
Abstract
Clinical and experimental studies support the notion that adrenergic stimulation and chronic stress affect inflammation, metabolism, and tumor growth. Eicosanoids are also known to heavily influence inflammation while regulating certain stress responses. However, additional work is needed to understand the full extent of interactions between the stress-related pathways and eicosanoids. Here, we review the potential influences that stress, inflammation, and metabolic pathways have on each other, in the context of eicosanoids. Understanding the intricacies of such interactions could provide insights on how systemic metabolic effects mediated by the stress pathways can be translated into therapies for cancer and other diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
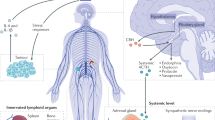
Psychological stress and environmental factors trigger a cascade of pathways in the central nervous system (CNS) and periphery, which subsequently activate stress responses in the autonomic nervous system (ANS) and hypothalamic-pituitary-adrenal (HPA) axis [1, 2]. Hypothalamic production of corticotrophin-releasing factor and arginine vasopressin mediates HPA responses by inducing adrenocorticotropic hormone (ACTH). ACTH mediates downstream release of glucocorticoids by the adrenal cortex, which plays a crucial role in regulating stress responses [3]. Activation of the sympathetic nervous system (SNS) and downstream release of catecholamines from sympathetic neurons and adrenal medulla form the ANS response cascade to stress. Activation of these pathways enable an individual to overcome threat and stressful situations. Stressed individuals are known to have higher levels of catecholamines along with ANS-associated effects on their respiratory, vascular, and cardiac systems [3]. However, chronic stress and the associated prolonged exposure to stress mediators (e.g., glucocorticoids and catecholamines) can have more profound effects on biological systems [4]. Epidemiological data show that social characteristics and chronic stress impact cancer progression. While some studies suggest that stressful life events and other psychosocial factors may correlate with increased risk of cancer [5], others have not found such links [6, 7]. Chronic stress and lack of social support increases breast cancer incidence by almost ninefold [8]. A meta-analysis of 165 studies concluded that psychosocial factors may be associated with cancer incidence and survival [9].
The role of psychosocial factors and stress in affecting disease outcome is an area of long-standing interest. In 1936, Hans Selye identified the activating influence of stress on physiological systems that not only transiently protect and restore the body, but also damage it. [10, 11]. Research over the years that followed has shown that stress impacts multiple biological processes, including inflammation, metabolism, and malignant progression [12]. Clinical and pre-clinical studies in various malignancies have shown that chronic psychological stress influences disease progression, relapse, and response to therapy [13,14,15,16,17,18]. Sustained adrenergic signaling mediated by increased norepinephrine levels drives both tumor inflammation and aberrant arachidonic acid metabolism in cancer [19]. Stress responses occurring via the ANS are also known to directly affect immune cell number and function [20].
Stress mediators and their receptors influence various pathways that modulate tumor biology. cAMP responsive element-binding (CREB) protein is activated by signaling pathways in response to stress hormones and promotes proliferation, migration, angiogenesis, and apoptosis evasion [21]. Viral infections play a major role in tumor initiation, and adrenergic or glucocorticoid-induced signaling activates many tumor-associated viruses [1]. Phospholipase Cγ and protein kinase Cα upregulation by norepinephrine promotes cell migration, and in vitro invasiveness of ovarian cancer and nasopharyngeal carcinoma also increases in the presence of norepinephrine [22, 23]. Catecholamines activate signal transducer and activator of transcription factor-3 (STAT3), which contributes to tumor growth, progression, angiogenesis, and apoptosis suppression [1, 24]. Neuroendocrine stress also activates hormones such as prolactin, which is known to promote tumor growth and cell survival in breast and other cancers [1].
Bioactive lipids have been recognized to promote processes associated with malignant progression. For example, upregulation of sphingosine kinase 1 (SPHK1) in gastric cancer patients is associated with poor survival and increased progression [25]. The eicosanoid PGE2 promotes colorectal cancer progression and inhibits activity of anti-tumor therapy like celecoxib in mouse models [26, 27]. Similarly, lipids are also known to promote chronic inflammation, a process that drives carcinogenesis. In this review, we discuss the complex interactions between inflammation, stress, and metabolic changes, with an emphasis on arachidonic acid metabolism and eicosanoids.
2 Stress and metabolism
Arachidonic acid is an essential dietary fatty acid and its derivatives have an important influence on cancer and inflammation. Arachidonic acid metabolism via cyclooxygenase (COX), lipoxygenase (LOX), and P450 epoxygenase pathways produces eicosanoids, including prostanoids, leukotrienes, and thromboxanes [28] (Fig. 1). Cyclooxygenase exists in two isoforms, namely COX-1 and COX-2. While COX-1 is a constitutive enzyme in most cells in basal state, COX-2 is present in insignificant amounts which is inducible by cytokines and other inflammatory stimuli [28]. Importantly, COX-2 overexpression led to spontaneous development of pancreatic ductal adenocarcinoma in mice, which regressed completely upon COX-2 selective inhibitor (celecoxib) treatment [29].

Biosynthesis of eicosanoids. Arachidonic acid metabolism is initiated by phospholipase A which is activated by various chemical and mechanical stimuli, leading to arachidonic acid generation. Free arachidonic acid is metabolized by two main enzymes, cyclooxygenases (COX), and lipoxygenases (LOX). COX enzymes (COX-1 and COX-2) catalyze the formation of PGH2, which is further converted to various prostanoids (prostaglandins, prostacyclins, and thromboxanes). LOX enzymes (5-LOX, 12-LOX, and 15-LOX) mediate the synthesis of leukotrienes, hydroxyeicosatetraenoic acid (HETE) and lipoxins
Non-steroidal anti-inflammatory drugs (NSAIDs) are effective inhibitors of the COX pathway, with most NSAIDs being non-selective COX-1 and COX-2 inhibitors. LOX pathway mainly operates in the lung, white blood cells, and platelets; its most significant metabolites are leukotrienes [30]. Epidemiological and clinical evidence implicates COX and LOX pathways in chronic inflammation and carcinogenesis caused by aberrant arachidonic acid metabolism [31, 32]. Many population-based epidemiological studies, focused mainly on colorectal cancer, showed a lower incidence upon long-term NSAID use [33, 34]. A comprehensive meta-analysis of published work from 1980 to 2008 showed an inverse correlation between long-term NSAID use and incidence of colon, breast, prostate, and lung cancers [35, 36]. Detailed analysis showed that regular intake of NSAIDs caused risk reductions of up to 43% in colon cancer, 28% in lung cancer, 27% in prostate cancer, and 25% in breast cancer [35]. Among the various arachidonic acid metabolites, prostaglandin E2 (PGE2) is the most widely investigated, especially in the context of the chronic stress. It is a bioactive arachidonic acid-derivative cyclooxygenase metabolite which exerts its activity via four G-protein-coupled receptors (EP1, EP2, EP3, and EP4) and produced abundantly in the brain in response to stress and COX-2 dysregulation. Studies have shown that PGE2 regulates stress responses, immunity, and inflammatory pathways [37, 38].
2.1 Glucocorticoid influences
Studies have shown glucocorticoids to have a suppressive effect on eicosanoid synthesis. Therapeutic doses of the corticosteroid drug prednisone suppress eicosanoids synthesized by both COX and LOX, including PGE2, PGF2, LTB4, and TXB2 in human macrophage-enriched bronchoalveolar lavage cells but not in other cells which express steroid receptors [39]. Endogenous glucocorticoids typically do not affect constitutive COX expression but inhibit inducible COX which is expressed only in the context of inflammation. This suggests that there is a balance between glucocorticoids and COX levels in basal state and any disruptions to this balance could result in inflammation [40]. Similar effects are seen in glial cells and with the corticosteroid drug dexamethasone in human blood monocytes [41, 42]. Abrogation of inflammatory response by glucocorticoids is also observed in zebrafish in response to stress caused by tail vein amputation along with decrease in leukotriene B4 (LTB4) synthesis, an effect that is also seen upon treatment with the synthetic glucocorticoid beclomethasone [43]. Proteasome and eicosanoid profiling of human fibroblasts show that inflammation upregulates a number of eicosanoids synthesized by COX and LOX, such as PGE1, PGE2, TXB2, 11-HETE, and 12-HETE. The upregulated eicosanoid biosynthesis is inhibited by dexamethasone in both fibroblasts and human mesenchymal stem cells [44]. These findings suggest that certain stress mediators activated by components of the CNS suppress eicosanoid production in a pro-inflammatory environment.
2.2 Activation of HPA axis
Excessive/prolonged physical or psychological strain (stress) induced by various stimuli (e.g., physical, mental, or emotional) can manifest itself physically by illness-associated stress responses such as fever, lethargy, anorexia, hyperalgesia, and glucocorticoid secretion [45]. The primary physical stimuli for illness-associated responses is stress induced systemic inflammation, which can be efficiently replicated in various animal models. Many stress stimuli at the end culminate in PGE2 synthesis. Experimental findings support the involvement of the PGE2-synthesizing COX2 enzyme in response to illness as well as the upregulation of this enzyme by illness-associated stimuli [46, 47]. Studies in mice have shown that COX1 is involved in early responses to illness-related stress, particularly in HPA axis activation [46, 48, 49]. Systemic NSAID administration blocks HPA activation by illness-induced stress, substantiating the fact that HPA activation is associated with a COX-dependent pathway [50]. Injection of COX1 inhibitor SC-560 into the CNS abrogates initial (ACTH) release in response to stress, suggesting the presence of a synergistic relationship between arachidonic acid metabolism and pituitary function [49]. Additionally, EP1 and EP3 deficient mice have impaired ACTH release, thereby establishing the importance of prostaglandins in HPA-mediated stress response [51]. EP3 is also expressed in catecholaminergic neuron groups and these sites are thought to be involved in illness-induced HPA activation [52]. These findings collectively suggest that certain CNS responses to illness-induced stress are governed by eicosanoid-dependent mechanisms [45].
2.3 Psychological stress response
Under psychological stress, PGE2 signaling via its receptor EP1 plays an important role in regulating impulsive behavior in vivo, with EP1 deficiency resulting in impulsive aggression and deficits in social behavior in response to psychological stress. Some of these aggressive responses can be mimicked by administering EP1 antagonists to wild-type mice [53]. COX2 expression in cortical pyramidal neurons increases under psychological stressors like restraint and forced-swim stress [54, 55]. Increase in the catecholamine norepinephrine caused by psychosocial stress enhances PGE2 synthesis via PTGS2 upregulation, which promotes formation of a pro-inflammatory and pro-metastatic microenvironment in ovarian cancer [19]. These studies indicate the presence of a direct relationship between psychological stress and prostaglandin synthesis; the underlying mechanisms remain elusive but are being investigated. Similarly, high levels of PGE2 correlate with poor prognosis in many malignancies including colon, lung, and breast cancer [56,57,58].
3 Stress, inflammation, and the role of eicosanoids
Apart from the well-studied stress-induced tumor angiogenesis program, another area of interest is the role of tumor infiltration of autonomous nervous system projections (tumor innervation) in tumor growth. The role of stress mediators and adrenergic signaling in inflammation has been widely studied, especially in the context of tumor angiogenesis. In addition to enhanced tumor angiogenesis, tumor innervation might further enhance tumorigenesis by enriching the tumor microenvironment with catecholamines released by the newly formed nerve endings. We and others have found that in response to catecholamines, tumor cells release neurotrophins (BDNF/NGF), which leads to increased tumor innervation and growth in restraint stress tumor models [59, 60]. There is evidence to show that behavioral stress contributes to an increase in catecholaminergic nerve fibers within lymphoid organs in primates, an indication of a long-term regulatory influence of stress on immune responses [61]. Norepinephrine, the ANS stress response mediator, possesses potent pro-inflammatory properties. High concentration of norepinephrine in circulation caused by cutaneous injury and skin damage leads to sustained inflammation and impaired wound healing [62, 63]. Post-ganglionic neurons innervate all primary and secondary lymphoid organs where norepinephrine is stored within the nerve terminals [64, 65]. In response to CNS activation by psychological stress, norepinephrine is released from the nerve terminals and interacts with immune cells via adrenergic receptors (AR) present on the immune cells. The level of receptor expression varies with immune cell type, activation state, and cytokines [20], which could explain the heterogeneity in response of different immune cells to stress mediators.
Stress-induced upregulation of catecholamines and glucocorticoids in general impairs antigen presentation, T cell proliferation, and attenuate humoral and cell-mediated immunity [2]. However, the actions of stress mediators on individual components of the immune system are quite varied. Norepinephrine increases dendritic cell migration in stress-induced CD8+ T cell response but antigen-presenting ability and delayed-type hypersensitivity are greatly reduced [66,67,68]. Adrenergic receptor stimulation of dendritic cells promotes Th2-associated inflammation and Th17 differentiation [69, 70]. In vitro exposure to norepinephrine or ADRB2 agonists decreases IL-2 secretion from naïve murine CD4+ T cells. Under Th1-promoting culture conditions and pathogens, norepinephrine induces naïve T cells to differentiate into Th1 cells which in turn produce high IFNγ levels in response to norepinephrine [71, 72]. Suppressive phenotype of Treg and Th17 cells is enhanced by ADRB2 agonists and norepinephrine by upregulating CTLA-4, leading to decrease in IL-2 and inflammatory response [73, 74]. The effect of stress mediators on activity of CD8+ T cells remains unclear; however, psychological stress is known to upregulate CD8+ T cell number in humans [75, 76]. In macrophages, exposure to catecholamines and sympathomimetic molecules via β-adrenergic receptor 2 (ADRB2) abrogates production of pro-inflammatory TNF-α [77,78,79,80], whereas TNF-α production is greatly increased by stimulation of α-adrenergic receptors [81,82,83].
Various studies have shown that stress promotes recruitment of tumor-associated macrophages which produce pro-inflammatory and pro-angiogenic factors in large quantities including vascular endothelial growth factor and matrix metalloproteinases [84, 85]. Stress can also activate immune suppressor cells like myeloid-derived suppressor cells (MDSCs) which can greatly hinder CD8+ T cell function [86,87,88,89]. Besides psychological stress, other forms of stress also adversely affect the immune system. For example, forced swim and surgical stress suppress NK cell activity in syngeneic F344 rat models of leukemia, mammary, and colon carcinomas while causing leukemia-related mortality and mammary carcinoma metastasis [90]. Excessive prostaglandin and catecholamine levels have been shown to contribute to postoperative immune suppression, suggesting the presence of an interaction between CNS activity and eicosanoid metabolism. Perioperative combination treatment with COX-2 inhibitors and β-blockers reduces lung tumor retention in mammary carcinoma models, while β-blockade prevents NK cell attenuation [91]. Studies on 38 early-stage breast cancer patients found that perioperative treatment with the ADRB2 antagonist propranolol and the COX-2 inhibitor etodolac inhibits metastasis-associated molecular pathways as well as recurrence of disease [92].
There is growing evidence to demonstrate that eicosanoids, mainly prostanoids and leukotrienes, are involved in cross-talk between immune components and epithelial cells in cancer. Under conditions of chronic inflammation, normal or transformed epithelial cells and resident immune cells secrete pro-inflammatory eicosanoids and cytokines which recruit additional leukocytes to the site. This leads to increased infiltration of dysregulated immune cells, loss of epithelial integrity, and upregulation of inflammatory mediators [93]. PGE2 and elevated COX2 levels are known to increase neutrophil infiltration and exacerbate inflammation in inflammatory bowel disease (IBD), thereby increasing the risk for colon carcinogenesis. In IBD, inflammatory Th17 expansion is enhanced by PGE2 via IL-23 derived from dendritic cells [94]. IL-23 derived from peripheral blood mononuclear cells and naïve T cells promotes Th17 expansion in IBD, a process that is strongly mediated by PGE2 [95, 96]. PGE2 is also elevated in blood and tumor tissues of colon cancer patients, indicating a role for prostaglandins in inflammation-induced malignancy [97]. Murine colon cancer models with COX2 knockout or treated with COX2 inhibitors exhibit reduced carcinogenesis, suggesting that the COX2-PGE2 pathway could serve as a therapeutic target [98]. In line with this, the positive feedback between COX-2 and PGE2 is essential for expression of CXCR4 in cancer-associated MDSCs and the production of its ligand of CXCL2 in ovarian cancer [99]. Similarly, aspirin-PC inhibits cell proliferation, angiogenesis, and increased apoptosis in ovarian cancer cells and mouse models [100]. PGE2 also functions as an immunosuppressive factor and contributed to the microenvironment of ovarian cancer by compromising the Toll-like receptor-mediated dendritic cell activation [101]. In animal models of chronic inflammation, PGE2 promotes inflammation via Th1 and Th17cells, thereby aggravating inflammation [102]. While most research on the involvement of eicosanoids in inflammation, immune system, and cancer is focused on COX-2-mediated pathways, LOXs and their metabolites also appear to play a role in inflammation, immune modulation, and associated cancer. The leukotriene LTB4 is also a potent attractant of neutrophils, eosinophils, T cells, and macrophages to sites of inflammation, via Rac-Erk signaling [103,104,105,106]. In BALB/c mouse models of asthma, leukotrienes are known to mediate airway inflammation by increasing eosinophil infiltration [107]. Low affinity LTB4 receptor (BLT2) and LTB4 can also contribute to the chemoresistance; co-treatment with a BLT2 inhibitor substantially reduced resistance to cisplatin in the SKOV-3 cell tumor model [108]. Expression of 5-LOX was much higher in the human ovarian tumor tissues with highly infiltrated tumor-associated macrophages and macrophages treated with 5-LOX metabolites led to increased expression of MMP-7 through the p38 pathway [109].
There is considerable evidence to demonstrate the therapeutic potential of targeting the interaction between nerves with immune cells to improve immunity, particularly in cancer. Use of host cell-targeted immune therapies such as CTLA-4 and PD-1 blockade heavily rely on the presence of functional T cells, establishing the need for therapies which prevent immune suppression in the microenvironment. As described above, there is extensive experimental data to suggest that use of β-blockers could be an effective strategy to improve host immune responses. Similarly, NSAIDs that target COX and LOX pathways could be included in the therapeutic regimen to treat tumor-associated inflammation. Despite the seemingly independent pro-inflammatory actions of biobehavioral pathways and eicosanoids, there could be an underlying synergism between these processes in inducing inflammation, an interaction that has not been fully characterized yet due to biological complexity. Continued research in these areas is expected to uncover some of the mechanisms of interactions between CNS responses and eicosanoids in promoting inflammation.
4 Discussion
Multiple pre-clinical and clinical studies have shown that psychosocial factors, depression, and stress in humans affect many biological processes. Many of these studies specifically link SNS activation to malignant progression, metabolism dysregulation, and inflammation. Stress-inducible metabolic factors, such as COX2, modulate stress responses in illness and are also involved in activation of components of the CNS under stress. Stress mediators such as norepinephrine directly influence immune cell number and activity at a site, subsequently affecting inflammation. The studies outlined in this review show that stress can shape the immune environment and metabolism of tumors and normal cells, highlighting the importance of deeper understanding of the interactions between these processes in driving disease (Fig. 2).

Stress and eicosanoids directly influence inflammation. However, the relationship between stress and eicosanoids is not well established. Eicosanoids activate certain components of CNS-mediated stress response pathway, while stress mediators are known to influence synthesis of certain eicosanoids. It is possible that stress mediators and eicosanoid-synthesizing enzymes influence each other, should their basal levels be disrupted. However, additional research is needed to elucidate the exact pathways and mediators involved in this interaction
While the effects of adrenergic signaling and stress on crucial tumor metabolic pathways have been extensively investigated [110, 111], interactions between stress and eicosanoids are not fully understood. Arachidonic acid pathway and eicosanoids respond to different types of stress stimuli and there could be responses to adrenergic signaling and psychological stress. It is important to uncover the underlying interactions between these processes in order to expand the understanding of disease development and progression.
References
Antoni, M. H., Lutgendorf, S. K., Cole, S. W., Dhabhar, F. S., Sephton, S. E., McDonald, P. G., Stefanek, M., & Sood, A. K. (2006). The influence of bio-behavioural factors on tumour biology: pathways and mechanisms. Nature Reviews. Cancer, 6(3), 240–248.
Glaser, R., & Kiecolt-Glaser, J. K. (2005). Stress-induced immune dysfunction: implications for health. Nature Reviews. Immunology, 5(3), 243–251.
Charmandari, E., Tsigos, C., & Chrousos, G. (2005). Endocrinology of the stress response. Annual Review of Physiology, 67, 259–284.
McEwen, B. S. (2002). Sex, stress and the hippocampus: allostasis, allostatic load and the aging process. Neurobiology of Aging, 23(5), 921–939.
Penninx, B. W., et al. (1998). Chronically depressed mood and cancer risk in older persons. Journal of the National Cancer Institute, 90(24), 1888–1893.
Duijts, S. F., Zeegers, M. P., & Borne, B. V. (2003). The association between stressful life events and breast cancer risk: a meta-analysis. International Journal of Cancer, 107(6), 1023–1029.
Bleiker, E. M., et al. (2008). Personality factors and breast cancer risk: a 13-year follow-up. Journal of the National Cancer Institute, 100(3), 213–218.
Price, M. A., Tennant, C. C., Smith, R. C., Butow, P. N., Kennedy, S. J., Kossoff, M. B., & Dunn, S. M. (2001). The role of psychosocial factors in the development of breast carcinoma: part I. The cancer prone personality. Cancer, 91(4), 679–685.
Chida, Y., Hamer, M., Wardle, J., & Steptoe, A. (2008). Do stress-related psychosocial factors contribute to cancer incidence and survival? Nature Clinical Practice. Oncology, 5(8), 466–475.
Selye, H. (1998). A syndrome produced by diverse nocuous agents. 1936. The Journal of Neuropsychiatry and Clinical Neurosciences, 10(2), 230–231.
Reiche, E. M., Nunes, S. O., & Morimoto, H. K. (2004). Stress, depression, the immune system, and cancer. The Lancet Oncology, 5(10), 617–625.
Cole, S. W., Nagaraja, A. S., Lutgendorf, S. K., Green, P. A., & Sood, A. K. (2015). Sympathetic nervous system regulation of the tumour microenvironment. Nature Reviews. Cancer, 15(9), 563–572.
Hassan, S., Karpova, Y., Baiz, D., Yancey, D., Pullikuth, A., Flores, A., Register, T., Cline, J. M., D’Agostino R Jr, Danial, N., Datta, S. R., & Kulik, G. (2013). Behavioral stress accelerates prostate cancer development in mice. The Journal of Clinical Investigation, 123(2), 874–886.
Kruk, J., & Aboul-Enein, H. Y. (2004). Psychological stress and the risk of breast cancer: a case-control study. Cancer Detection and Prevention, 28(6), 399–408.
Lutgendorf, S. K., de Geest, K., Bender, D., Ahmed, A., Goodheart, M. J., Dahmoush, L., Zimmerman, M. B., Penedo, F. J., Lucci III, J. A., Ganjei-Azar, P., Thaker, P. H., Mendez, L., Lubaroff, D. M., Slavich, G. M., Cole, S. W., & Sood, A. K. (2012). Social influences on clinical outcomes of patients with ovarian cancer. Journal of Clinical Oncology, 30(23), 2885–2890.
Ramirez, A. J., Craig, T. K., Watson, J. P., Fentiman, I. S., North, W. R., & Rubens, R. D. (1989). Stress and relapse of breast cancer. BMJ, 298(6669), 291–293.
Thaker, P. H., Han, L. Y., Kamat, A. A., Arevalo, J. M., Takahashi, R., Lu, C., Jennings, N. B., Armaiz-Pena, G., Bankson, J. A., Ravoori, M., Merritt, W. M., Lin, Y. G., Mangala, L. S., Kim, T. J., Coleman, R. L., Landen, C. N., Li, Y., Felix, E., Sanguino, A. M., Newman, R. A., Lloyd, M., Gershenson, D. M., Kundra, V., Lopez-Berestein, G., Lutgendorf, S. K., Cole, S. W., & Sood, A. K. (2006). Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nature Medicine, 12(8), 939–944.
Wang, H. M., Liao, Z. X., Komaki, R., Welsh, J. W., O’Reilly, M. S., Chang, J. Y., Zhuang, Y., Levy, L. B., Lu, C., & Gomez, D. R. (2013). Improved survival outcomes with the incidental use of beta-blockers among patients with non-small-cell lung cancer treated with definitive radiation therapy. Annals of Oncology, 24(5), 1312–1319.
Nagaraja, A. S., Dorniak, P. L., Sadaoui, N. C., Kang, Y., Lin, T., Armaiz-Pena, G., Wu, S. Y., Rupaimoole, R., Allen, J. K., Gharpure, K. M., Pradeep, S., Zand, B., Previs, R. A., Hansen, J. M., Ivan, C., Rodriguez-Aguayo, C., Yang, P., Lopez-Berestein, G., Lutgendorf, S. K., Cole, S. W., & Sood, A. K. (2016). Sustained adrenergic signaling leads to increased metastasis in ovarian cancer via increased PGE2 synthesis. Oncogene, 35(18), 2390–2397.
Sanders, V. M., & Straub, R. H. (2002). Norepinephrine, the beta-adrenergic receptor, and immunity. Brain, Behavior, and Immunity, 16(4), 290–332.
Jean, D., & Bar-Eli, M. (2000). Regulation of tumor growth and metastasis of human melanoma by the CREB transcription factor family. Molecular and Cellular Biochemistry, 212(1–2), 19–28.
Sood, A. K., Bhatty, R., Kamat, A. A., Landen, C. N., Han, L., Thaker, P. H., Li, Y., Gershenson, D. M., Lutgendorf, S., & Cole, S. W. (2006). Stress hormone-mediated invasion of ovarian cancer cells. Clinical Cancer Research, 12(2), 369–375.
Yang, E. V., Sood, A. K., Chen, M., Li, Y., Eubank, T. D., Marsh, C. B., Jewell, S., Flavahan, N. A., Morrison, C., Yeh, P. E., Lemeshow, S., & Glaser, R. (2006). Norepinephrine up-regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)-2, and MMP-9 in nasopharyngeal carcinoma tumor cells. Cancer Research, 66(21), 10357–10364.
Landen Jr., C. N., et al. (2007). Neuroendocrine modulation of signal transducer and activator of transcription-3 in ovarian cancer. Cancer Research, 67(21), 10389–10396.
Li, W., Yu, C. P., Xia, J. T., Zhang, L., Weng, G. X., Zheng, H. Q., Kong, Q. L., Hu, L. J., Zeng, M. S., Zeng, Y. X., Li, M., Li, J., & Song, L. B. (2009). Sphingosine kinase 1 is associated with gastric cancer progression and poor survival of patients. Clinical Cancer Research, 15(4), 1393–1399.
Hansen-Petrik, M. B., McEntee, M., Jull, B., Shi, H., Zemel, M. B., & Whelan, J. (2002). Prostaglandin E(2) protects intestinal tumors from nonsteroidal anti-inflammatory drug-induced regression in Apc(min/+) mice. Cancer Research, 62(2), 403–408.
Yan, M., Myung, S. J., Fink, S. P., Lawrence, E., Lutterbaugh, J., Yang, P., Zhou, X., Liu, D., Rerko, R. M., Willis, J., Dawson, D., Tai, H. H., Barnholtz-Sloan, J. S., Newman, R. A., Bertagnolli, M. M., & Markowitz, S. D. (2009). 15-Hydroxyprostaglandin dehydrogenase inactivation as a mechanism of resistance to celecoxib chemoprevention of colon tumors. Proceedings of the National Academy of Sciences of the United States of America, 106(23), 9409–9413.
Wang, D., & Dubois, R. N. (2010). Eicosanoids and cancer. Nature Reviews. Cancer, 10(3), 181–193.
Colby, J. K., et al. (2008). Progressive metaplastic and dysplastic changes in mouse pancreas induced by cyclooxygenase-2 overexpression. Neoplasia, 10(8), 782–796.
Lewis, R. A., Austen, K. F., & Soberman, R. J. (1990). Leukotrienes and other products of the 5-lipoxygenase pathway. Biochemistry and relation to pathobiology in human diseases. The New England Journal of Medicine, 323(10), 645–655.
Agarwal, S., Reddy, G. V., & Reddanna, P. (2009). Eicosanoids in inflammation and cancer: the role of COX-2. Expert Review of Clinical Immunology, 5(2), 145–165.
Harizi, H., Corcuff, J. B., & Gualde, N. (2008). Arachidonic-acid-derived eicosanoids: roles in biology and immunopathology. Trends in Molecular Medicine, 14(10), 461–469.
Ruder, E. H., Laiyemo, A. O., Graubard, B. I., Hollenbeck, A. R., Schatzkin, A., & Cross, A. J. (2011). Non-steroidal anti-inflammatory drugs and colorectal cancer risk in a large, prospective cohort. The American Journal of Gastroenterology, 106(7), 1340–1350.
Kuo, C. N., Pan, J. J., Huang, Y. W., Tsai, H. J., & Chang, W. C. (2018). Association between nonsteroidal anti-inflammatory drugs and colorectal cancer: a population-based case-control study. Cancer Epidemiology, Biomarkers & Prevention.
Harris, R. E. (2009). Cyclooxygenase-2 (cox-2) blockade in the chemoprevention of cancers of the colon, breast, prostate, and lung. Inflammopharmacology, 17(2), 55–67.
Gurpinar, E., Grizzle, W. E., & Piazza, G. A. (2014). NSAIDs inhibit tumorigenesis, but how? Clinical Cancer Research, 20(5), 1104–1113.
Narumiya, S. (2007). Physiology and pathophysiology of prostanoid receptors. Proceedings of the Japan Academy. Series B, Physical and Biological Sciences, 83(9–10), 296–319.
Brown, J. R., & DuBois, R. N. (2005). COX-2: a molecular target for colorectal cancer prevention. Journal of Clinical Oncology, 23(12), 2840–2855.
Sebaldt, R. J., Sheller, J. R., Oates, J. A., Roberts, L. J., & FitzGerald, G. A. (1990). Inhibition of eicosanoid biosynthesis by glucocorticoids in humans. Proceedings of the National Academy of Sciences of the United States of America, 87(18), 6974–6978.
Masferrer, J. L., Seibert, K., Zweifel, B., & Needleman, P. (1992). Endogenous glucocorticoids regulate an inducible cyclooxygenase enzyme. Proceedings of the National Academy of Sciences of the United States of America, 89(9), 3917–3921.
Brenner, T., Boneh, A., Shohami, E., Abramsky, O., & Weidenfeld, J. (1992). Glucocorticoid regulation of eicosanoid production by glial cells under basal and stimulated conditions. Journal of Neuroimmunology, 40(2–3), 273–279.
Fu, J. Y., Masferrer, J. L., Seibert, K., Raz, A., & Needleman, P. (1990). The induction and suppression of prostaglandin H2 synthase (cyclooxygenase) in human monocytes. The Journal of Biological Chemistry, 265(28), 16737–16740.
Chatzopoulou, A., Heijmans, J. P. M., Burgerhout, E., Oskam, N., Spaink, H. P., Meijer, A. H., & Schaaf, M. J. M. (2016). Glucocorticoid-induced attenuation of the inflammatory response in zebrafish. Endocrinology, 157(7), 2772–2784.
Tahir, A., Bileck, A., Muqaku, B., Niederstaetter, L., Kreutz, D., Mayer, R. L., Wolrab, D., Meier, S. M., Slany, A., & Gerner, C. (2017). Combined proteome and eicosanoid profiling approach for revealing implications of human fibroblasts in chronic inflammation. Analytical Chemistry, 89(3), 1945–1954.
Furuyashiki, T., & Narumiya, S. (2011). Stress responses: the contribution of prostaglandin E(2) and its receptors. Nature Reviews. Endocrinology, 7(3), 163–175.
Elander, L., Engstrom, L., Ruud, J., Mackerlova, L., Jakobsson, P. J., Engblom, D., Nilsberth, C., & Blomqvist, A. (2009). Inducible prostaglandin E2 synthesis interacts in a temporally supplementary sequence with constitutive prostaglandin-synthesizing enzymes in creating the hypothalamic-pituitary-adrenal axis response to immune challenge. The Journal of Neuroscience, 29(5), 1404–1413.
Elmquist, J. K., Breder, C. D., Sherin, J. E., Scammell, T. E., Hickey, W. F., Dewitt, D., & Saper, C. B. (1997). Intravenous lipopolysaccharide induces cyclooxygenase 2-like immunoreactivity in rat brain perivascular microglia and meningeal macrophages. The Journal of Comparative Neurology, 381(2), 119–129.
Elander, L., Ruud, J., Korotkova, M., Jakobsson, P. J., & Blomqvist, A. (2010). Cyclooxygenase-1 mediates the immediate corticosterone response to peripheral immune challenge induced by lipopolysaccharide. Neuroscience Letters, 470(1), 10–12.
Garcia-Bueno, B., Serrats, J., & Sawchenko, P. E. (2009). Cerebrovascular cyclooxygenase-1 expression, regulation, and role in hypothalamic-pituitary-adrenal axis activation by inflammatory stimuli. The Journal of Neuroscience, 29(41), 12970–12981.
Turnbull, A. V., & Rivier, C. L. (1999). Regulation of the hypothalamic-pituitary-adrenal axis by cytokines: actions and mechanisms of action. Physiological Reviews, 79(1), 1–71.
Matsuoka, Y., Furuyashiki, T., Bito, H., Ushikubi, F., Tanaka, Y., Kobayashi, T., Muro, S., Satoh, N., Kayahara, T., Higashi, M., Mizoguchi, A., Shichi, H., Fukuda, Y., Nakao, K., & Narumiya, S. (2003). Impaired adrenocorticotropic hormone response to bacterial endotoxin in mice deficient in prostaglandin E receptor EP1 and EP3 subtypes. Proceedings of the National Academy of Sciences of the United States of America, 100(7), 4132–4137.
Ericsson, A., Arias, C., & Sawchenko, P. E. (1997). Evidence for an intramedullary prostaglandin-dependent mechanism in the activation of stress-related neuroendocrine circuitry by intravenous interleukin-1. The Journal of Neuroscience, 17(18), 7166–7179.
Matsuoka, Y., Furuyashiki, T., Yamada, K., Nagai, T., Bito, H., Tanaka, Y., Kitaoka, S., Ushikubi, F., Nabeshima, T., & Narumiya, S. (2005). Prostaglandin E receptor EP1 controls impulsive behavior under stress. Proceedings of the National Academy of Sciences of the United States of America, 102(44), 16066–16071.
Garcia-Bueno, B., et al. (2008). Stress mediators regulate brain prostaglandin synthesis and peroxisome proliferator-activated receptor-gamma activation after stress in rats. Endocrinology, 149(4), 1969–1978.
Yamagata, K., Andreasson, K. I., Kaufmann, W. E., Barnes, C. A., & Worley, P. F. (1993). Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron, 11(2), 371–386.
McLemore, T. L., et al. (1988). Profiles of prostaglandin biosynthesis in normal lung and tumor tissue from lung cancer patients. Cancer Research, 48(11), 3140–3147.
Rigas, B., Goldman, I. S., & Levine, L. (1993). Altered eicosanoid levels in human colon cancer. The Journal of Laboratory and Clinical Medicine, 122(5), 518–523.
Wang, D., & Dubois, R. N. (2004). Cyclooxygenase-2: a potential target in breast cancer. Seminars in Oncology, 31(1 Suppl 3), 64–73.
Allen, J. K., Armaiz-Pena, G. N., Nagaraja, A. S., Sadaoui, N. C., Ortiz, T., Dood, R., Ozcan, M., Herder, D. M., Haemerrle, M., Gharpure, K. M., Rupaimoole, R., Previs, R., Wu, S. Y., Pradeep, S., Xu, X., Dong Han, H., Zand, B., Dalton, H. J., Taylor, M., Hu, W., Bottsford-Miller, J., Moreno-Smith, M., Kang, Y., Mangala, L. S., Rodriguez-Aguayo, C., Sehgal, V., Spaeth, E. L., Ram, P. T., Wong, S. T. C., Marini, F. C., Lopez-Berestein, G., Cole, S. W., Lutgendorf, S. K., diBiasi, M., & Sood, A. K. (2018). Sustained adrenergic signaling promotes intratumoral innervation through BDNF induction. In Cancer Res (p. canres.1701.2016).
Renz, B. W., et al. (2018). β2 Adrenergic-neurotrophin feedforward loop promotes pancreatic cancer. Cancer Cell, 33(1), 75–90 e7.
Sloan, E. K., Capitanio, J. P., & Cole, S. W. (2008). Stress-induced remodeling of lymphoid innervation. Brain, Behavior, and Immunity, 22(1), 15–21.
Gosain, A., Jones, S. B., Shankar, R., Gamelli, R. L., & DiPietro, L. A. (2006). Norepinephrine modulates the inflammatory and proliferative phases of wound healing. The Journal of Trauma, 60(4), 736–744.
Sivamani, R. K., Pullar, C. E., Manabat-Hidalgo, C. G., Rocke, D. M., Carlsen, R. C., Greenhalgh, D. G., & Isseroff, R. R. (2009). Stress-mediated increases in systemic and local epinephrine impair skin wound healing: potential new indication for beta blockers. PLoS Medicine, 6(1), e12.
Felten, D. L., et al. (1985). Noradrenergic and peptidergic innervation of lymphoid tissue. Journal of Immunology, 135(2 Suppl), 755s–765s.
Felten, S. Y., & Olschowka, J. (1987). Noradrenergic sympathetic innervation of the spleen: II. Tyrosine hydroxylase (TH)-positive nerve terminals form synapticlike contacts on lymphocytes in the splenic white pulp. Journal of Neuroscience Research, 18(1), 37–48.
Maestroni, G. J., & Mazzola, P. (2003). Langerhans cells beta 2-adrenoceptors: role in migration, cytokine production, Th priming and contact hypersensitivity. Journal of Neuroimmunology, 144(1–2), 91–99.
Saint-Mezard, P., Chavagnac, C., Bosset, S., Ionescu, M., Peyron, E., Kaiserlian, D., Nicolas, J. F., & Berard, F. (2003). Psychological stress exerts an adjuvant effect on skin dendritic cell functions in vivo. Journal of Immunology, 171(8), 4073–4080.
Seiffert, K., Hosoi, J., Torii, H., Ozawa, H., Ding, W., Campton, K., Wagner, J. A., & Granstein, R. D. (2002). Catecholamines inhibit the antigen-presenting capability of epidermal Langerhans cells. Journal of Immunology, 168(12), 6128–6135.
Manni, M., Granstein, R. D., & Maestroni, G. (2011). β2-Adrenergic agonists bias TLR-2 and NOD2 activated dendritic cells towards inducing an IL-17 immune response. Cytokine, 55(3), 380–386.
Yanagawa, Y., Matsumoto, M., & Togashi, H. (2011). Adrenoceptor-mediated enhancement of interleukin-33 production by dendritic cells. Brain, Behavior, and Immunity, 25(7), 1427–1433.
Alaniz, R. C., Thomas, S. A., Perez-Melgosa, M., Mueller, K., Farr, A. G., Palmiter, R. D., & Wilson, C. B. (1999). Dopamine beta-hydroxylase deficiency impairs cellular immunity. Proceedings of the National Academy of Sciences of the United States of America, 96(5), 2274–2278.
Swanson, M. A., Lee, W. T., & Sanders, V. M. (2001). IFN-gamma production by Th1 cells generated from naive CD4+ T cells exposed to norepinephrine. Journal of Immunology, 166(1), 232–240.
Guereschi, M. G., Araujo, L. P., Maricato, J. T., Takenaka, M. C., Nascimento, V. M., Vivanco, B. C., Reis, V. O., Keller, A. C., Brum, P. C., & Basso, A. S. (2013). Beta2-adrenergic receptor signaling in CD4+ Foxp3+ regulatory T cells enhances their suppressive function in a PKA-dependent manner. European Journal of Immunology, 43(4), 1001–1012.
Vida, G., et al. (2011). β2-Adrenoreceptors of regulatory lymphocytes are essential for vagal neuromodulation of the innate immune system. The FASEB Journal, 25(12), 4476–4485.
Benschop, R. J., Jacobs, R., Sommer, B., Schürmeyer, T. H., Raab, J. R., Schmidt, R. E., & Schedlowski, M. (1996). Modulation of the immunologic response to acute stress in humans by beta-blockade or benzodiazepines. The FASEB Journal, 10(4), 517–524.
Mathews, P. M., et al. (1983). Enhancement of natural cytotoxicity by beta-endorphin. Journal of Immunology, 130(4), 1658–1662.
Deng, J., Muthu, K., Gamelli, R., Shankar, R., & Jones, S. B. (2004). Adrenergic modulation of splenic macrophage cytokine release in polymicrobial sepsis. American Journal of Physiology. Cell Physiology, 287(3), C730–C736.
Elenkov, I. J., et al. (1996). Modulatory effects of glucocorticoids and catecholamines on human interleukin-12 and interleukin-10 production: clinical implications. Proceedings of the Association of American Physicians, 108(5), 374–381.
Hetier, E., Ayala, J., Bousseau, A., & Prochiantz, A. (1991). Modulation of interleukin-1 and tumor necrosis factor expression by beta-adrenergic agonists in mouse ameboid microglial cells. Experimental Brain Research, 86(2), 407–413.
Panina-Bordignon, P., Mazzeo, D., Lucia, P. D., D’Ambrosio, D., Lang, R., Fabbri, L., Self, C., & Sinigaglia, F. (1997). Beta2-agonists prevent Th1 development by selective inhibition of interleukin 12. The Journal of Clinical Investigation, 100(6), 1513–1519.
Heijnen, C. J., van der Voort, C. R., Wulffraat, N., van der Net, J., Kuis, W., & Kavelaars, A. (1996). Functional alpha 1-adrenergic receptors on leukocytes of patients with polyarticular juvenile rheumatoid arthritis. Journal of Neuroimmunology, 71(1–2), 223–226.
Spengler, R. N., et al. (1990). Stimulation of alpha-adrenergic receptor augments the production of macrophage-derived tumor necrosis factor. Journal of Immunology, 145(5), 1430–1434.
Szelenyi, J., Kiss, J. P., & Vizi, E. S. (2000). Differential involvement of sympathetic nervous system and immune system in the modulation of TNF-alpha production by alpha2- and beta-adrenoceptors in mice. Journal of Neuroimmunology, 103(1), 34–40.
Madden, K. S., Szpunar, M. J., & Brown, E. B. (2011). Beta-Adrenergic receptors (beta-AR) regulate VEGF and IL-6 production by divergent pathways in high beta-AR-expressing breast cancer cell lines. Breast Cancer Research and Treatment, 130(3), 747–758.
Sloan, E. K., Priceman, S. J., Cox, B. F., Yu, S., Pimentel, M. A., Tangkanangnukul, V., Arevalo, J. M. G., Morizono, K., Karanikolas, B. D. W., Wu, L., Sood, A. K., & Cole, S. W. (2010). The sympathetic nervous system induces a metastatic switch in primary breast cancer. Cancer Research, 70(18), 7042–7052.
Waight, J. D., Netherby, C., Hensen, M. L., Miller, A., Hu, Q., Liu, S., Bogner, P. N., Farren, M. R., Lee, K. P., Liu, K., & Abrams, S. I. (2013). Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. The Journal of Clinical Investigation, 123(10), 4464–4478.
Gabrilovich, D. I., & Nagaraj, S. (2009). Myeloid-derived suppressor cells as regulators of the immune system. Nature Reviews. Immunology, 9(3), 162–174.
Ostrand-Rosenberg, S., Sinha, P., Beury, D. W., & Clements, V. K. (2012). Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Seminars in Cancer Biology, 22(4), 275–281.
Jin, J., Wang, X., Wang, Q., Guo, X., Cao, J., Zhang, X., Zhu, T., Zhang, D., Wang, W., Wang, J., Shen, B., Gao, X., Shi, Y., & Zhang, J. (2013). Chronic psychological stress induces the accumulation of myeloid-derived suppressor cells in mice. PLoS One, 8(9), e74497.
Ben-Eliyahu, S., et al. (1999). Evidence that stress and surgical interventions promote tumor development by suppressing natural killer cell activity. International Journal of Cancer, 80(6), 880–888.
Benish, M., Bartal, I., Goldfarb, Y., Levi, B., Avraham, R., Raz, A., & Ben-Eliyahu, S. (2008). Perioperative use of beta-blockers and COX-2 inhibitors may improve immune competence and reduce the risk of tumor metastasis. Annals of Surgical Oncology, 15(7), 2042–2052.
Shaashua, L., Shabat-Simon, M., Haldar, R., Matzner, P., Zmora, O., Shabtai, M., Sharon, E., Allweis, T., Barshack, I., Hayman, L., Arevalo, J., Ma, J., Horowitz, M., Cole, S., & Ben-Eliyahu, S. (2017). Perioperative COX-2 and beta-adrenergic blockade improves metastatic biomarkers in breast cancer patients in a phase-II randomized trial. Clinical Cancer Research, 23(16), 4651–4661.
Noonan, D. M., de Lerma Barbaro, A., Vannini, N., Mortara, L., & Albini, A. (2008). Inflammation, inflammatory cells and angiogenesis: decisions and indecisions. Cancer Metastasis Reviews, 27(1), 31–40.
Sheibanie, A. F., Yen, J. H., Khayrullina, T., Emig, F., Zhang, M., Tuma, R., & Ganea, D. (2007). The proinflammatory effect of prostaglandin E2 in experimental inflammatory bowel disease is mediated through the IL-23->IL-17 axis. Journal of Immunology, 178(12), 8138–8147.
Boniface, K., Bak-Jensen, K. S., Li, Y., Blumenschein, W. M., McGeachy, M. J., McClanahan, T. K., McKenzie, B. S., Kastelein, R. A., Cua, D. J., & de Waal Malefyt, R. (2009). Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. The Journal of Experimental Medicine, 206(3), 535–548.
Chizzolini, C., Chicheportiche, R., Alvarez, M., de Rham, C., Roux-Lombard, P., Ferrari-Lacraz, S., & Dayer, J. M. (2008). Prostaglandin E2 synergistically with interleukin-23 favors human Th17 expansion. Blood, 112(9), 3696–3703.
Prescott, S. M., & Fitzpatrick, F. A. (2000). Cyclooxygenase-2 and carcinogenesis. Biochimica et Biophysica Acta, 1470(2), M69–M78.
Oshima, M., Dinchuk, J. E., Kargman, S. L., Oshima, H., Hancock, B., Kwong, E., Trzaskos, J. M., Evans, J. F., & Taketo, M. M. (1996). Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell, 87(5), 803–809.
Obermajer, N., Wong, J. L., Edwards, R. P., Odunsi, K., Moysich, K., & Kalinski, P. (2012). PGE(2)-driven induction and maintenance of cancer-associated myeloid-derived suppressor cells. Immunological Investigations, 41(6–7), 635–657.
Huang, Y., Lichtenberger, L. M., Taylor, M., Bottsford-Miller, J. N., Haemmerle, M., Wagner, M. J., Lyons, Y., Pradeep, S., Hu, W., Previs, R. A., Hansen, J. M., Fang, D., Dorniak, P. L., Filant, J., Dial, E. J., Shen, F., Hatakeyama, H., & Sood, A. K. (2016). Antitumor and antiangiogenic effects of aspirin-PC in ovarian cancer. Molecular Cancer Therapeutics, 15(12), 2894–2904.
Brencicova, E., Jagger, A. L., Evans, H. G., Georgouli, M., Laios, A., Attard Montalto, S., Mehra, G., Spencer, J., Ahmed, A. A., Raju-Kankipati, S., Taams, L. S., & Diebold, S. S. (2017). Interleukin-10 and prostaglandin E2 have complementary but distinct suppressive effects on Toll-like receptor-mediated dendritic cell activation in ovarian carcinoma. PLoS One, 12(4), e0175712.
Yao, C., Sakata, D., Esaki, Y., Li, Y., Matsuoka, T., Kuroiwa, K., Sugimoto, Y., & Narumiya, S. (2009). Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nature Medicine, 15(6), 633–640.
Bomalaski, J. S., Dundee, D., Brophy, L., & Clark, M. A. (1990). Leukotriene B4 modulates phospholipid methylation and chemotaxis in human polymorphonuclear leukocytes. Journal of Leukocyte Biology, 47(1), 1–12.
Haribabu, B., Verghese, M. W., Steeber, D. A., Sellars, D. D., Bock, C. B., & Snyderman, R. (2000). Targeted disruption of the leukotriene B(4) receptor in mice reveals its role in inflammation and platelet-activating factor-induced anaphylaxis. The Journal of Experimental Medicine, 192(3), 433–438.
Islam, S. A., Thomas, S. Y., Hess, C., Medoff, B. D., Means, T. K., Brander, C., Lilly, C. M., Tager, A. M., & Luster, A. D. (2006). The leukotriene B4 lipid chemoattractant receptor BLT1 defines antigen-primed T cells in humans. Blood, 107(2), 444–453.
Woo, C. H., You, H. J., Cho, S. H., Eom, Y. W., Chun, J. S., Yoo, Y. J., & Kim, J. H. (2002). Leukotriene B(4) stimulates Rac-ERK cascade to generate reactive oxygen species that mediates chemotaxis. The Journal of Biological Chemistry, 277(10), 8572–8578.
Henderson Jr., W. R., et al. (1996). The importance of leukotrienes in airway inflammation in a mouse model of asthma. The Journal of Experimental Medicine, 184(4), 1483–1494.
Park, J., Park, S. Y., & Kim, J. H. (2016). Leukotriene B4 receptor-2 contributes to chemoresistance of SK-OV-3 ovarian cancer cells through activation of signal transducer and activator of transcription-3-linked cascade. Biochimica et Biophysica Acta, 1863(2), 236–243.
Wen, Z., Liu, H., Li, M., Li, B., Gao, W., Shao, Q., Fan, B., Zhao, F., Wang, Q., Xie, Q., Yang, Y., Yu, J., & Qu, X. (2015). Increased metabolites of 5-lipoxygenase from hypoxic ovarian cancer cells promote tumor-associated macrophage infiltration. Oncogene, 34(10), 1241–1252.
Fruhbeck, G., et al. (2001). The adipocyte: a model for integration of endocrine and metabolic signaling in energy metabolism regulation. American Journal of Physiology. Endocrinology and Metabolism, 280(6), E827–E847.
Fearon, K. C., Glass, D. J., & Guttridge, D. C. (2012). Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metabolism, 16(2), 153–166.
Funding
This work is supported, in part, by the National Institutes of Health (CA016672, CA109298, CA193249, UH3TR000943, P50 CA217685, P50 CA083639, R35 CA209904), Ovarian Cancer Research Fund, Inc. (Program Project Development Grant), the Blanton-Davis Ovarian Cancer Research Program, the American Cancer Society Research Professor Award, and the Frank McGraw Memorial Chair in Cancer Research (A.K.S.).
Author information
Authors and Affiliations
Corresponding author
Additional information
Sujanitha Umamaheswaran and Santosh K. Dasari are co-first authors
Rights and permissions
About this article

Cite this article
Umamaheswaran, S., Dasari, S.K., Yang, P. et al. Stress, inflammation, and eicosanoids: an emerging perspective. Cancer Metastasis Rev 37, 203–211 (2018). https://doi.org/10.1007/s10555-018-9741-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-018-9741-1