A., Li, J. H., et al. (2019). Hepatocytes direct the formation of a pro-metastatic niche in the liver. Nature, 567(7747), 249–252. https://doi.org/10.1038/s41586-019-1004-y
Sethi, V., Kurtom, S., Tarique, M., Lavania, S., Malchiodi, Z., Hellmund, L., et al. (2018). Gut microbiota promotes tumor growth in mice by modulating immune response. Gastroenterology, 155(1), 33–37. https://doi.org/10.1053/j.gastro.2018.04.001
Bhattacharjee, S., Hamberger, F., Ravichandra, A., Miller, M., Nair, A., Affo, S., et al. (2021). Tumor restriction by type I collagen opposes tumor-promoting effects of cancer-associated fibroblasts. The Journal of Clinical Investigation, 131(11). https://doi.org/10.1172/JCI146987
Biffi, G., & Tuveson, D. A. (2021). Diversity and biology of cancer-associated fibroblasts. Physiological Reviews, 101(1), 147–176. https://doi.org/10.1152/physrev.00048.2019
Barbazan, J., & Matic Vignjevic, D. (2019). Cancer associated fibroblasts: Is the force the path to the dark side? Current Opinion in Cell Biology, 56, 71–79. https://doi.org/10.1016/j.ceb.2018.09.002
Qi, M., Fan, S., Huang, M., Pan, J., Li, Y., Miao, Q., et al. (2022). Targeting FAPα-expressing hepatic stellate cells overcomes resistance to antiangiogenics in colorectal cancer liver metastasis models. The Journal of Clinical Investigation, 132. https://doi.org/10.1172/JCI157399
Lampi, M. C., & Reinhart-King, C. A. (2018). Targeting extracellular matrix stiffness to attenuate disease: From molecular mechanisms to clinical trials. Science Translational Medicine, 10(422). https://doi.org/10.1126/scitranslmed.aao0475
Kalli, M., & Stylianopoulos, T. (2018). Defining the role of solid stress and matrix stiffness in cancer cell proliferation and metastasis. Frontiers in Oncology, 8, 55. https://doi.org/10.3389/fonc.2018.00055
Burnier, J. V., Wang, N., Michel, R. P., Hassanain, M., Li, S., Lu, Y., et al. (2011). Type IV collagen-initiated signals provide survival and growth cues required for liver metastasis. Oncogene, 30(35), 3766–3783. https://doi.org/10.1038/onc.2011.89
Nystrom, H., Tavelin, B., Bjorklund, M., Naredi, P., & Sund, M. (2015). Improved tumour marker sensitivity in detecting colorectal liver metastases by combined type IV collagen and CEA measurement. Tumour Biology, 36(12), 9839–9847. https://doi.org/10.1007/s13277-015-3729-z
Shen, Y., Wang, X., Lu, J., Salfenmoser, M., Wirsik, N. M., Schleussner, N., et al. (2020). Reduction of liver metastasis stiffness improves response to bevacizumab in metastatic colorectal cancer. Cancer Cell, 37(6), 800–817. https://doi.org/10.1016/j.ccell.2020.05.005
Liu, L., Mayes, P. A., Eastman, S., Shi, H., Yadavilli, S., Zhang, T., et al. (2015). The BRAF and MEK inhibitors dabrafenib and trametinib: Effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clinical Cancer Research, 21(7), 1639–1651. https://doi.org/10.1158/1078-0432.CCR-14-2339
Pfirschke, C., Engblom, C., Rickelt, S., Cortez-Retamozo, V., Garris, C., Pucci, F., et al. (2016). Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity, 44(2), 343–354. https://doi.org/10.1016/j.immuni.2015.11.024
Zhang, L., Zhu, Z., Yan, H., Wang, W., Wu, Z., Zhang, F., et al. (2021). Creatine promotes cancer metastasis through activation of Smad2/3. Cell Metabolism, 33(6), 1111–1123. https://doi.org/10.1016/j.cmet.2021.03.009
Le, D. T., Durham, J. N., Smith, K. N., Wang, H., Bartlett, B. R., Aulakh, L. K., et al. (2017). Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science, 357(6349), 409–413. https://doi.org/10.1126/science.aan6733
Ebert, P. J. R., Cheung, J., Yang, Y., McNamara, E., Hong, R., Moskalenko, M., et al. (2016). MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity, 44(3), 609–621. https://doi.org/10.1016/j.immuni.2016.01.024
Hodi, F. S., Lawrence, D., Lezcano, C., Wu, X., Zhou, J., Sasada, T., et al. (2014). Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunology Research, 2(7), 632–642. https://doi.org/10.1158/2326-6066.CIR-14-0053
Funding
This work was supported by (1) National Natural Science Foundation of China (82073246); (2) Sichuan Science and Technology Program (2022JDRC0049, 2022YFS0175); (3) 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYGD20006).
Author information
Authors and Affiliations
Contributions
Hai-Ning Chen and Zong-Guang Zhou designed the article, Qiu-Luo Liu and Huijie Zhou performed the literature search and drafted the work, Hai-Ning Chen and Zong-Guang Zhou revised the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liu, QL., Zhou, H., Zhou, ZG. et al. Colorectal cancer liver metastasis: genomic evolution and crosstalk with the liver microenvironment. Cancer Metastasis Rev 42, 575–587 (2023). https://doi.org/10.1007/s10555-023-10107-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-023-10107-0