
Abstract
Gastrointestinal stromal tumors (GIST) are rare neoplasms arising from the interstitial cell of Cajal in the gastrointestinal tract. Two thirds of GIST in adult patients have c-Kit mutation and smaller fractions have platelet derived growth factor receptor alpha (PDGFRA) mutation. Surgery is the only curative treatment for localized disease. Imatinib improves survival when used adjuvantly and in advanced disease. Several targeted therapies have also improved survival in GIST patients after progression on imatinib including sunitinib and regorafenib. Recently, United States Federal and Drug Administration (FDA) approved two new tyrosine kinase inhibitors for the treatment of heavily pretreated advanced/unresectable GIST including avapritinib (a selective inhibitor for PDGFRA exon 18 mutation including D842V mutations) and ripretinib (a broad-spectrum kinase inhibitor of c-Kit and PDGFRA). In this article, we will provide a comprehensive review of GIST including the current standard of care treatment and exploring future paradigm shifts in therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Gastrointestinal stromal tumors (GIST) are the most common of the rare non-epithelial neoplasms of gastrointestinal tract, accounting for 0.1–3% of gastrointestinal malignancies [1,2,3,4]. They were considered smooth muscle sarcomas until being recognized as a distinct disease in 1990s [5]. Interstitial cell of Cajal (ICC), also called gastrointestinal pacemaker cell, regulates the gastrointestinal tract peristalsis and is proposed to be the cell of origin for GIST [6].
GIST occurs mainly in adults with a median age at diagnosis of 60–65 years [7, 8]. The true incidence of GIST varies because of relative lack of unified diagnostic criteria. It ranges from 7 to 15 case per million population per year [3, 8,9,10,11,12], although studies from autopsies suggested a much higher incidence [13, 14]. Majority of the new cases are sporadic and only 5% of the patients have an associated familial autosomal dominant syndrome [15]. The most common genetic syndromes are type-1 neurofibromatosis (NF-1), Carney’s triad, familial GIST syndromes with germline c-Kit/platelet derived growth factor receptor alpha (PDGFRA) mutations, and Carney–Stratakis syndrome [7, 16]. Pediatric GISTs are rare tumors that have distinct features; they tend to be part of syndromes with female predilection, more indolent in nature, and majority have no c-KIT or PDGFRA mutations [17, 18].
2 Pathology
GISTs are sub-epithelial growing tumors [6] with three categories of cellular morphology: spindle cell type (70%), epithelioid type (20%), and mixed type (10%) [19, 20]. Spindle cell type is characterized by uniform cells with eosinophilic cytoplasm, ovoid nuclei, and juxta nuclear vacuoles [21]. Epithelioid cell type has round cells and round nuclei with vesicular chromatin [22, 23]. It is called epithelioid due to nested appearance that may be confused with epithelial or melanocytic neoplasms. The mixed type presents either as a transition between spindle and epithelioid type or mixture of both types. There are distinctive immunohistochemical markers for GISTs. Kit (CD117) overexpression is found in 90% of GISTs and does not necessarily correlate with c-Kit mutation. DOG-1 (Discovered on GIST-1) and PKC theta (Protein Kinase C theta) are two immunohistochemical markers of GIST that are expressed irrespective of Kit/PDGFRA mutations [24,25,26,27]. Other markers commonly expressed are CD34 that is positive in 70% of GISTs, and desmin in 25%, and less commonly smooth muscle actin (SMA), which is seen in less than 5% of GIST [28].
3 Molecular pathogenesis OF GIST
Most GIST tumors are characterized by oncogenic mutations in Kit or PDGFRA genes. Different mutations in exon loci of tyrosine kinase genes are associated with clinical features and may predict disease behavior [29].
3.1 Kit mutation
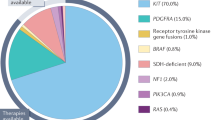
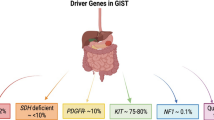
The gain-of-function mutations in Kit gene, first described by Hirota et al. in 1998, are found in 70–80% of GISTs [30,31,32]. Kit is part of tyrosine kinase III family that includes PDGFRA, PDGFRB, FLT3 and macrophage colony stimulating factor receptor (CSF1R). Kit mutation leads to autonomous activity of the enzyme without binding to its ligand stem cell factor (SCF). This induces activation of downstream signals including MAPK, AKT, S6k, STAT1, STAT3, PI3K/mTOR pathway and ETV1 [33, 34]. ETV1 is part of ETS family of transcription factors, acts with Kit to regulate genes that promote GIST growth [35]. Mutations affect different exons of Kit gene as missense mutations, insertions, or deletions, most frequently in exon 11 (67%) followed by exon 9 (10%), exon 13 (1%), and exon 17 (1%) [36].
3.2 PDGFRA mutation
PDGFRA tyrosine Kinase mutations are mutually exclusive of Kit mutations [37]. They most commonly occur in exon 18 (6% of all GISTs) that codes for the activation loop. The most frequent mutation is in exon 18.D842V, which accounts for 60–70% of PDGFRA mutated GISTs [31, 38]. Other exons affected are 12 (0.7% of GISTs) which codes the juxta-membrane domain, and 14 (0.1% of GISTs) which codes for the ATP binding domain [30, 37, 39, 40]. These gain-of function mutations lead to activation of downstream signals in a pattern similar to that of Kit mutated GIST. Activated signal transduction molecules include MAPK, AKT, STAT1 and STAT3 [39]. Clinically, PDGFRA mutated GISTs almost exclusively develop in the stomach, specifically with exon 18 mutation [49]. They often have epithelioid morphology, particularly with exons 18 and 12 mutations. They also show low mitotic activity, and are expected to follow an indolent course [41, 42].
3.3 Wild type KIT/wild type PDGFRA
Ten to fifteen percent of GISTs lack tyrosine kinase mutations, and are described as Kit/PDGFRA wild type (WT) [43]. In this subset, 10–20% have succinate dehydrogenase deficiency (SDH) which is characterized by loss of beta subunit of SDH enzyme [44], and 15% have RAS pathway mutations, which can involve RAS/BRAF [45]. The other 50% of Kit/PDGFRA wild type GISTs have neither SDH deficiency, nor RAS pathway mutations, and hence called quadruple-WT GISTs or true-WT GISTs (account for 5% of all cases of GISTs) [46]. GISTs diagnosed in younger patients are more commonly associated with WT-variant. Eighty-five percent of GIST diagnosed in patients under 23 years are WT [37] [47]. WT-GIST can be sporadic or associated with syndromes, such as NF-1. Twenty-five percent of NF-1 patients develop GIST, which tends to be WT variant [15, 48]. Carney-Stratakis syndrome and Carney’s triad are associated with SDH-deficient WT-GIST [49]. Of the WT GISTs, tumors with SDH mutations tend to occur in gastric location and to be more predominant in females. SDHB loss on IHC with no SDH mutation is more common in pediatrics and young adults, occurs more in gastric location, and is exclusively found in female patients [50].
4 Diagnosis of GIST
The site of tumor in gastrointestinal tract has an association impacts clinical presentation; 60% of GISTs arise in stomach, 30% in small intestine, and they less commonly arise in duodenum (5%), rectum and colon (<5%), esophagus and appendix (<1%) [7, 19, 51]. The most common clinical presentation of GIST is gastrointestinal (GI) bleeding. Other symptoms include abdominal pain/discomfort sometimes presenting as an acute abdomen. GIST can also be an incidental diagnosis [52] [53]. Paraneoplastic syndrome has also been reported as an initial presentation [54].
Computed tomography (CT) scan is the preferred radiographic modality for diagnosis. It usually describes GIST as a solid mass with smooth contour and bright enhancement following intravenous iodine-based contrast. Magnetic resonance imaging (MRI) is an alternative for patients unable to receive intravenous iodine contrast [55]. Esophagogastroduodenoscopy (EGD) is typically performed in patients who present with GI bleeding. It is an important diagnostic tool in gastric GIST especially when combined with endoscopic ultrasound (EUS). EUS appearance is typically described as smooth homogenous, and hypoechoic lesion that mostly originates from the muscularis propria [56]. EUS guided fine needle aspiration (FNA) may not be sufficient to confirm the diagnosis [57]. One study reported a high yield of EUS FNA biopsies and high accuracy in diagnosis of GISTs, with sensitivity of 82%, specificity of 100% and overall accuracy of 86%. Factors thought to increase the diagnostic yield of the FNA were larger lesion size, gastric location, and on-site cytopathology expertise [57]. In potentially resectable GISTs, definitive diagnosis should be obtained by EUS guided FNA biopsy over percutaneous biopsy, a recommendation supported by National Comprehensive Cancer Network (NCCN) guidelines [58], to limit the theoretical risk of peritoneal seeding. Snare biopsies are not preferred due to risk of perforation which carries a worse prognosis in GIST [59]. It is noteworthy that a tissue diagnosis is not always required preoperatively, unless neoadjuvant treatment is considered to downsize the tumor or in the setting of suspected metastatic disease [58]. Positron emission tomography (PET) scan using fluorodeoxyglucose (FDG) can be helpful for localizing a GIST primary or to clarify uncertain findings on CT scan [60, 61]. PET scan is superior to cross-sectional imaging in detection of early response to therapy [62].
Molecular subtyping is essential after diagnosis of GIST. Tumors that lack c-kit and PDGFRA mutations should be tested for SDH status [63]. Loss of SDHB expression on immunohistochemistry (IHC) is an accurate diagnostic marker for SDH deficiency [64,65,66]. Patients with deficient SDH should have tumor and germline sequencing done, to evaluate for SDHX germline mutations that are associated with increased risk of other tumors, such as paragangliomas and pheochromocytomas [67]. Patients with SDH-competent GISTs should also be tested for BRAF- and NFI mutations [68].
5 Prognostic factors
GISTs carry a variable risk of recurrence and metastasis and recent risk stratification models classify GISTs accordingly [69,70,71]. Complete resection of tumor in surgically treated GISTs was a significant variable that affects overall survival in multiple studies [72,73,74,75]. The main factors that determine prognosis of localized GIST are tumor location, size and mitotic rate. Three retrospective studies from the Armed Forces Institute of Pathology (AFIP) contributed to a large portion of prognostic information in literature [52, 53, 76]. Small tumors less than 5 cm with low mitotic index (<5/50 HPF) have low metastatic potential, and tumors larger than 10 cm with high mitotic index (>5/50 HPF) had high metastatic potential [52]. This is regardless of the primary tumor site. However, risk assessment in small tumors with high mitotic index and large tumors with low mitotic index is largely dependent on tumor location. Gastric GISTs that are either >10 cm with low mitotic index or <5 cm with high mitotic index carry a relatively low risk of metastasis [52], while small intestine GISTs with similar features have a relatively higher risk of metastasis [53]. Colorectal GISTs have comparable risk with small intestine GISTs, while tumors diagnosed outside GI tract have more frequent relapses [77, 78]. Tumor rupture, whether spontaneous or induced surgically, negatively affects disease free survival [79, 80].
Molecular characteristics of the tumor also impact on survival. Despite prior reports in literature of worse survival in Kit-Mutated GISTs [81], other reports revealed potential improved survival in tumors that harbor Kit mutation [82]. More recent reports reveal a prognostic value of specific Kit mutations or deletions: exon 9 mutation [83] and exon 11 deletions of codons 557 to 558 [31, 84, 85] have been associated with poor prognosis and more aggressive disease course. The US-Finnish B2222 phase II study using imatinib proved better objective response rates, event-free survival, and overall survival rates in patients with exon 11 mutations compared to patients with exon 9 mutations or with no KIT mutation identified [86].
6 Treatment of GIST
6.1 Localized GIST and the role of surgery
The most common initial treatment for non-metastatic GIST tumors is surgical resection. The goal of surgery is potential cure and/or palliation if the primary tumor is causing symptoms, most commonly pain, bleeding, and/or obstruction. Non-operative, endoscopic surveillance may play a role in the management of small (< 2 cm in size), asymptomatic, histologically non-aggressive tumors [87], especially in elderly patients with severe comorbidities. The frequency of EUS surveillance is controversial, but the most common approach is to repeat EUS yearly [88]. Beyond these small benign lesions, the great majority of localized tumors should be considered for resection.
The goal of curative surgery is a margin-negative resection. As opposed to adenocarcinomas, this can often be achieved with a relatively, organ-sparing approach (especially in the stomach). Tumors in the small bowel or colorectum, and especially those arising in the esophagus or duodenum, may require more radical resection. Since lymphatic metastases are rare [89], regional lymphadenectomy is not indicated unless the lymph nodes are grossly involved. Of note, lymph node involvement is more common in SDH-mutant GISTs [90]. En bloc resection of adjacent organs may sometimes be necessary for large tumors with direct invasion into these structures. Tumor rupture should be avoided during surgery as this event is linked to local recurrence [60, 87].
Although controversial, positive microscopic margins appear not negatively impact survival [91]. The issue of laparoscopic vs open approach would largely be dependent on tumor size, extent of involvement of surrounding structures, and surgeon’s personal experience [60].
6.2 Locally advanced and metastatic GIST
6.2.1 Role of systemic therapy
GIST is typically chemotherapy-resistant compared to other soft tissue tumors [89, 92, 93]. The mainstay of initial systemic therapy is imatinib, which is also the only drug indicated in neoadjuvant and adjuvant settings. However, around 20–30% of patients with GIST who do not express Kit mutation will experience treatment failure on imatinib [31, 32].
6.3 Use of imatinib in neoadjuvant setting
Neoadjuvant treatment with imatinib is used to reduce tumor size and potentially decrease the extent of the surgery. However, major radiological responses per RECIST criteria are uncommon despite the more frequent pathological responses. In the RTOG 0132/ACRIN 6665 phase II clinical trial, partial responses were observed in 7% and stable disease in 83% of patients with primary GIST treated with imatinib 600 mg daily [94]. It did appear that preoperative imatinib therapy was feasible and safe as it did not result in increased postoperative complications.
Neoadjuvant therapy does appear to be associated with improvement in resectability and need for less extensive resection. Fiore et al. reported a median tumor size reduction of 34% with use of preoperative imatinib in 15 patients. Three patients initially considered unresectable underwent complete surgery and 7 patients with initial indication for extensive surgery were more conservatively operated on [95]. The phase II APOLLON study administered neoadjuvant imatinib to 41 patients with “locally advanced but potentially resectable” GISTs. Patients received 400 mg daily for 6 months prior to resection. Treatment response was evaluated by 18F-FDG PET/CT scan 2 months from starting treatment. Thirty-four out of 41 patients (83%) underwent resection after a median of 200 days, with 30/34 achieving R0 resection. No patient received postop imatinib, and the m3-year PFS was 85.2% [96]. An Asian multinational phase II study specifically recruited patients with gastric GISTs ≥ 10cm who were treated with 6–9 months of neoadjuvant imatinib. The primary end point, R0 resection rate was 91%. Patients in this cohort had 98% 2-year PFS and 89% 2-year OS at a median follow-up time of 32 months [97].
MD Anderson Cancer Center reported a phase II clinical trial of preoperative and postoperative imatinib that showed evidence of rapid radiographic response and tumor cell apoptosis in response to imatinib [98]. A subgroup analysis of the prospective trial BFR14 in patients with non-metastatic GIST treated with neoadjuvant imatinib was undertaken. Patients who had resection also received adjuvant imatinib and there was significant improvement in OS and DFS in this subgroup. Overall survival of patients who did not undergo resection was close to those with metastatic GIST [99].
In conclusion, imatinib in the neoadjuvant setting is mainly reserved for patients who may initially have been thought unresectable and in whom reduction in tumor size might result in conversion to resectability. It may also be indicated if downsizing the primary tumor may decrease the extent of the surgery, and hence, the perioperative morbidity. Figure 1 shows an example of response to imatinib neoadjuvant therapy at time of surgical resection.

Fifty-five year old woman initially diagnosed with colonic GIST 9 years ago (Fig. 1a). She underwent 3 months of neoadjuvant imatinib therapy. Note necrosis within the tumor after treatment (white box Fig. 1b). Figure 1c and 1d show the anterior and posterior aspects of the specimen, and the necrotic area is clearly also seen within the white box in Fig. 1d. She is currently without evidence of recurrence
6.4 Imatinib in adjuvant setting
In the pre-imatinib era, 50% of patients who underwent surgical resection of GISTs, experienced recurrence within 5 years with a 5-year survival rate of 50% [89, 100, 101]. The use of adjuvant imatinib significantly improved the outcomes for GIST patients with intermediate or high risk of recurrence.
ACOSOG Z9000 trial was the phase II trial of adjuvant imatinib for GIST. Patients with high risk of recurrence after surgical resection received imatinib 400 mg daily for 1 year. They reported 1-, 3-, and 5-year OS rates of 99, 97, and 83%, respectively. Relapse-free survival (RFS) rates at 1-, 3-, and 5 years were 96, 60, and 40% respectively, with a median follow-up time of 7.7 months [102]. Multivariate analysis of this study revealed a lower RFS with larger tumor size, small bowel tumors, high mitotic index, Kit exon 9 mutation, and older age.
ACOSOG subsequently conducted a phase III trial that compared one year of adjuvant imatinib to placebo. RFS remained superior in imatinib arm (hazard ratio, 0.6; 95% CI, 0.43 to 0.75; Cox model–adjusted P < 0.001). It confirmed the lower RFS with large tumor size, small bowel location, and high mitotic rate, but not tumor genomics. Imatinib did not seem to affect OS [103]. Patients with Kit Exon 11 deletion of any type had better RFS with imatinib. This was not demonstrated in patients with Kit exon 11 insertions or point mutation, Kit exon 9 mutation, PDGFRA mutation, or wild-type tumor.
The optimal duration of therapy was further studied by the European Organization for Research and Treatment of Cancer (EORTC) 62024 trial that compared 2 years of 400 mg daily imatinib post resection with observation in 835 patients with intermediate- or high-risk GIST. After 4.7 years median follow-up, the primary end point, imatinib failure-free survival, did not show statistically significant difference between the two arms (87% in the imatinib arm versus 84% in the control arm, hazard ratio, 0.79; 98.5% CI, 0.50 to 1.25; P = .21); however, RFS favored imatinib arm at 3 years (84% compared to 66%, log-rank P < .001) and 5 years (69% compared to 63%, log-rank P < .001) [104].
A phase III trial was conducted by the Scandinavian Sarcoma Group XVIII/AIO comparing 3-year to 1-year adjuvant imatinib for patients with Kit-positive GIST who had R0/R1 resection with high risk features of recurrence. Dose of imatinib was 400 mg daily. Patients who received imatinib for 3 years had a statistically significant improvement in RFS (hazard ratio [HR], 0.46; 95% CI, 0.32–0.65; P < .001; 5-year RFS, 65.6% vs 47.9%, respectively) and OS (HR, 0.45; 95% CI, 0.22–0.89; P = .02; 5-year survival, 92.0% vs 81.7%) compared to 1 year imatinib arm [105].
Joensuu et al. conducted a second planned analysis of their Scandinavian Sarcoma Group XVIII/AIO trial to investigate whether the survival benefits of 3 years of imatinib have persisted. At a median follow-up of 90 months, the patients who received 3 year of adjuvant imatinib had better RFS (5-year RFS rate was 71.1% versus 52.3%, hazard ratio [HR], 0.60; 95% CI 0.44 to 0.81; P < .001) and OS (91.9% versus 85.3% (HR, 0.60; 95% CI, 0.37 to 0.97; P = .036) compared to those patients that received only 1 year of adjuvant imatinib [106]. This analysis also showed that most of the benefit was seen in a subgroup of GISTs patients with Kit exon 11 mutation.
Subsequently, the same group published an exploratory analysis focused on predictive role of Kit and PDGFRA mutations in patients with GIST treated with adjuvant imatinib. It concluded that patients with Kit exon 11 deletion or insertion-deletion on codons 557 and/or 558 had improved RFS when treated with imatinib for 3 years compared to 1 year. This improved RFS was not found in exon 11 substitution mutations, exon 9 mutations, PDGFRA mutation (including D842V mutation), or Wild-type GISTs [107].
NCCN Guidelines recommend 3-year adjuvant imatinib treatment for high-risk GIST after the surgical resection. However, the question that developed subsequently was if extending the adjuvant imatinib treatment to 5 years would have a better impact on RFS and/or OS. PERSIST-5 clinical trial tried to answer this question on a smaller scale using a single arm phase II trial that enrolled 91 patients with high risk Kit-positive primary GIST. The 5- and 8-year estimated RFS rates were 90% (95% CI, 80–95) and 81% (95% CI, 62–91), respectively. The 5-year OS rate was 95% (95% CI, 86–99) [108]. These findings lead to a phase III clinical trial (NCT02413736), aiming to confirm the results on a larger scale with randomization between 3 vs 5 years of adjuvant imatinib.
6.5 Imatinib as first line treatment of metastatic GIST
The anti-tumor effect, safety, and tolerability of imatinib in patients with metastatic or unresectable GIST were determined in patients randomized to receive either 400 mg or 600 mg imatinib daily. Treatment induced a sustained objective response in more than half of the patients treated with either 400 mg or 600 mg [109]. The EORTC phase I and phase II trials, revealed that the highest feasible dose of imatinib was 800 mg daily [110, 111]. Another EORTC study randomized 946 patients to 400 mg imatinib either once- or twice daily. It showed that patients who received imatinib 400 mg twice daily had superior PFS than those who received imatinib once daily. There was no difference in the response rate between the two doses [112]. .The phase III trial, S0033, also randomized patients to 400 mg once daily versus 400 mg twice daily. The median progression-free survival was 18 months in patients receiving once daily dose versus 20 months for those received twice daily dose. Median overall survival was 55 months in patients received the lower dose compared to 51 months in patients received the higher dose. There were also high-grade toxicities in the high-dose arm [113].
The randomized EORTIC 62005 trial described Kit exon 9 mutation as the strongest adverse risk factor for progression and death in patients, and reported a significant improvement is PFS and reduction of relative risk in patients who have Kit exon 9 mutation treated with higher dose (800 mg/day) [32]. The North American phase III SWOG S0033/CALGB 150105 study also showed improvement in response rate in patients with exon 9 mutation treated with 800 mg daily of imatinib compared to those who received 400 mg daily [114]. A meta-analysis of 1, 640 patients a from the two large randomized studies (EORTIC 62005 and III SWOG S0033/CALGB 150105 ) confirmed a small PFS advantage in patients with exon 9 mutation who received high dose Imatinib but without meaningful OS benefit [115].
6.6 Imatinib resistance
Two types of resistance described: primary resistance, in which the tumor progresses during the first six months of treatment, and secondary resistance, which is characterized by tumor progression after six month of being controlled with imatinib treatment. Primary imatinib resistance is mostly seen in GISTs with exon 9 mutation treated with 400 mg imatinib daily, tumors that are Kit- and PDGFRA wild type—mainly SDH deficient GISTs, and those with PDGFRA exon 18 D842V mutation [86] [114, 116]. Secondary imatinib resistance likely develops from clonal evolution and/or polyclonal secondary mutations in Kit [117,118,119].
6.7 Treatment options for patients with GIST
Table 1 and Table 3 outline the targeted agents, both FDA approved and non-FDA approved, that are available to patients with GIST. Clinical trials leading to approval of agents for GIST have been summarized in Table 2. FDA approved agents have variable degree of inhibiting tyrosine kinases with some similarity in their mechanism of action. Figure 2 shows the mechanisms of action of targeted drugs in GIST.

Mechanism of action of FDA approved agents for patients with GIST
-
1.1.
Treatment beyond imatinib
6.7.1 Sunitinib
Sunitinib is an oral tyrosine kinase inhibitor of platelet-derived growth factor receptors (PDGFRA and PDGFRB) and stem cell factor receptor (KIT) that is approved for the treatment of patients with gastrointestinal stromal tumor after disease progression on or intolerance to imatinib. Sunitinib is usually used in the second-line setting. Approval was granted after a phase III clinical trial (NCT00075218) showed an improved median time to progression in comparison to placebo. The side effects profile was noticed to be broader than that of imatinib, most likely secondary to wide inhibition of other tyrosine kinases including vascular endothelial growth factor receptors (VEGFR1, VEGFR2 and VEGFR3), Fms-like tyrosine kinase-3 (FLT3), colony stimulating factor receptor Type 1 (CSF-1R), and the glial cell-line derived neurotrophic factor receptor encoded by RET oncogene [120, 121].
6.7.2 Regorafenib
Regorafenib, multi-kinase inhibitor targeting KIT and PDGFR and other receptors showed significant activity in patients with advanced GIST who progressed after failure of both imatinib and sunitinib in early phase clinical trials [122]. Regorafenib was examined in a phase III clinical trial against placebo and showed a median PFS of 4.8 months in comparison to 0.9 months in the placebo arm with no major changes in OS because of cross over. This led to FDA approval of regorafenib in the third-line setting [123]. Long-term follow-up of one of the initial phase II trials showed a benefit in patients with primary KIT exon 11 mutations and those with SDH-deficient GIST [124]. Also, in a phase II trial, regorafenib showed prolonged PFS in patients previously treated with imatinib and sunitinib who developed secondary mutations of exon 17 [125]. Regorafenib is being tested against avapritinib (BLU-285) in a phase III clinical trial (NCT03465722) as a second or third line and in a phase II clinical trial (NCT02638766) in patients with wild-type GIST.
6.7.3 Avapritinib (BLU-285)
Avapritinib, a selective inhibitor of mutated KIT and PDGFRA, was approved by the FDA after the results of the NAVIGATOR phase I trial (NCT02508532). In the dose-expansion part, 20 patients with a PDGFRA D842V-mutant GIST, and 36 patients with a PDGFRA D842V-mutant GIST were enrolled. At data cutoff, 49 (88%; 95% CI 76–95) of 56 patients had an overall response, with 5 (9%) complete responses and 44 (79%) partial responses. No dose-limiting toxicities were observed. Both duration of response (DOR) and median progression-free survival (PFS) were not reached at the time of data cutoff [126]. The role of avapritinib will be determined in the phase III VOYAGER trial (NCT03465722) which is testing regorafenib vs. avapritinib in GIST patients with KIT or PDGFRA in third-line setting.
6.7.4 Ripretinib
Ripretinib, with a dual mechanism of action, is a switch-control tyrosine kinase inhibitor with broad activity against primary and drug-resistant KIT/ PDGFRA mutants and an inhibitor of all known activation loop mutations [127]. The phase III trial (NCT03353753) and (NCT03673501) led to the approval of ripretinib for patients with advanced GIST after third-line of treatment [128,129,130]. Median progression-free survival was 6.3 months (95% CI 4.6–6.9) for ripretinib vs. 1.0 months (0.9–1.7) for placebo (HR 0.15 (95% CI 0.09–0.25) with P<0.0001). Ripretinib is being evaluated in an ongoing phase III study (INTRIGUE) in second-line treatment compared with sunitinib (NCT03673501).
7 Experimental agents
Resistance to approved agents is a common clinical dilemma that leads to clinicians to encourage patients to be enrolled in clinical trials once the patient progresses. A list of non-FDA approved agents and some selected clinical trials is provided in Table 3.
7.1 Cabozantinib (XL184)
Cabozantinib, a potent inhibitor of tyrosine kinases targeting MET, VEGFR2 and RET showed antitumor activity in preclinical models through inhibition of tumor growth, proliferation, and angiogenesis, in both imatinib-sensitive and imatinib-resistant models [131]. Phase II trial, EORTC 1317, met its primary endpoint, with median progression-free survival of 6.0 months; this supports further exploration of the role of cabozantinib in GIST in more advanced trials [132].
7.2 Nilotinib
Niltoinib, a selective potent tyrosine kinase inhibitor targeting KIT and PDGFRA, was reported in few case reports to have some response that may reach up to 12 months in patients with advanced GIST [133, 134]. Some benefits were noticed in phase I clinical trials [135]. Nilotinib was also tested more in phase II and III clinical trials but the results were not suggestive of major benefits [136,137,138,139]. However, it was suggested that the role of nilotinib be re-tested before it is discarded for use as a treatment for GIST [140]. A phase IV clinical trial (NCT01735955), whose purpose is to allow continued use of nilotinib in patients who benefited from it, may give more insight about its role in GIST. The estimated date of reporting the results is April 2023.
7.3 Ponatinib
Ponatinib, which inhibits multiple kinases including KIT, is FDA approved for Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) as well as chronic myeloid leukemia (CML). Ponatinib showed activity against KIT mutated refractory GIST in an in vivo setting [141]. It showed some benefits in patients with advanced GIST in a clinical phase II trial (NCT01874665) [142]. And it is currently evaluated currently in a phase II clinical trial to evaluate its role in patients with metastatic and/or unresectable GIST after prior failure or intolerability of Imatinib (NCT03171389). The results of the later study may guide further directions in this regard.
7.4 Pazopanib
Pazopanib, an inhibitor of the VEGFR, PDGFR and stem cell receptor c-KIT, was tested in phase II clinical trial in France. Eighty-one patients were enrolled, and the trial achieved a 40% disease control rate at 4 months using Pazopanib in patients with refractory GIST [143]. This finding was questioned, especially in light of a previous phase II trial in the USA that enrolled 25 patients with a median number of 3 previous agents, showing a median PFS of 1.9 months (95% CI 1.6–5.2), and the a median OS of 10.7 months (95% CI 3.9–NR) [144, 145].
7.5 Masitinib
Masitinib is a highly selective tyrosine kinase inhibitor with activity against wild-type and mutant KIT (exons 9 and 11) [146]. Masitinib was tested to establish safety and efficacy in phase I/II studies in the first-line setting and after the failure of imatinib. Median PFS was 27.2 months in a study population of 30 patients; the response rate was 20% according to RECIST criteria and 84.6% according to FDG-PET response criteria [147, 148]. A phase III clinical trial was initiated to compare the efficacy of masitinib to sunitinib after progression on imatinib (NCT01694277). Masitinib seems to have the potential to obtain approval if the results of this phase III trial are consistent with the findings from the phase I/II studies.
7.6 Crenolanib
Crenolanib, a potent inhibitor of imatinib resistant PDGFRA (including the PDGFRA D842V mutation), is being actively studied in a placebo-controlled phase III trial that completed accrual in August 2020 (NCT02847429) [149]. The previous phase III trial was launched after preclinical studies, while phase I/II clinical trials showed that Crenolanib is a potent inhibitor of Imatinib resistant PDGFRA, including the PDGFRA D842V mutation and plays a potential role in GIST patients. Crenolanib demonstrated a 31% clinical benefit rate with two patients achieving partial response and three patients maintaining stable disease in 16 evaluable and heavily previously treated patients with the PDGFRA D842V mutation [150].
7.7 Sorafenib
Sorafenib, another tyrosine kinase inhibitor of KIT and PDGFRA-mutant kinase in drug-sensitive and drug-resistant GIST, engages in dose-dependent inhibition of GIST tumors [151]. Clinically, two phase II clinical trials and three retrospective analyses showed that sorafenib played some role in the third- or fourth-line settings, mostly after progression after imatinib and Sunitinib [152,153,154,155,156]. One of these phase II clinical trials enrolled 38 patients. It showed a partial response of 13%, while stable disease was 55%, median PFS was 5.2 months (95% CI: 3.4, 7.4), and median OS was 11.6 months [157].
7.8 mTOR inhibitors
The oral mTOR inhibitor everolimus (RAD001) has shown efficacy in GIST refractory to imatinib and sunitinib in a phase I/II clinical trial in which 37% of the patients were progression-free for at least 4 months and 36% achieved stable disease (SD). The most common adverse events were diarrhea, nausea and fatigue [158]. In another phase II study, 9 patients out of 27 showed SD at 16 weeks with controllable adverse events such as anemia, diarrhea, nausea etc. [159]. Another mTOR inhibitor, sirolimus has also demonstrated a benefit in combination with tyrosine kinase inhibitors including Imatinib in a limited number of GIST patients with PDGFRA-D842V mutations. The combination was relatively well tolerated except for skin toxicity in one patient [160, 161].
8 Other options for patients with GIST
Several other systemic therapies are actively being investigated in patients with GIST including immunotherapy, cytotoxic chemotherapy, novel tyrosine kinase inhibitors, binimetinib, selinexor, and few others.
8.1 Immunotherapy
Because inflammation has been implicated in patients with GIST, immunotherapy is being investigated in patients with GIST [162]. Examples include the testing of anti-PD-1 antibody, pembrolizumab, nivolumab with or without ipilimumab, avelumab with axitinib, and vaccine therapy. A durable response was observed in a highly refractory metastatic KIT/PDGFRA wild-type GIST following treatment with nivolumab in one reported case [163]. Future directions in immunotherapy for patients with GIST are summarized in Table 4.
8.2 Cytotoxic chemotherapy
A phase II clinical trial is investigating the role of paclitaxel in patients with advanced and/or metastatic GIST after the failure of at least imatinib and sunitinib in patients with low P-glycoprotein expression (NCT03944304).
8.3 Novel tyrosine kinase inhibitors
8.3.1 Anlotinib
Anlotinib, a multi-targeted tyrosine kinase inhibitor with activity against c-KIT, VEGFR, PDGFR, and FGFR inhibitor, which showed broad activity against soft tissue sarcoma and GIST with D842V, D816H, V560G, and V654A mutations is being studied in a phase II trial in patients with advanced GIST after the failure of Imatinib (NCT04106024)
8.3.2 PLX9486 in combination with PLX3397 or sunitinib
The novel tyrosine kinases inhibitor, PLX9486, is being studied in combination with another novel tyrosine kinase inhibitor, PLX3397 or with Sunitinib in a phase I/II trials in patients with locally advanced, unresectable, or metastatic GIST (NCT02401815).
8.4 Temozolomide (TMZ)
Temozolomide (TMZ), an oral alkylating agent approved by the FDA for glioblastoma multiforme (GBM) and refractory anaplastic astrocytoma cancers, is being investigated in phase II clinical trial in patients with SDH-mutant/deficient gastrointestinal stromal tumor (NCT03556384).
8.5 Binimetinib in combination with pexidartinib or imatinib
Binimetinib, a reversible inhibitor of mitogen-activated extracellular signal regulated kinase 1 (MEK1) and MEK2 activity [164], which is approved for unresectable or metastatic melanoma with a BRAF V600E or V600K mutation, is being studied in a phase I study in combination with pexidartinib (a tyrosine kinase inhibitor that targets colony stimulating factor 1 receptor (CSF1R), KIT proto-oncogene receptor tyrosine kinase (KIT), and FMS-like tyrosine kinase 3 (FLT3) harboring an internal tandem duplication (ITD) mutation and approved for patients with symptomatic tenosynovial giant cell tumor (TGCT)), in patients with advanced gastrointestinal stromal tumor (GIST) (NCT03158103).
Binimetinib is also being investigated with imatinib [165]. A phase Ib/II study of MEK162 (binimetinib) in combination with imatinib is actively recruiting patients with untreated advanced GIST to test its effect of combining binimetinib and imatinib in the first line setting (NCT01991379).
8.6 Selinexor
Selinexor, which is FDA approved for patients with relapsed or refractory multiple myeloma, inhibits nuclear export of tumor suppressor proteins (TSPs), growth regulators, and mRNAs of oncogenic proteins by blocking exportin 1 (XPO1). Selinexor is being examined in phase I/II clinical trials in combination with imatinib in patients with metastatic or unresectable GIST (NCT04138381)
8.7 Bispecific antibody
A bispecific antibody, XmAb18087, against somatostatin receptor 2 (SSTR2) and CD3 that showed in vitro and in vivo activity in GIST via the stimulation of target-dependent T-cell activation is being studied in a phase I study in patients with advanced GIST (NCT03411915).
8.8 Antibody-drug conjugate (ADC)
Monotherapy with DS-6157a, an antibody-drug conjugate, is being studied in a phase I first-in-human study in patients with gastrointestinal stromal tumor (GIST) (NCT04276415).
9 Conclusion
GIST represents the first success story in targeted therapy in patients with non-hematological malignancies. Despite its rarity it is treatable with a high degree of success even in the metastatic setting. Major driver mutation is that of Kit followed by PDGFR. Imatinib revolutionized the outcome of such patients who respond very poorly to conventional cytotoxic therapy. At this time, patients with advanced disease expect prolonged times of disease control when treated with imatinib as first line therapy. Imatinib also markedly improved the outcome of patients with localized disease who had resection of primary tumor but with high risk of disease recurrence and death. Patients who become either resistant or intolerant to imatinib may benefit from an increasing number of oral agents that include sunitinib, regorafenib, avapritinib, and ripretinib. All these drugs are administered orally and are generally well tolerated. Better understanding of the molecular biology of GIST is currently translated to clinical practice to better manage patients. The identification of the molecular profile of a given patient’s tumor can provide prognostic information and also guide treatment choices. All this leads to better personalized therapies. There are a number of ongoing clinical trials testing newer agents including immunotherapy that may further improve the outcome of patients with drug resistant disease.
References
Miettinen, M., & Lasota, J. (2001). Gastrointestinal stromal tumors-Definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Archiv.
Miettinen, M., Sarlomo-Rikala, M., & Lasota, J. (1999). Gastrointestinal stromal tumors: Recent advances in understanding of their biology. Human Pathology.
Nilsson, B., Bümming, P., Meis-Kindblom, J. M., et al. (2005). Gastrointestinal stromal tumors: The incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era-A population-based study in western Sweden. Cancer.
Szucs, Z., Thway, K., Fisher, C., et al. (2017). Molecular subtypes of gastrointestinal stromal tumors and their prognostic and therapeutic implications. Oncol: Futur.
Newman, P. L., Wadden, C., & Fletcher, C. D. M. (1991). Gastrointestinal stromal tumours: Correlation of immunophenotype with clinicopathological features. The Journal of Pathology.
Sircar, K., Hewlett, B. R., Huizinga, J. D., Chorneyko, K., Berezin, I., & Riddell, R. H. (1999). Interstitial cells of cajal as precursors of gastrointestinal stromal tumors. The American Journal of Surgical Pathology.
Kollár, A., Aguiar, P. N., Forones, N. M., & De Mello, R. A. (2019). Gastrointestinal stromal tumor (GIST): Diagnosis and treatment. In International Manual of Oncology Practice.
Ma, G. L., Murphy, J. D., Martinez, M. E., & Sicklick, J. K. (2015). Epidemiology of gastrointestinal stromal tumors in the era of histology codes: Results of a population-based study. Cancer Epidemiology, Biomarkers & Prevention.
Tran, T., Davila, J. A., & El-Serag, H. B. (2005). The epidemiology of malignant gastrointestinal stromal tumors: An analysis of 1, 458 cases from 1992 to 2000. The American Journal of Gastroenterology.
Tryggvason, G., Gíslason, H. G., Magnússon, M. K., & Jónasson, J. G. (2005). Gastrointestinal stromal tumors in Iceland, 1990-2003: The Icelandic GIST study, a population-based incidence and pathologic risk stratification study. International Journal of Cancer.
Goettsch, W. G., Bos, S. D., Breekveldt-Postma, N., Casparie, M., Herings, R. M. C., & Hogendoorn, P. C. W. (2005). Incidence of gastrointestinal stromal tumours is underestimated: Results of a nation-wide study. European Journal of Cancer.
Tzen, C. Y., Wang, J. H., Huang, Y. J., et al. (2007). Incidence of gastrointestinal stromal tumor: A retrospective study based on immunohistochemical and mutational analyses. Digestive Diseases and Sciences.
Kawanowa, K., Sakuma, Y., Sakurai, S., et al. (2006). High incidence of microscopic gastrointestinal stromal tumors in the stomach. Human Pathology.
Agaimy, A., Wünsch, P. H., Hofstaedter, F., et al. (2007). Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c-KIT mutations. The American Journal of Surgical Pathology.
Mussi, C., Schildhaus, H. U., Gronchi, A., Wardelmann, E., & Hohenberger, P. (2008). Therapeutic consequences from molecular biology for gastrointestinal stromal tumor patients affected by neurofibromatosis type 1. Clinical Cancer Research.
Comandone, A., Gasperoni, S., Manetti, R., & Tonelli, P. (2013). Gastrointestinal stromal tumors: Surgical and medical therapy. In What’s New in Surgical Oncology.
Agaram, N. P., Laquaglia, M. P., Ustun, B., et al. (2008). Molecular characterization of pediatric gastrointestinal stromal tumors. Clinical Cancer Research.
Pappo, A. S., & Janeway, K. A. (2009). Pediatric gastrointestinal stromal tumors. Hematology/Oncology Clinics of North America.
Cenaj, O., Jo, V. Y., & Doyle, L. A. (2017). Surgical pathology of gastrointestinal stromal tumors: Correlation with clinical and molecular subtypes. In Gastrointestinal stromal tumors.
Nishida, T., Goto, O., Raut, C. P., & Yahagi, N. (2016). Diagnostic and treatment strategy for small gastrointestinal stromal tumors. Cancer.
Fletcher, C. D. M., Berman, J. J., Corless, C., et al. (2002). Diagnosis of gastrointestinal stromal tumors: A consensus approach. Human Pathology.
Foo, W. C., Liegl-Atzwanger, B., & Lazar, A. J. (2012). Pathology of gastrointestinal stromal tumors. Clinical Medicine Insights Pathology.
Medeiros, F., Corless, C. L., Duensing, A., et al. (2004). KIT-negative gastrointestinal stromal tumors: Proof of concept and therapeutic implications. The American Journal of Surgical Pathology.
Novelli, M., Rossi, S., Rodriguez-Justo, M., et al. (2010). DOG1 and CD117 are the antibodies of choice in the diagnosis of gastrointestinal stromal tumours. Histopathology.
Miettinen, M., Wang, Z. F., & Lasota, J. (2009). DOG1 antibody in the differential diagnosis of gastrointestinal stromal tumors: A study of 1840 cases. The American Journal of Surgical Pathology.
West, R. B., Corless, C. L., Chen, X., et al. (2004). The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. The American Journal of Pathology.
Kang, G. H., Srivastava, A., Kim, Y. E., et al. (2011). DOG1 and PKC-are useful in the diagnosis of KIT-negative gastrointestinal stromal tumors. Modern Pathology.
Miettinen, M., & Lasota, J. (2006). Gastrointestinal stromal tumors: Review on morphology, molecular pathology, prognosis, and differential diagnosis. Archives of Pathology & Laboratory Medicine.
Anderson, W., O’Sullivan, B., Hughes, F., et al. (2017). Microscopic gastrointestinal stromal tumours: a clinical and molecular study of 13 cases. Histopathology.
Hirota, S., Isozaki, K., Moriyama, Y., et al. (1998). Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science (80- ).
Wozniak, A., Rutkowski, P., Piskorz, A., et al. (2012). Prognostic value of KIT/PDGFRA mutations in gastrointestinal stromal tumours (GIST): Polish Clinical GIST Registry experience. Annals of Oncology.
Debiec-Rychter, M., Sciot, R., Le Cesne, A., et al. (2006). KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. European Journal of Cancer.
Rossi, F., Ehlers, I., Agosti, V., et al. (2006). Oncogenic Kit signaling and therapeutic intervention in a mouse model of gastrointestinal stromal tumor. Proceedings of the National Academy of Sciences of the United States of America.
Chi, P., Chen, Y., Zhang, L., et al. (2010). ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature.
Ran, L., Sirota, I., Cao, Z., et al. (2015). Combined inhibition of MAP kinase and KIT signaling synergistically destabilizes ETV1 and suppresses GIST tumor growth. Cancer Discovery.
Wardelmann, E., Losen, I., Hans, V., et al. (2003). Deletion of Trp-557 and Lys-558 in the juxtamembrane domain of the c-kit protooncogene is associated with metastatic behavior of gastrointestinal stromal tumors. International Journal of Cancer.
Heinrich, M. C., Corless, C. L., Duensing, A., et al. (2003). PDGFRA activating mutations in gastrointestinal stromal tumors. Science (80- ).
Wozniak, A., Rutkowski, P., Schöffski, P., et al. (2014). Tumor genotype is an independent prognostic factor in primary gastrointestinal stromal tumors of gastric origin: A European multicenter analysis based on ConticaGIST. Clinical Cancer Research.
Corless, C. L., & Heinrich, M. C. (2008). Molecular pathobiology of gastrointestinal stromal sarcomas. Annual Review of Pathology: Mechanisms of Disease.
Niinuma, T., Suzuki, H., & Sugai, T. (2018). Molecular characterization and pathogenesis of gastrointestinal stromal tumor. Translation of Gastroenterology and Hepatology.
Lasota, J., Dansonka-Mieszkowska, A., Sobin, L. H., & Miettinen, M. (2004). A great majority of GISTs with PDGFRA mutations represent gastric tumors of low or no malignant potential. Laboratory Investigation.
Sakurai, S., Hasegawa, T., Sakuma, Y., et al. (2004). Myxoid epithelioid gastrointestinal stromal tumor (GIST) with mast cell infiltrations: A subtype of GIST with mutations of platelet-derived growth factor receptor alpha gene. Human Pathology.
Corless, C. L., Fletcher, J. A., & Heinrich, M. C. (2004). Biology of gastrointestinal stromal tumors. Journal of Clinical Oncology.
Gill, A. J., Chou, A., Vilain, R., et al. (2010). Immunohistochemistry for SDHB divides gastrointestinal stromal tumors (GISTs) into 2 distinct types. The American Journal of Surgical Pathology.
Daniels, M., Lurkin, I., Pauli, R., et al. (2011). Spectrum of KIT/PDGFRA/BRAF mutations and Phosphatidylinositol-3-Kinase pathway gene alterations in gastrointestinal stromal tumors (GIST). Cancer Letters.
Pantaleo, M. A., Nannini, M., Corless, C. L., & Heinrich, M. C. (2015). Quadruple wild-type (WT) GIST: Defining the subset of GIST that lacks abnormalities of KIT, PDGFRA, SDH, or RAS signaling pathways. Cancer Medicine.
Janeway, K. A., Liegl, B., Harlow, A., et al. (2007). Pediatric KIT-wild-type and platelet-derived growth factor receptor α-wild-type gastrointestinal stromal tumors share KIT activation but not mechanisms of genetic progression with adult gastrointestinal stromal tumors. Cancer Research.
Kinoshita, K., Hirota, S., Isozaki, K., et al. (2004). Absence of c-kit gene mutations in gastrointestinal stromal tumours from neurofibromatosis type I patients. The Journal of Pathology.
Stratakis, C. A., & Carney, J. A. (2009). The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (Carney triad), and the dyad of paragangliomas and gastric stromal sarcomas (Carney-Stratakis syndrome): Molecular genetics and clinical implications. Journal of Internal Medicine.
Belinsky, M. G., Rink, L., & von Mehren, M. (2013). Succinate dehydrogenase deficiency in pediatric and adult gastrointestinal stromal tumors. Frontiers in Oncology.
Roberts, P. J., & Eisenberg, B. (2002). Clinical presentation of gastrointestinal stromal tumors and treatment of operable disease. European Journal of Cancer.
Miettinen, M., Sobin, L. H., & Lasota, J. (2005). Gastrointestinal stromal tumors of the stomach: A clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. The American Journal of Surgical Pathology.
Miettinen, M., Makhlouf, H., Sobin, L. H., & Lasota, J. (2006). Gastrointestinal stromal tumors of the jejunum and ileum: A clinicopathologic, immunohistochemical, and molecular genetic study of 906 cases before imatinib with long-term follow-up. The American Journal of Surgical Pathology.
Maynard, M. A., Marino-Enriquez, A., Fletcher, J. A., et al. (2014). Thyroid hormone inactivation in gastrointestinal stromal tumors. The New England Journal of Medicine.
Scarpa, M., Bertin, M., Ruffolo, C., Polese, L., D’Amico, D. F., & Angriman, I. (2008). A systematic review on the clinical diagnosis of gastrointestinal stromal tumors. Journal of Surgical Oncology.
Hwang, J. H., Rulyak, S. D., & Kimmey, M. B. (2006). American Gastroenterological Association Institute technical review on the management of gastric subepithelial masses. Gastroenterology.
Watson, R. R., Binmoeller, K. F., Hamerski, C. M., et al. (2011). Yield and performance characteristics of endoscopic ultrasound-guided fine needle aspiration for diagnosing upper GI tract stromal tumors. Digestive Diseases and Sciences.
Mehren, M. V., Randall, R. L., Benjamin, R. S., et al. (2018). Soft tissue sarcoma, version 2.2018: Clinical practice guidelines in oncology. Journal National Comprehensive Cancer Network.
Tio, T. L., Tytgat, G. N. J., & den Hartog Jager, F. C. A. (1990). Endoscopic ultrasonography for the evaluation of smooth muscle tumors in the upper gastrointestinal tract: An experience with 42 cases. Gastrointestinal Endoscopy.
Demetri, G. D., Benjamin, R. S., Blanke, C. D., et al. (2007). NCCN task force report: Management of patients with Gastrointestinal Stromal Tumor (GIST)-Update of the NCCN clinical practice guidelines. Journal National Comprehensive Cancer Network.
Gayed, I., Vu, T., Iyer, R., et al. (2004). The role of 18F-FDG PET in staging and early prediction of response to therapy of recurrent gastrointestinal stromal tumors. Journal of Nuclear Medicine.
Kamiyama, Y., Aihara, R., Nakabayashi, T., et al. (2005). 18F-fluorodeoxyglucose positron emission tomography: Useful technique for predicting malignant potential of gastrointestinal stromal tumors. World Journal of Surgery.
Nishida, T. (2017). Therapeutic strategies for wild-type gastrointestinal stromal tumor: Is it different from KIT or PDGFRA-mutated GISTs? Translation of Gastroenterology and Hepatology.
Miettinen, M., Wang, Z. F., Sarlomo-Rikala, M., Osuch, C., Rutkowski, P., & Lasota, J. (2011). Succinate dehydrogenase-deficient GISTs: A clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric GISTs with predilection to young age. The American Journal of Surgical Pathology.
Haller, F., Moskalev, E. A., Faucz, F. R., et al. (2014). Aberrant DNA hypermethylation of SDHC: A novel mechanism of tumor development in Carney triad. Endocrine-Related Cancer.
Gaal, J., Stratakis, C. A., Carney, J. A., et al. (2011). SDHB immunohistochemistry: A useful tool in the diagnosis of Carney-Stratakis and Carney triad gastrointestinal stromal tumors. Modern Pathology.
Jasperson, K. W., Kohlmann, W., Gammon, A., et al. (2014). Role of rapid sequence whole-body MRI screening in SDH-associated hereditary paraganglioma families. Familial Cancer.
Agaram, N. P., Wong, G. C., Guo, T., et al. (2008). Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes, Chromosomes and Cancer.
Miettinen, M., & Lasota, J. (2006). Gastrointestinal stromal tumors: Pathology and prognosis at different sites. Seminars in Diagnostic Pathology.
Rubin, B. P., Blanke, C. D., Demetri, G. D., et al. (2010). Protocol for the examination of specimens from patients with Gastrointestinal Stromal Tumor. Archives of Pathology & Laboratory Medicine.
Huang, H. Y., Li, C. F., Huang, W. W., et al. (2007). A modification of NIH consensus criteria to better distinguish the highly lethal subset of primary localized gastrointestinal stromal tumors: A subdivision of the original high-risk group on the basis of outcome. Surgery.
Crosby, J. A., Catton, C. N., Davis, A., et al. (2001). Malignant gastrointestinal stromal tumors of the small intestine: A review of 50 cases from a prospective database. Annals of Surgical Oncology.
Besana-Ciani, I., Boni, L., Dionigi, G., Benevento, A., & Dionigi, R. (2003). Outcome and long term results of surgical resection for gastrointestinal stromal tumors (GIST). Scandinavian Journal of Surgery.
Langer, C., Gunawan, B., Schüler, P., Huber, W., Füzesi, L., & Becker, H. (2003). Prognostic factors influencing surgical management and outcome of gastrointestinal stromal tumours. The British Journal of Surgery.
Carboni, F., Carlini, M., Scardamaglia, F., et al. (2003). Gastrointestinal stromal tumors of the stomach. A ten-year surgical experience. Journal of Experimental & Clinical Cancer Research.
Emory, T. S., Sobin, L. H., Lukes, L., Lee, D. H., & O’Leary, T. J. (1999). Prognosis of gastrointestinal smooth-muscle (stromal) tumors: Dependence on anatomic site. The American Journal of Surgical Pathology.
Joensuu, H., Vehtari, A., Riihimäki, J., et al. (2012). Risk of recurrence of gastrointestinal stromal tumour after surgery: An analysis of pooled population-based cohorts. The Lancet Oncology.
Kukar, M., Kapil, A., Papenfuss, W., Groman, A., Grobmyer, S. R., & Hochwald, S. N. (2015). Gastrointestinal stromal tumors (GISTs) at uncommon locations: A large population based analysis. Journal of Surgical Oncology.
Hølmebakk, T., Bjerkehagen, B., Boye, K., Bruland, Stoldt, S., & Sundby Hall, K. (2016). Definition and clinical significance of tumour rupture in gastrointestinal stromal tumours of the small intestine. The British Journal of Surgery.
Joensuu, H. (2008). Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Human Pathology.
Kim, T. W., Lee, H., Kang, Y. K., et al. (2004). Prognostic significance of c-kit mutation in localized gastrointestinal stromal tumors. Clinical Cancer Research.
Taniguchi, M., Nishida, T., Hirota, S., et al. (1999). Effect of c-kit mutation on prognosis of gastrointestinal stromal tumors. Cancer Research.
Antonescu, C. R., Sommer, G., Sarran, L., et al. (2003). Association of KIT exon 9 mutations with nongastric primary site and aggressive behavior: KIT mutation analysis and clinical correlates of 120 gastrointestinal stromal tumors. Clinical Cancer Research.
Emile, J. F., Théou, N., Tabone, S., et al. (2004). Clinicopathologic, phenotypic, and genotypic characteristics of gastrointestinal mesenchymal tumors. Clinical Gastroenterology and Hepatology.
Martín, J., Poveda, A., Llombart-Bosch, A., et al. (2005). Deletions affecting codons 557-558 of the c-KIT gene indicate a poor prognosis in patients with completely resected gastrointestinal stromal tumors: A study by the Spanish Group for Sarcoma Research (GEIS). Journal of Clinical Oncology.
Heinrich, M. C., Corless, C. L., Demetri, G. D., et al. (2003). Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. Journal of Clinical Oncology.
Sepe, P. S., & Brugge, W. R. (2009). A guide for the diagnosis and management of gastrointestinal stromal cell tumors. Nature Reviews. Gastroenterology & Hepatology.
Hwang, J. H., & Kimmey, M. B. (2004). The incidental upper gastrointestinal subepithelial mass. Gastroenterology.
DeMatteo, R. P., Lewis, J. J., Leung, D., Mudan, S. S., Woodruff, J. M., & Brennan, M. F. (2000). Two hundred gastrointestinal stromal tumors: Recurrence patterns and prognostic factors for survival. Annals of Surgery.
Boikos, S. A., Pappo, A. S., Killian, J. K., et al. (2016). Molecular subtypes of KIT/PDGFRA wild-type gastrointestinal stromal tumors a report from the national institutes of health gastrointestinal stromal tumor clinic. JAMA Oncology.
Catena, F., Di Battista, M., Ansaloni, L., et al. (2012). Microscopic margins of resection influence primary gastrointestinal stromal tumor survival. Onkologie.
Edmonson, J. H., Marks, R. S., Buckner, J. C., & Mahoney, M. R. (2002). Contrast of response to dacarbazine, mitomycin, doxorubicin, and cisplatin (DMAP) plus GM-CSF between patients with advanced malignant gastrointestinal stromal tumors and patients with other advanced leiomyosarcomas. Cancer Investigation.
Plaat, B. E. C., Hollema, H. H., Molenaar, W. M., et al. (2000). Soft tissue leiomyosarcomas and malignant gastrointestinal stromal tumors: Differences in clinical outcome and expression of multidrug resistance proteins. Journal of Clinical Oncology.
Eisenberg, B. L., Harris, J., Blanke, C. D., et al. (2009). Phase II trial of neoadjuvant/adjuvant imatinib mesylate (IM) for advanced primary and metastatic/recurrent operable gastrointestinal stromal tumor (GIST): Early results of RTOG 0132/ACRIN 6665. Journal of Surgical Oncology.
Fiore, M., Palassini, E., Fumagalli, E., et al. (2009). Preoperative imatinib mesylate for unresectable or locally advanced primary gastrointestinal stromal tumors (GIST). European Journal of Surgical Oncology.
Hohenberger, P., Langer, C., Wendtner, C. M., et al. (2012). Neoadjuvant treatment of locally advanced GIST: Results of APOLLON, a prospective, open label phase II study in KIT- or PDGFRA-positive tumors. JCO.
Kurokawa, Y., Yang, H. K., Cho, H., et al. (2017). Phase II study of neoadjuvant imatinib in large gastrointestinal stromal tumours of the stomach. British Journal of Cancer.
McAuliffe, J. C., Hunt, K. K., Lazar, A. J. F., et al. (2009). A randomized, phase II study of preoperative plus postoperative imatinib in GIST: Evidence of rapid radiographic response and temporal induction of tumor cell apoptosis. Annals of Surgical Oncology.
Blesius, A., Cassier, P. A., Bertucci, F., et al. (2011). Neoadjuvant imatinib in patients with locally advanced non metastatic GIST in the prospective BFR14 trial. BMC Cancer.
Eisenberg, B. L., & Judson, I. (2004). Surgery and imatinib in the management of GIST: Emerging approaches to adjuvant and neoadjuvant therapy. Annals of Surgical Oncology.
Gold, J. S., & DeMatteo, R. P. (2006). Combined surgical and molecular therapy: The gastrointestinal stromal tumor model. Annals of Surgery.
DeMatteo, R. P., Ballman, K. V., Antonescu, C. R., et al. (2013). Long-term results of adjuvant imatinib mesylate in localized, high-risk, primary gastrointestinal stromal tumor: ACOSOG Z9000 (Alliance) intergroup phase 2 trial. Annals of Surgery.
Corless, C. L., Ballman, K. V., Antonescu, C. R., et al. (2014). Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: The ACOSOG Z9001 trial. Journal of Clinical Oncology.
Casali, P. G., Le Cesne, A., Velasco, A. P., et al. (2015). Time to definitive failure to the first tyrosine kinase inhibitor in localized GI stromal tumors treated with imatinib as an adjuvant: A European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Intergroup Randomized Tr. Journal of Clinical Oncology.
Joensuu, H., Eriksson, M., Hall, K. S., et al. (2012). One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: A randomized trial. JAMA - Journal of American Medicine Association.
Joensuu, H., Eriksson, M., Sundby Hall, K., et al. (2016). Adjuvant imatinib for high-risk GI stromal tumor: Analysis of a randomized trial. Journal of Clinical Oncology.
Joensuu, H., Wardelmann, E., Sihto, H., et al. (2017). Effect of KIT and PDGFRA mutations on survival in patients with gastrointestinal stromal tumors treated with adjuvant imatinib: An exploratory analysis of a randomized clinical trial. JAMA Oncology.
Raut, C. P., Espat, N. J., Maki, R. G., et al. (2017). Extended treatment with adjuvant imatinib (IM) for patients (pts) with high-risk primary gastrointestinal stromal tumor (GIST): The PERSIST-5 study. Journal of Clinical Oncology.
Demetri, G. D., Von Mehren, M., Blanke, C. D., et al. (2002). Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. The New England Journal of Medicine.
Van Oosterom, A. T., Judson, I., Verweij, J., et al. (2001). Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: A phase I study. Lancet.
Verweij, J., Van Oosterom, A., Blay, J. Y., et al. (2003). Imatinib mesylate (STI-571 Glivec®, GleevecTM) is an active agent for gastrointestinal stromal tumours, but does not yield responses in other soft-tissue sarcomas that are unselected for a molecular target: Results from an EORTC Soft Tissue and Bone Sarcom. European Journal of Cancer.
Verweij, J., Casali, P. G., Zalcberg, J., et al. (2004). Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: Randomised trial. Lancet.
Blanke, C. D., Rankin, C., Demetri, G. D., et al. (2008). Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. Journal of Clinical Oncology.
Heinrich, M. C., Owzar, K., Corless, C. L., et al. (2008). Correlation of kinase genotype and clinical outcome in the North American intergroup phase III trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 study by cancer and leukemia group B and southwest oncology gr. Journal of Clinical Oncology.
Van Glabbeke, M. (2010). Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: A meta-analysis of 1, 640 patients. Journal of Clinical Oncology.
Heinrich, M. C., Maki, R. G., Corless, C. L., et al. (2008). Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. Journal of Clinical Oncology.
Antonescu, C. R., Besmer, P., Guo, T., et al. (2005). Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clinical Cancer Research.
Desai, J., Shankar, S., Heinrich, M. C., et al. (2007). Clonal evolution of resistance to imatinib in patients with metastatic gastrointestinal stromal tumors. Clinical Cancer Research.
Wardelmann, E., Merkelbach-Bruse, S., Pauls, K., et al. (2006). Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clinical Cancer Research.
George, S., Blay, J. Y., Casali, P. G., et al. (2009). Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. European Journal of Cancer.
Demetri, G. D., van Oosterom, A. T., Garrett, C. R., et al. (2006). Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet.
George, S., Wang, Q., Heinrich, M. C., et al. (2012). Efficacy and safety of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of imatinib and sunitinib: A multicenter phase II trial. Journal of Clinical Oncology.
Demetri, G. D., Reichardt, P., Kang, Y. K., et al. (2013). Effi cacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet.
Ben-Ami, E., Barysauskas, C. M., von Mehren, M., et al. (2016). Long-term follow-up results of the multicenter phase II trial of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of standard tyrosine kinase inhibitor therapy. Annals of Oncology.
Yeh, C. N., Chen, M. H., Chen, Y. Y., et al. (2017). A phase II trial of regorafenib in patients with metastatic and/ or a unresectable gastrointestinal stromal tumor harboring secondary mutations of exon 17. Oncotarget.
Heinrich, M. C., Jones, R. L., von Mehren, M., et al. (2020). Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): A multicentre, open-label, phase 1 trial. Lancet Oncol [Internet], 21(7), 935–946. https://doi.org/10.1016/S1470-2045(20)30269-2.
Smith, B. D., Kaufman, M. D., Lu, W. P., et al. (2019). Ripretinib (DCC-2618) is a switch control kinase inhibitor of a broad spectrum of oncogenic and drug-resistant KIT and PDGFRA variants. Cancer Cell.
George, S., Heinrich, M., Chi, P., et al. (2018). Initial results of phase I study of DCC-2618, a broad-spectrum KIT and PDGFRa inhibitor, in patients (pts) with gastrointestinal stromal tumor (GIST) by number of prior regimens. Annals Oncology Official: Journal Euro Society Medicine Oncology.
von Mehren, M., Serrano, C., Bauer, S., et al. (2019). INVICTUS: A phase III, interventional, double-blind, placebo-controlled study to assess the safety and efficacy of ripretinib as ≥ 4th-line therapy in patients with advanced gastrointestinal stromal tumors (GIST) who have received treatment with prior ant. Annals of Oncology.
Nemunaitis, J., Bauer, S., Blay, J. Y., et al. (2019). Intrigue: Phase III study of ripretinib versus sunitinib in advanced gastrointestinal stromal tumor after imatinib. Future Oncology.
Gebreyohannes, Y. K., Schöffski, P., Van Looy, T., et al. (2016). Cabozantinib is active against human gastrointestinal stromal tumor xenografts carrying different KIT mutations. Molecular Cancer Therapeutics.
Schoffski, P., Mir, O., Kasper, B., et al. (2019). Activity and safety of cabozantinib in patients with gastrointestinal stromal tumor after failure of imatinib and sunitinib: EORTC phase II trial 1317 CaboGIST. Journal of Clinical Oncology.
Manley, P. W., Stiefl, N., Cowan-Jacob, S. W., et al. (2010). Structural resemblances and comparisons of the relative pharmacological properties of imatinib and nilotinib. Bioorganic and Medicinal Chemistry.
Ozaslan, E., Ozkan, M., Bozkurt, O., et al. (2015). Are Rogerofenib and Nilotinib effective for advanced gastrointestinal stromal tumor (GIST) patients who have already been given main treatments? Asian Pacific Journal of Cancer Prevention.
Demetri, G. D., Casali, P. G., Blay, J. Y., et al. (2009). A phase I study of single-agent nilotinib or in combination with imatinib in patients with imatinib-resistant gastrointestinal stromal tumors. Clinical Cancer Research.
Cauchi, C., Somaiah, N., Engstrom, P. F., et al. (2012). Evaluation of nilotinib in advanced GIST previously treated with imatinib and sunitinib. Cancer Chemotherapy and Pharmacology.
Reichardt, P., Blay, J. Y., Gelderblom, H., et al. (2012). Phase III study of nilotinib versus best supportive care with or without a TKI in patients with gastrointestinal stromal tumors resistant to or intolerant of imatinib and sunitinib. Annals of Oncology.
Sawaki, A., Nishida, T., Doi, T., et al. (2011). Phase 2 study of nilotinib as third-line therapy for patients with gastrointestinal stromal tumor. Cancer.
Blay, J. Y., Shen, L., Kang, Y. K., et al. (2015). Nilotinib versus imatinib as first-line therapy for patients with unresectable or metastatic gastrointestinal stromal tumours (ENESTg1): A randomised phase 3 trial. The Lancet Oncology.
del Muro, X. G. (2015). Nilotinib, imatinib, and GIST therapy. The Lancet Oncology.
Garner, A. P., Gozgit, J. M., Anjum, R., et al. (2014). Ponatinib inhibits polyclonal drug-resistant KIT oncoproteins and shows therapeutic potential in heavily pretreated gastrointestinal stromal tumor (GIST) patients. Clinical Cancer Research.
Heinrich, M. C., von Mehren, M., & Demetri, G. D. (2014). et al., A phase 2 study of ponatinib in patients (pts) with advanced gastrointestinal stromal tumors (GIST) after failure of tyrosine kinase inhibitor (TKI) therapy: Initial report. Journal of Clinical Oncology.
Mir, O., Cropet, C., Toulmonde, M., et al. (2016). Pazopanib plus best supportive care versus best supportive care alone in advanced gastrointestinal stromal tumours resistant to imatinib and sunitinib (PAZOGIST): A randomised, multicentre, open-label phase 2 trial. The Lancet Oncology.
Nishida, T., & Doi, T. (2016). Pazopanib for both GIST and soft-tissue sarcoma. The Lancet Oncology.
Ganjoo, K. N., Villalobos, V. M., Kamaya, A., et al. (2014). A multicenter phase II study of pazopanib in patients with advanced gastrointestinal stromal tumors (GIST) following failure of at least imatinib and sunitinib. Annals of Oncology.
Dubreuil, P., Letard, S., Ciufolini, M., et al. (2009). Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS One.
Soria, J. C., Massard, C., Magné, N., et al. (2009). Phase 1 dose-escalation study of oral tyrosine kinase inhibitor masitinib in advanced and/or metastatic solid cancers. European Journal of Cancer.
Le Cesne, A., Blay, J., Bui, N. B., et al. (2009). Masatinib mesylate in imatinib-naive locally advanced or metastatic gastrointestinal stromal tumor (GIST): Results of the French Sarcoma Group phase II trial. Journal of Clinical Oncology.
Blay, J.-Y., Heinrich, M. C., Hohenberger, P., et al. (2017). A randomized, double-blind, placebo-controlled, phase III study of crenolanib in advanced or metastatic GIST patients bearing a D842V mutation in PDGFRA: The CrenoGIST study. Journal of Clinical Oncology.
Heinrich, M. C., Griffith, D., McKinley, A., et al. (2012). Crenolanib inhibits the drug-resistant PDGFRA D842V mutation associated with imatinib-resistant gastrointestinal stromal tumors. Clinical Cancer Research.
Huynh, H., Lee, J. W. J., Chow, P. K. H., et al. (2009). Sorafenib induces growth suppression in mouse models of gastrointestinal stromal tumor. Molecular Cancer Therapeutics.
Wiebe, L., Kasza, K. E., Maki, R. G., et al. (2008). Activity of sorafenib (SOR) in patients (pts) with imatinib (IM) and sunitinib (SU)-resistant (RES) gastrointestinal stromal tumors (GIST): A phase II trial of the University of Chicago Phase II Consortium. Journal of Clinical Oncology.
Park, S. H., Ryu, M. H., Ryoo, B. Y., et al. (2012). Sorafenib in patients with metastatic gastrointestinal stromal tumors who failed two or more prior tyrosine kinase inhibitors: A phase II study of Korean gastrointestinal stromal tumors study group. Investigational New Drugs.
Campbell, N. P., Wroblewski, K., Maki, R. G., et al. (2011). Final results of a University of Chicago phase II consortium trial of sorafenib (SOR) in patients (pts) with imatinib (IM)- and sunitinib (SU)-resistant (RES) gastrointestinal stromal tumors (GIST). Journal of Clinical Oncology.
Brzozowska, M., Wierzba, W., Szafraniec-Buryło, S., et al. (2019). Real-world evidence of patient outcome following treatment of advanced gastrointestinal stromal tumor (GIST) with imatinib, sunitinib, and sorafenib in publicly funded health care in Poland. Medical Science Monitor.
Rutkowski, P., Jagielska, B., Andrzejuk, J., et al. (2017). The analysis of the long-term outcomes of sorafenib therapy in routine practice in imatinib and sunitinib resistant gastrointestinal stromal tumors (GIST). Wspolczesna Onkology.
Singeltary, B., Ghose, A., Sussman, J., Choe, K., & Olowokure, O. (2014). Durable response with a combination of imatinib and sorafenib in KIT exon 17 mutant gastrointestinal stromal tumor. Journal of Gastrointest Oncology.
Schöffski, P., Reichardt, P., Blay, J. Y., et al. (2010). A phase I-II study of everolimus (RAD001) in combination with imatinib in patients with imatinib-resistant gastrointestinal stromal tumors. Annals of Oncology.
Hohenberger, P., Bauer, S., Grunwald, V., et al. (2010). Multicenter, single-arm, two-stage phase II trial of everolimus (RAD001) with imatinib in imatinib-resistant patients (pts) with advanced GIST. Journal of Clinical Oncology.
Piovesan, C., Fumagalli, E., Coco, P., et al. (2009). Response to sirolimus in combination to tirosine kinase inhibitors (TKI) in three cases of PDGFRA-D842V metastatic gastrointestinal stromal tumor (GIST). Journal of Clinical Oncology.
Palassini, E., Fumagalli, E., Coco, P., et al. (2008). Combination of PKC412 and sirolimus in a metastatic patient with PDGFRA-D842V gastrointestinal stromal tumor (GIST). Journal of Clinical Oncology.
Tan, Y., Trent, J. C., Wilky, B. A., Kerr, D. A., & Rosenberg, A. E. (2017). Current status of immunotherapy for gastrointestinal stromal tumor. Cancer Gene Therapy.
Schroeder, B. A., Kohli, K., O’Malley, R. B., et al. Durable tumor regression in highly refractory metastatic KIT/PDGFRA wild-type GIST following treatment with nivolumab. Oncoimmunology [Internet] 2020;9(1). Available from https://doi.org/10.1080/2162402X.2019.1710064.
Rosenbaum, E., Kelly, C., D’Angelo, S. P., et al. (2019). A phase I study of binimetinib (MEK162) combined with pexidartinib (PLX3397) in patients with advanced gastrointestinal stromal tumor. Oncologist.
Chi, P., Qin, L.-X., D’Angelo, S. P., et al. (2015). A phase Ib/II study of MEK162 (binimetinib [BINI]) in combination with imatinib in patients with advanced gastrointestinal stromal tumor (GIST). Journal of Clinical Oncology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Informed consent Specific informed consent is not required. Images and radiography are de-identified. Patient in the case included in the article has signed an informed consent preoperatively for possible use of images and pictures for research.
Conflict of interest
Asfar Azmi—funding from Janssen and Karyopharm Therapeutics Inc. Consultant for Guidepoint and GLG. Speaker bureau at Karyopharm Therapeutics Inc. All other authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Al-Share, B., Alloghbi, A., Al Hallak, M.N. et al. Gastrointestinal stromal tumor: a review of current and emerging therapies. Cancer Metastasis Rev 40, 625–641 (2021). https://doi.org/10.1007/s10555-021-09961-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-021-09961-7