
Abstract
The integrin “very late antigen-4” (VLA-4) is expressed by numerous cells of hematopoietic origin and possesses a key function in the cellular immune response, e.g., by mediating leukocyte tethering, rolling, binding, and finally transmigration of the vascular wall at inflammatory sites. Thus, VLA-4 is a valuable target in medical sciences to interfere with pathological inflammations. In addition, leukemic cells and different solid tumors, which express VLA-4, make use of these adhesive functions and confer VLA-4 a progressive role in the metastatic spread. With a growing insight into the molecular mechanisms for creating a tumor-friendly microenvironment at metastatic sites and various tumor host interactions, the multiple functions of VLA-4 became evident recently, e.g., in leukocyte recruitment to micrometastases, the protection of tumors from immune surveillance, or contribution to a chemoresistance. Nevertheless, despite accumulating evidence for several functions of VLA-4 in tumorigenicity, a therapeutic interference with VLA-4 in cancer sciences has not been developed yet to the clinical level, undoubtedly by a marked impact on the physiological immune response. This review gives an up to date insight into the multiple functional role of VLA-4 in cancer and introduces this integrin as a promising target worthwhile to attract attention in biomedical cancer research.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Molecular background and physiological function of VLA-4
Integrins are cell surface receptors which are expressed by almost all cell types capable to mediate cellular contacts to other cells and to the extracellular matrix (ECM) or constituents of the blood plasma [1–4]. All integrins consist of non-covalently linked α and β subunits. In vertebrates, 18 α and 8 β subunits have been identified, forming 24 α/β pairs [5, 6]. Each integrin subunit consists of a large extracellular domain, a single-spanning transmembrane domain, and a short cytoplasmic tail [7]. Integrins are implicated in cell adhesion and migration, embryogenesis, and several other different biological processes [8–11]. The term “integrin” refers to the ability to integrate contacts with the cell microenvironment to intracellular signals, also known as outside-in signaling. Vice versa, the binding of adaptor molecules or signaling components to the integrin cytoplasmic tail can alter ligand affinity by conformational changes in the integrin structure [7, 12]. Hence, integrins provide cells a general capability to adapt to multiple physiological situations.
The functional classification of integrins into different groups is generally conducted according to the β subunits. Whereas β1 integrins recognize components of the ECM, β2 integrins are expressed by almost all leukocytes, displaying their crucial role in leukocyte trafficking to the sites of inflammation. As an exception, the integrin very late antigen-4 (VLA-4) (α4β1, CD49d/CD29) has a dual role, since this leukocyte integrin mediates not only the recruitment of leukocytes to site of inflammation but also the binding to fibronectin or osteopontin as components of the ECM [13, 14].
VLA-4 was identified by Hemler and colleagues in 1987 together with VLA-3 and VLA-5 [15]. Due to its very late appearance on T cells after activation, the whole family of proteins was termed VLA-very late antigen. Subsequently, VLA-4 was found to be widely distributed on hematopoietic cells like thymocytes, peripheral blood lymphocytes, monocytes, activated T and B lymphoblastoid, and myeloid cell lines [16]. Sequence studies revealed that the α4 subunit consists of 999 amino acids containing three potential cationic binding sites but lacks an inserted I- or A-domain [17, 18]. VLA-4 recognizes LDV-related protein sequences in its ligands, which is comprised in vascular cell adhesion molecule-1 (VCAM-1) or fibronectin, the two best characterized interaction partners [14, 19, 20]. Further ligands for VLA-4 are osteopontin [13], ADAM7 and ADAM28 (a disintegrin and metalloprotease) [21], the LPS receptor CD14 [22], or the Lutheran blood group antigen/basal cell adhesion molecule (BCAM) [23, 24]. VLA-4 binding to mucosal addressin cell adhesion molecule-1 (MadCAM) is described controversial [25–27].
VLA-4 is generally regarded as one of the key integrins mediating tethering, rolling, firm adhesion, and the transendothelial migration of leukocytes to inflamed tissues [28–32]. Furthermore, VLA-4 is involved in immunological synapse formation, phagosome maturation in macrophages, embryogenesis, and angiogenesis in the central nervous system [8, 33–37].
The capacity of VLA-4 to conduct cell tethering or rolling before finally mediating firm adhesion under shear conditions is, in contrast to, e.g., the leukocyte integrin LFA-1 (lymphocyte function-associated antigen-1, αLβ2) [38, 39], regulated by different adhesive states in which VLA-4 is potentially existent [40]. Chigaev et al. described the existence of six different VLA-4 conformational states, varying in ligand affinity, molecular extension, and beta subunit hybrid domain exposure. Ligand binding affinity and molecular extension were apparently regulated independently either by cytoplasmatic Ca2+ concentrations or in a diacylglycerol-dependent manner. Whereas binding affinity primarily accounts for the duration of ligand interaction, molecular unbending contributes to improved capture affinity [18, 41, 42]. The different states of activation are regulated by a multitude of signals, varying from extracellular shear stress [43] to the activation of different G protein-coupled receptors, which can increase or downregulate VLA-4 affinity [44, 45]. Also, a direct binding of monomeric C-reactive protein (mCRP), cysteine-rich protein 61 (Cyr61), or allosteric modulation by the chemokine fractalkine can have an impact on VLA-4 affinity [46–48].
Intracellular, a lot of signaling molecules were identified, which potentially participate in VLA-4 affinity regulation. Besides the phosphatidylinositol-4,5-bisphosphate 3-kinases γ and δ, the Src kinase p56, or B-Raf, also the small GTPase Rap1, seems to interfere in VLA-4 activation [49–53]. The adaptor molecules talin-1, paxillin, and kindlin-3 connecting the cytoskeleton with the VLA-4 subunits were described to regulate adhesion strengthening [54–57].
In addition to the outstanding physiological role of VLA-4,a plenty of excellent reviews provide surveys of the involvement of VLA-4 next to other integrins in diverse pathophysiological situations. This review shall give a broad overview of the implication of the integrin VLA-4 in tumorigenicity and cancer metastasis, a synopsis, which is to the best of our knowledge currently not available.
2 Contribution of tumor cell expressed VLA-4 to metastasis
The interaction of tumor cells (TCs) with blood components and endothelial cells (ECs) is indispensable for a successful TC extravasation into the surrounding parenchyma and finally the establishment of a metastatic niche [58]. First reports described an increase in melanoma EC interaction due to EC activation with inflammatory mediators like TNFα or IL-1, without shedding light on the molecular basis of this phenomenon [59, 60]. Shortly after, VLA-4 was discovered also on leukemia, lymphoma, osteosarcoma, and kidney carcinoma cells, and the process of leukocyte rolling and adhesion could be conferred to circulating TCs utilizing the same panel of adhesion receptors [61–63]. In in vitro studies, endothelial activation by either IL-1, IL-4, bacterial endotoxin, or TNFα resulted in an increased VCAM-1 expression and augmented TC adhesion to ECs [59, 61, 62, 64]. On the other hand, the treatment of melanoma cells either with TNFα or laser radiation augmented α4 and β1 integrin subunit expression which led to an elevated adhesion to collagen and invasion through fibronectin layers [65, 66]. By comparing different melanoma cell lines varying in VLA-4 affinities, Klemke and colleagues revealed that cells which display high-affinity interactions between VLA-4 and endothelial VCAM-1 possess an enhanced transendothelial migration [67].
These investigations convey the first hints that the VLA-4/VCAM-1 interaction crucially contributes to metastasis. Okahara and coworkers conducted the first in vivo experimental conformation by illustrating an enhanced number of metastatic foci in the lungs of mice that received TNFα before B16-BL6 melanoma cell inoculation. Antibodies against VLA-4 or VCAM-1 alleviated the number of lung metastases [68]. Further studies revealed an enhancement of lung metastasis after IL-1 application or TNFα release due to surgical stress, which elevated VCAM-1 expression on the lung epithelium [69, 70].
Cardones et al. demonstrated an upregulation of VLA-4 affinity on melanoma cells by CXCR4 activation, which increased melanoma cell adhesion and the number of lesions in the lungs of mice [71]. Recent data from Rebhun imply that VLA-4 expressing B16F1 melanoma cells metastasize to lymphatic tissues due to constitutively expressed VCAM-1 on the lymphatic endothelial cells [72]. All these studies embrace the given fact of a constitutive VCAM-1 expression at the site of metastasis offering a hospitable microenvironment to the TCs, which is triggered by inflammatory mediators (Fig. 1).

Interaction of VLA-4 expressing TCs with endothelial VCAM-1. TCs entering the blood are immediately surrounded by platelets and different leukocytes. This clot can become stuck in tight vessels and finally enable the TCs to transmigrate in the surrounding tissue to establish metastatic foci. TCs expressing VLA-4 have the additional opportunity to adhere to endothelial expressed VCAM-1 and subsequently leave the blood flow in a VLA-4/VCAM-1 facilitated manner
Nonetheless, under pathophysiological conditions, an endothelial VCAM-1 expression can also be induced by TC host cell contacts [73–75] or reduced shear stress [76], whereas high shear rates have anti-inflammatory effects [77–79]. After VLA-4 mediated TC adhesion to the endothelium, TCs are able to extravasate and proliferate in the adjacent tissue.
In a model of spontaneous melanoma metastasis, which is quite closer to the pathological situation than the common experimental metastasis assays, Quian et al. detected an inverse correlation between VLA-4 expression and the number of metastatic foci in the lungs. A high VLA-4 expression on B16F10 cells strongly reduced lung metastases formation and Matrigel invasion [80]. Further studies provided molecular explanations for these findings illustrating homophilic interactions between VLA-4 molecules, which generated VLA-4-mediated cell aggregation and reduced metalloproteinase secretion [81–87]. Hence, single cells in a tumor with high VLA-4 density are hindered to leave the tumor network and emigrate to different organs. These results are supported by Hart and Schadendorf, investigating the VLA-4 expression of melanocyte strains, naevi, primary malignant melanoma, and melanoma metastases. They found a significant increase in VLA-4 expression on primary melanomas which correlated with the development of metastases. Also, a negative association of VLA-4 expression and disease-free periods were feasible. In comparison to the VLA-4 increase on melanoma metastases, a decrease in VLA-1, VLA-2, and VLA-6 was detectable [81, 88–91].
Concluding, on primary/early melanoma lesions, VLA-4 seems to keep the cells together, whereas in a later phase, VLA-4 is able to support steps of the metastatic cascade by interaction with endothelial VCAM-1. Nonetheless, the fact that also VLA-4 negative cells can be highly metastatic shows the existence of different, abundant pathways mediating the critical steps of tumor cell adhesion to and transmigration through the endothelium.
3 How VLA-4 expressed by leukocytes contributes to cancer metastasis
The VLA-4 expression by TCs confers them the ability of vascular adhesion and transmigration out of the blood flow, as outlined above. Besides VLA-4 on TCs, VLA-4 expressed by myeloid cells contributes to tumorigenicity and metastasis in different manners which shall be reconsidered in this chapter.
The VLA-4 counterreceptor VCAM-1, which is usually expressed on inflamed endothelial cells, is also aberrantly expressed by several cancer cells, e.g., breast, renal, malignant mesothelioma cells, or gastric carcinomas [92–96]. Thus, VCAM-1 belongs, e.g., to the site-specific metastasis gene signature for breast cancer [97–99], since the aberrant VCAM-1 expression confers several advantages to the breast cancer cells either metastasizing to the lungs or the bones. In the lung tissue, the TCs interact with VLA-4 positive tumor-associated macrophages, which leads to a VCAM-1 clustering and subsequently a phosphorylation and binding of the adaptor molecule Ezrin to the VCAM-1 cytoplasmic tail. Ezrin in turn activates phosphoinositide-3-kinase (PI3K) finally leading to an Akt (protein kinase B) activation, which conveys strong pro-survival signals to the TCs [92, 100]. In the bone marrow, indolent breast cancer micrometastases awake from dormancy and attract VLA-4 positive osteoclast progenitor cells by the novel secretion of VCAM-1 in a NF-κB-driven manner [100–102]. The osteoclast progenitor cells adhere to the breast cancer cells in a VLA-4 dependent fashion, mature to osteoclasts, and resorb the bone matrix, which leads to a discharge of growth factors. These growth factors in turn activate the TCs to support osteoclast progenitor maturation. Hence, the VLA-4/VCAM-1 interaction incites the vicious cycle of breast cancer bone metastasis and osteolysis.
Wu and coworkers proposed to study a TC immune escape and created a human papillomavirus-16 E7-expressing cancer cell line and a mouse vaccine containing E7 [93, 103–105]. Mice vaccinated with E7 showed a reduced tumor growth and an augmented number of CD4+ and CD8+ T cells. Finally, TCs were isolated, which exhibited a complete resistance against vaccine-induced immune response. Gene expression analysis revealed a strong VCAM-1 upregulation in resistant cells compared to susceptible TCs. Due to the fact that CD8+ cells express high levels of VLA-4, the authors proposed that CD8+ T cells migrate away from the TCs in a VLA-4 dependent mode, and the contact between TC MHC receptors and T cell receptors is alleviated [93, 105]. It is a well-known fact that VLA-4 can foster cell migration either directly or via the transregulation of the integrin αLβ2 [32, 106–109].
Next to the direct interaction between VLA-4 positive host cells with VCAM-1 expressing TCs, myeloid cells are crucially involved in the creation of permissive metastatic niches. Lyden et al. impressively demonstrated that Vascular Endothelial Growth Factor Receptor-1 (VEGFR1)- and VLA-4 positive hematopoietic bone marrow-derived cells (BMDCs) instigate a pre-metastatic niche [110]. Mice were implanted with either Lewis lung carcinoma (LLC) or B16 melanoma cells, and by day 14, before a metastasis of the tumors occurred, BMDC clusters appeared in the lungs to prepare a pre-metastatic surrounding (Fig. 2). Also, the administration of melanoma-conditioned media mobilized immature BMDCs from the bone marrow, which created a conducive microenvironment in the lungs for incoming tumor cells. The sites of BMDC clusters were tumor-type specific, LLC cells dictated cluster formation mainly to the lungs and liver, whereas B16 melanoma cells induced clusters in the liver, testis, spleen, and kidney. Following TC implantation, but before BMDC arrival, an augmented fibronectin expression by fibroblast-like stromal cells in adjacency to the future metastatic lesion was detectable (from day 3 on), which supported VLA-4+-BMDCs adhesion and cluster formation. For successful TC engraftment in the pre-metastatic niche, a matrix metalloproteinase 9 and SDF-1 (CXCL12) expression was observed in a completely formed cluster, which may attract CXCR4+ TCs to the pre-metastatic niche. Hence, an aggregation of VLA-4 and VEGFR1-positive hematopoietic bone marrow progenitor cells can allude to a pre-metastatic nodule.

Involvement of VLA-4 expressing myeloid cells in the establishment of a metastatic niche. A primary tumor secretes soluble factors, which stimulate fibroblast-like stromal cells to express fibronectin. Bone marrow-derived cells adhere to fibronectin via VLA-4, create a cluster and thereby a conducive microenvironment for TCs arriving in the pre-metastatic niche posterior (1). Cells of the myeloid lineage are also recruited to the early metastatic nodule in a VCAM-1- and VAP-1-dependent manner providing a permissive metastatic environment (2). In a major tumor, an abundance of myeloid cells is found within the tumor tissue comprising several pro-tumorigenic effects like immune suppression. VLA-4 is crucially involved in the attraction of different myeloid cells to the tumor vasculature (3) and is also entangled in the transition of M1 macrophages to a M2 phenotype (4)
Besides CXCR4, also CCR7 is a chemokine receptor often expressed on TCs, navigating the way to the organ of final destination [111–114]. Recently, also CCL2 (Monocyte Chemoattractant Protein-1, MCP-1) was associated with cancer metastasis, recruiting myeloid cells to the nascent metastatic nodules [115, 116]. Colon cancer cells, which metastasize to the liver, recruited a distinct myeloid subset of cells (CD11b/Gr1mid) 14 days after TC inoculation to hepatic metastatic foci by the secretion of CCL2, which subsequently supported TC survival and proliferation [117, 118]. Similar observations were made for breast cancer cells engrafting in lung tissue, which attracted inflammatory monocytes and afterwards macrophages in a CCL2-dependent manner [119, 120]. Macrophage depletion inhibited TC pulmonary seeding and growth. Muschel et al. revealed a tissue factor-dependent clot formation of melanoma and breast cancer cells in the lungs of mice. This clot immediately activated the TC adjacent ECs to an elevated VCAM-1 and vascular adhesion protein-1 (VAP-1) expression [121, 122]. The subsequent myeloid cell recruitment was crucially related to VCAM-1 and VAP-1 expression in TC vicinity, referring to a strict VLA-4 dependency of the recruiting mechanism.
Thus, also shortly after TC engraftment in the lungs, the distinct populations of myeloid cells/macrophages participate in the creation of a supportive niche indispensable for early steps of metastasis.
In the later stage of cancer progression, an abundance of different cells of the myeloid lineage are detectable within tumors and metastatic lesions, supporting angiogenesis and tumor growth and mediate immune suppression in the tumor microenvironment [123–126]. The mechanisms, how myeloid cells traffic into the tumor microenvironment, have not been elucidated for a long time. Varner confirmed that VLA-4 is the key integrin for recruiting monocytes to tumors and thereby acts as an essential factor for tumor angiogenesis. The blocking of VLA-4 reduced the number of macrophages in Lewis lung carcinoma tumors and colon carcinomas [127]. Later on, the γ-subtype of PI3K was shown to activate VLA-4 on myeloid cells in a Pap1a-, RIAM-, talin-, and paxillin-dependent fashion, which is a prerequisite for myeloid cell trafficking to tumor microenvironment [50].
Durden and colleagues could identify a unique role of VLA-4 on tumor-associated macrophages in the transition of macrophages from M1 to a M2 metastatic phenotype. VLA-4 transmits signals from the ECM to activate PI3K, which subsequently activates the small GTPase Rac2. Rac2 in turn initiates the M1 to M2 phenotype shift (Fig. 2, right). VLA-4 with a point mutation in the α4 cytoplasmic tail had reduced levels of Rac2-GTP levels and mice with this mutation exhibited decreased tumor growth [128].
Concluding, the direct interaction between aberrantly VCAM-1 expressing TCs and VLA-4 positive host cells can provide proliferative and pro-survival signals to the TCs and additionally mediate a hospitable environment for the growing tumor in the bone tissue. Beside this, cells of the myeloid lineage are recruited in a VLA-4 dependent mode by the primary tumor to the potential sites of metastasis, when TCs have not yet arrived, and generate a permissive niche for incoming TCs. Also, in the very early phase of the development of metastatic foci, TCs recruit myeloid cells in CCL2 and VCAM-1-related manner which facilitates TC proliferation and survival. Once the metastatic foci are grown, an abundance of myeloid cells is detectable in the tumor tissue. Here, VLA-4 is involved in the macrophage recruitment and shift from a M1 to an immunosuppressive M2 type. Thus, VLA-4 positive myeloid cells conduct pro-tumorigenic signals in nearly every stadium of cancer progression.
4 Role of VLA-4 in angiogenesis and lymphangiogenesis
Tumors exceeding a size bigger than 1–2 mm3 are obliged to obtain access to the vascular system to ensure the continuous supply of oxygen and nutrition [129]. In 1939, the first pictures of tumor implant neovascularization were recorded in the Sandison-Clark rabbit ear chamber assay by Ide and colleagues [130]. Later on, the angiogenic processes of wound healing and tumor vascularization were described in more detail and differences thereof could be delineated [131]. Folkman et al. finally inaugurated the hypothesis that “tumor angiogenesis factors” are mitogenic to capillary endothelial cells and proposed angiogenesis as a target to inhibit the growth of solid tumors [129, 132, 133]. The implication of integrins in angiogenesis and tumor angiogenesis has been subject of intensive research and is reviewed in several brilliant articles [134, 135]. VLA-4 contributes to tumor angiogenesis in three different modes. This multitude of capacities is unique within the class of integrins.
First, VLA-4 is expressed on bone marrow-derived progenitor cells/stem cells and promotes progenitor cell homing to the tumor periphery (Fig. 3). Although the existence of endothelial precursor subpopulations in bone marrow-derived cells has for a long time not been recognized, recent studies have identified a common Lin−/Sca1+ endothelial progenitor cell lineage which is capable to differentiate into all hematopoietic cell lineages as well as ECs [136–140]. After progenitor cell homing to VCAM-1 or fibronectin in tumor periphery in a VLA-4 dependent fashion, the progenitor cells can differentiate into endothelial cells, thereby participating in angiogenesis. Other integrins or Lin+ progenitor cells did not contribute to vessel formation [141, 142]. Up to 16 % of tumor neovasculature can originate from bone marrow-derived cells, as Ruzinova revealed in 2003 [143].

Contribution of VLA-4 to angiogenesis. VLA-4 expressing endothelial progenitor cells bind to VCAM-1 and fibronectin in the tumor periphery, differentiate into endothelial cells, and are involved in the formation of new blood vessels in the tumor (1). Furthermore, monocytes or myeloid cells enter the tumor with the contribution of VLA-4 and promote angiogenesis, immune suppression, and finally TC survival (2). VLA-4 is upregulated on proliferating endothelial cells, while VCAM-1 is expressed on pericytes surrounding newly formed vessels, but not on quiescent ones (3). Consequently, the VLA-4/VCAM-1 interplay contributes to a stable embedding of the blood vessels into the tissues
Second, monocytes or myeloid cells in general are recruited in numerousness into tumor tissue and are capable to emit either angiogenic or angiostatic signals [123, 144]. The finding that various myeloid cell types like hemangiocytes [110, 139, 145], monocytes [115, 146], TIE2-expressing monocytes [147, 148], immature dendritic cells [149], myeloid-derived suppressor cells [125, 150], neutrophils [124, 151], eosinophils [152], or immature mast cells [153] are present in tumor tissue is well-known and extensively reviewed elsewhere [123]. Since myeloid cells express several different integrins as well as other adhesion receptors, it has long time been unclear how these cells might enter the tumor microenvironment from the blood circulation. Varner and coworkers finally revealed VLA-4 as the key integrin responsible for CD11b+/CD14+ monocytes trafficking into tumor tissue and promoting angiogenesis [127]. The authors propose VCAM-1 and CS-1 fibronectin as potential ligands for monocytes in the tumor tissue, notwithstanding other groups could show that angiogenic tumor vessels are in a state of anergy with reduced VCAM-1 and ICAM-1 expression due to VEGF and bFGF stimulation [154–158]. Hence, CS-1 fibronectin seems presumably to be the VLA-4 ligand in the leaky tumor vessel sprouts. The implication of VLA-4 in the recruitment of myeloid cells to solid tumors and their implication in angiogenesis should not be confounded with the recruitment of CD11b/Gr1mid myeloid cells to the nascent metastatic niche mentioned in the previous section.
In a third fashion, VLA-4 expressed on proliferating ECs participates in blood vessel formation. For a proper neovascularization of healthy as well as tumor tissue, the interaction between ECs and mural cells seems to play a fundamental role [159, 160]. Several studies proposed an enmeshment of VLA-4 and VCAM-1 in this reciprocity, but the studies’ foci were more related to soluble VCAM-1 and its chemotactic activity toward ECs under inflammatory conditions [161–164]. Varner highlighted a VLA-4 expression on proliferating but not on mature ECs due to VEGF-A stimulation [165]. VCAM-1 on the contrary was expressed by pericytes surrounding the ECs of neovessels in human breast carcinomas but not on pericytes of quiescent vessels. VLA-4 and VCAM-1 mediated the adhesion between ECs and pericytes in vitro and in vivo and a blockade of VLA-4 induced EC and pericyte apoptosis. Thus, EC-pericyte interactions induced by VLA-4 and VCAM-1 apparently facilitate the survival of both cell types during angiogenesis. The inhibition of this interaction reduced angiogenesis and tumor growth in vivo [165].
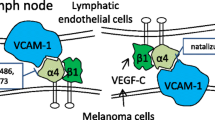
The investigation of lymphangiogenesis has long time been difficult due to the lack of distinct markers for lymphatic vessels. Since 1999, the specific lymphatic vessels markers Lyve-1 (a member of the CD44 family of proteins), Prox-1 (a transcription factor), podoplanin (a membrane-based glycoprotein), and CD31 were identified and opened the field for detailed investigations [166–169]. Several integrins have been implicated in the development of the lymphatic system or lymphangiogenesis under inflammatory conditions. Integrin α9β1 is required for a functional lymphatic development and lymphatic valve morphogenesis—mice deficient in α9β1 frequently die from chylothorax—but α9β1 is also important for VEGF-C-mediated lymphatic endothelial cell (LEC) motility, a process which is crucial for proper lymphangiogenesis [170–172]. Integrins α1β1, α2β1, and α5β1 have been investigated in their functional context of lymphangiogenesis of healing wounds and cornea inflammation [173–175]. VLA-4 in contrast was identified as a marker for proliferating LECs, whereas it is poorly expressed in normal lymphatic vessels. A strong VLA-4 upregulation could be detected in growing lymphatic vessels in tumor bearing mice or in mice treated with VEGF-C. Blocking VLA-4 either by mAbs, soluble VCAM-1, or knockdown approaches lead to an attenuated LEC migration and survival, mitigated adhesion to VCAM-1, reduced vessel branch point formation, and finally weakened lymphangiogenesis. Mice with VLA-4 knock-in mutations, in which VLA-4 is incapable to associate with intracellular paxillin and talin, revealed reduced lymphangiogenesis after tumor inoculations compared to wild-type mice [134, 135, 176, 177]. This confirms that VLA-4 signaling and not only VLA-4 expression is necessary for lymphangiogenesis. Later on, Varner and colleagues demonstrated that VEGF-C stimulates the PI3Kα/Akt signaling pathway in LEC which in turn activates VLA-4 to conduct lymphangiogenesis [178, 179]. Independent from lymphangiogenesis, the abundantly VLA-4 equipped lymphatic microenvironment serves as a harbor and site of accumulation for VCAM-1 expressing tumor cells [92, 100, 101]. Thus, VLA-4 contributes to metastasis via the lymphatic system in two different ways: fostering lymphangiogenesis and offering a harbor for VCAM-1 positive TCs.
Rebhun et al. in contrast detected an enhanced VCAM-1 expression on lymphatic vessels of melanoma bearing mice and could show that VLA-4 expressing B16 cells have a high affinity to these LECs. B16 cells with decreased VLA-4 expression displayed a reduced binding to LECs. The in vivo situation confirmed that cells with high VLA-4 levels share a higher tendency to establish metastatic foci in the lymph nodes than cell populations with poor VLA-4 expression [72]. These results have to be regarded carefully due to the fact that not only VLA-4 mediates cell adhesion but it is also capable to confer pro-survival signals in cells [180]. Accordingly, low VLA-4 surface levels could finally transmit pro-apoptotic signals in comparison to cells with upregulated VLA-4. Hence, the conclusion that VLA-4 fosters lymphatic metastasis merely by cell adhesion is possibly impermissible or too constricted.
5 Fusion of tumor cells with endothelial cells due to VLA-4 and VCAM-1 interaction
Fusion between cells of different entities is a well-known fact involved in reproduction and tissue formation and has also been used since the 1970s for the production of monoclonal antibodies [181].
Inflammatory conditions seem to be helpful to enhance heterotypic cell fusion [182–184]. In 2004, Mortensen et al. investigated the interaction between endothelial cells and breast cancer cells and observed a fusion of both to new hybrid cells, sharing properties of both parents [185]. Song and colleagues could reveal that the fusion between oral squamous cell carcinoma cells and ECs (HUVEC) depends on a VCAM-1/VLA-4 interaction. The activation of endothelial cells with TNFα augmented VCAM-1 expression and facilitated the subsequent fusion with VLA-4-positive carcinoma cells, whereas a blockade of VCAM-1 or VLA-4 with mAbs reduced cell fusion. These findings assign VCAM-1 a completely new mechanism in tumor cell fusion and metastasis [186].
6 VLA-4 mediated effects on chemoresistance
The development of chemoresistance is one of the worst complications in cancer treatment. Despite a primary response to chemotherapy, cancer patients finally become unresponsive to a broad quantity of cytostatic drugs. One mechanism of chemoresistance is cell adhesion-mediated drug resistance (CAM-DR) first described by Durand and Sutherland, who investigated irradiation effects on single Chinese hamster cells compared to agglomerates. They exhibited an enhanced repair of sublethal radiation damages in cells grown and irradiated in agglomerates [187]. Since that time, a lot of studies have been conducted revealing that integrin-dependent cell-cell or cell-ECM contacts mediate survival signals to various types of cancer cells and thus induce multiple resistance phenomena [188–192].
A small population of TCs surviving the initial chemotherapy, called minimal residual disease (MRD), are the predominant obstacle for a successful cancer treatment and a long-term survival [193, 194]. CAM-DR seems to account partially for MRD, and especially, VLA-4 appears to be involved in CAM-DR in multiple myeloma, lung, breast, and ovarian cancer [195–201]. Whereas in first studies, the transient VLA-4-mediated ECM binding and shielding effects in the matrix have been exposed correlating increased VLA-4 expression levels with a drug-resistant phenotype [196, 202, 203]; later studies were dedicated to reveal the signaling pathways in TCs leading to survival during chemotherapeutic drug application. Most findings are exemplarily summarized in Fig. 4.

VLA-4 promotes TC chemoresistance and survival. A plenty of mechanisms in different cells have been described by which VLA-4 maintains chemoprotection and survival. For blood-borne tumors like AML or ALL cells, a VLA-4-dependent interaction with bone marrow stromal cells confers chemoresistance to TCs by NF-κB activation in the stromal cells and subsequent secretion of several signaling molecules (IL8, IL6, and CCL2). A VLA-4-mediated upregulation of Bcl-2 and Bcl-XL in AML cells was responsible for an augmented survival when TC were cocultured with stromal cells. In fibroblasts, a VLA-4 ligation to fibronectin in turn phosphorylated FAK and led to a p53 suppression. In Jurkat cells, the α4 cytoplasmic tail was revealed to foster extracellular Ca2+ influx via L-type Ca2+ channels and doxorubicin efflux apparently responsible for the observed chemoresistance. A redox modulation of adjacent thiols in the exofacial domain of VLA-4 in AML cells by a tellurium compound restored chemosensitivity by mitigated Akt phosphorylation
Matsunaga et al. elucidated the phosphatidylinositol-3-kinase (PI-3K)/Akt/Bcl-2 pathway to assign resistance to apoptosis to cytarabine [195]. St Croix detected an upregulation of the cyclin-dependent kinase inhibitor p27Kip1 due to the three-dimensional culturing of human and mouse carcinoma cell lines [194, 204]. The involvement of a focal adhesion kinase (FAK) phosphorylation upon VLA-4 activation in the suppression of apoptosis is described controversially and not yet completely clarified. Fukai among others reported on an anti-apoptotic effect of phosphorylated FAK by Bcl-2 upregulation and p53 suppression [205, 206], whereas Damiano et al. could not detect an augmented tyrosine FAK phosphorylation due to fibronectin binding in a panel of myeloma cell lines [207]. Higashimoto unveiled a downregulation of Fas death receptor ligand by the adhesion of eosinophilic cells to fibronectin [208]. Further investigators focused on the Bcl-2 family of proteins involved in cell survival due to fibronectin ligation. De la Fuente detected an increased Bcl-2/Bax ratio in chronic lymphocytic leukemia cells [209], whereas Damiano could not prove changes in the expression of either pro- (Bcl-w, Mcl-1, Bik, Bak, Bad) or anti-apoptotic (Bcl-2, Bcl-XL) proteins in myeloma cells [196, 207]. Konopleva revealed an upregulation of Bcl-2 and Bcl-XL and an augmented survival in acute myeloid leukemia (AML) cells due to coculturing with stromal cells (Fig. 4) [210]. Further potential mechanisms leading to anti-apoptotic effects due to integrin ligation are reviewed excellently elsewhere [199].
Recently, Liu and coworkers created Jurkat cells which were originally deficient for the α4 VLA-4 chain and inserted a truncated α4 cytoplasmic motif connected to a carrier epitope in these cells. Cells carrying this membrane-proximal motif exhibited an adhesion-independent resistance, which did not require the β1-integrin as heterodimeric pair. Concomitantly, the authors detected an increased influx of extracellular Ca2+ by L-type Ca2+ channels, while a blockade of these channels restored chemosensitivity. Furthermore, these cells displayed similar levels of phosphorylated Akt like cells with wt α4 and also a binding of calreticulin to the inserted peptide motif was unchanged [211]. A resulting enhanced doxorubicin efflux out of the cells could serve as a functional reason for chemoresistance.
Layani-Bazar introduced a nontoxic tellurium compound, which restored chemosensitivity in AML cells by the redox modulation of adjacent thiols in the exofacial domain of VLA-4 [212]. Due to the treatment of AML cells with the tellurium compound, a decreased Akt phosphorylation after fibronectin ligation was exhibited. In contrast to the former mentioned study, the extracellular part of VLA-4 appears to be the key for an augmented chemoresistance.
Jacamo and colleagues could unveil a VLA-4/VCAM-1-dependent cross-talk between acute lymphoblastic leukemia (ALL) or AML cells with bone marrow mesenchymal stromal cells conferring a protection from chemotherapy-induced apoptosis [213]. Instead of focussing on alterations in the ALL or AML cells due to stromal cell interactions, they aimed on changes in bone marrow mesenchymal stromal cells and found a reciprocal NF-κB activation playing a pivotal role in chemoresistance. The gene expression analysis of stromal cells revealed an upregulation of VCAM-1, IL8, IL6, and CCL2 due to NF-κB nuclear translocation, whereas a NF-κB blockade reduced leukemia burden in vivo. The authors suggested a combination of soluble factor-mediated signals (IL1) and cell adhesion-mediated signals via the VLA-4/VCAM-1 axis as the key for ALL or AML cells to surmount chemotherapy.
Concluding, the VLA-4 ligation to fibronectin or VCAM-1 seems to confer chemoresistance to multiple tumor entities, whereas the exact mechanisms and molecular segments responsible for the observed sensitivity are far from being clear.
The application of VLA-4 inhibitors together with blunted cytostatic drugs could be an option to efficiently circumvent microenvironment-mediated chemoresistance and multidrug resistance.
7 Implications for a therapeutic VLA-4 targeting
VLA-4 is actively involved in various processes of cancer development and metastasis at different stages of disease. Consequently, VLA-4 appears as an attractive target for therapeutic approaches, or in term of diagnostic information. Concerning diagnosis, the VLA-4 subunit CD49d has been identified as a prognostic marker in chronic lymphocytic leukemia whereupon high CD49d expression correlated with significantly shorter treatment-free and overall patients’ survival than low CD49d levels [214–216].
However, concerning VLA-4-related treatment regimes, the utilization of VLA-4 inhibitors has not yet been established in clinical cancer practice. To pre-estimate the potential usefulness of therapeutic interventions aiming on VLA-4, one should take into account that VLA-4 is a versatile receptor on different hematopoietic cells contributing to the immunological balance. The blocking of VLA-4 induces immunosuppression which is an approved clinical principle for the treatment of chronic pathological inflammations using natalizumab, a mAb targeting VLA-4 in the course of relapsing- remitting multiple sclerosis and inflammatory bowel disease [217–220]. In several studies, natalizumab reduced the rate of clinical relapse in patients with multiple sclerosis and the risk of progression of disability [221, 222]. The impact on the immune system by this treatment became evident by serious adverse effects, e.g., the rare event of a multifocal leukoencephalopathy, a progressive and usually fatal complication by the activation of the polyoma JC virus in the immunosuppressed organisms [223]. However, considering a risk stratification for multifocal leukoencephalopathy onset upon natalizumab therapy, this antibody is a worthwhile contribution to therapy and confirms the clinical value of targeting VLA-4 [224, 225]. The impact of natalizumab on the immune status of a patient might also induce adverse effects for cancer development. In some subjects treated with natalizumab, a transformation of nevi to melanomas has been observed, which is in line with the findings that VLA-4 promotes homotypic interactions between melanoma cells, and a blockade of this interplay fosters melanoma spread and progression [80, 83, 226, 227].
Despite the nonexistence of clinical experience with VLA-4 inhibition for cancer treatment, a number of reports exist in the preclinical field, which confirms that an interference with VLA-4 functionality is beneficial to attenuate different processes of cancer development or growth. In a multiple myeloma mouse model, natalizumab or another anti-VLA-4 mAb, respectively, reduced multiple myeloma growth, VEGF secretion, angiogenesis, myeloma cell burden in blood and spleen, and finally prevented osteolysis of the bone marrow [197, 228, 229]. A clinical study on the efficiency of natalizumab in subjects with relapsed or refractory multiple myeloma has been terminated [230].
A recent report by Scalici et al. demonstrated that a VLA-4 blocking antibody is able to overcome the carboplatin resistance of ovarian cancer when co-administered with the cytostatic drug in a mouse model [201]. A VLA-4 blockade, next to other integrins involved in the CAM-DR phenomenon, appears as an extremely promising and upcoming field to foster the efficiency of a chemotherapeutic treatment.
The inhibition of VLA-4, e.g., by antibodies, has been reported in earlier studies of this field to directly affect the metastatic spread of melanoma cells in mouse models, obviously by interference with the melanoma/EC binding [68–71].
Beside VLA-4 antibodies, a multitude of small-molecule VLA-4 antagonists has been developed and partly included in clinical trials aiming on the therapy of asthma, multiple sclerosis, inflammatory bowel disease, and stem cell mobilization in combination with CXCR4 antagonists and G-CSF [231, 232]. Small-molecule inhibitors can roughly be classified in phenylalanine-based and urea-based structures [232–234]. One of these small-molecule inhibitors has been applied to demonstrate the use of VLA-4 inhibition, together with IL-1 and SDF-1 blockade in terms of reduced accumulation of tumor-associated macrophages to interfere with the promotion of angiogenic effects [235].
Heparin and low molecular weight heparin (LMWH) have been reported as potential inhibitors of VLA-4 functionality. A recent report demonstrated that the experimental metastasis of B16F10 melanoma cells in mice is strongly attenuated by a VLA-4 blocking function of a LMWH [236]. Therefore, the multiple findings on antimetastatic efficiency of heparin can, in dependence on tumor entity and model, partly be attributed to an impact on the various VLA-4 effects.
Concluding, the application of VLA-4 inhibitors in the course of cancer disease seems very plausible due to the general participation of VLA-4 in metastasis and cancer progression. However, cancer patients are routinely treated with cytostatic drugs, but this is frequently accompanied by a therapy limiting immune suppression. Thus, a further inhibition of the immune surveillance by blocking VLA-4 has to be regarded critically for a potential therapeutic approach. Nevertheless, the promises of targeting VLA-4 to sensitively affect certain processes in cancer development are worthwhile to consider in clinics, and the perspectives will further increase with growing insights into the underlying molecular mechanisms.
References
Hemler, M. E. (1990). VLA proteins in the integrin family: structures, functions, and their role on leukocytes. Annual Review of Immunology, 8, 365–400.
Kinashi, T. (2005). Intracellular signalling controlling integrin activation in lymphocytes. Nature Reviews Immunology, 5(7), 546–559.
Phillips, D. R., Fitzgerald, L. A., Charo, I. F., & Parise, L. V. (1987). The platelet membrane glycoprotein IIb/IIIa complex. Structure, function, and relationship to adhesive protein receptors in nucleated cells. Annals of the New York Academy of Sciences, 509, 177–187.
Phillips, D. R., Charo, I. F., Parise, L. V., & Fitzgerald, L. A. (1988). The platelet membrane glycoprotein IIb-IIIa complex. Blood, 71(4), 831–843.
Byron, A., Humphries, J. D., Craig, S. E., Knight, D., & Humphries, M. J. (2012). Proteomic analysis of α4β1 integrin adhesion complexes reveals α-subunit-dependent protein recruitment. Proteomics, 12(13), 2107–2114.
Humphries, J. D., Byron, A., & Humphries, M. J. (2006). Integrin ligands at a glance. Journal of Cell Science, 119(Pt 19), 3901–3903.
Luo, B.-H., & Springer, T. A. (2006). Integrin structures and conformational signaling. Current Opinion in Cell Biology, 18(5), 579–586.
Takada, Y., Strominger, J. L., & Hemler, M. E. (1987). The very late antigen family of heterodimers is part of a superfamily of molecules involved in adhesion and embryogenesis. Proceedings of the National Academy of Sciences of the United States of America, 84(10), 3239–3243.
Takada, Y., Ye, X., & Simon, S. (2007). The integrins. Genome Biology, 8(5), 215.
Hynes, R. O. (2002). Integrins: bidirectional, allosteric signaling machines. Cell, 110(6), 673–687.
Yang, J. T., Rayburn, H., & Hynes, R. O. (1995). Cell adhesion events mediated by alpha 4 integrins are essential in placental and cardiac development. Development (Cambridge, England), 121(2), 549–560.
Luo, B.-H., Carman, C. V., & Springer, T. A. (2007). Structural basis of integrin regulation and signaling. Annual Review of Immunology, 25, 619–647.
Bayless, K. J., Meininger, G. A., Scholtz, J. M., & Davis, G. E. (1998). Osteopontin is a ligand for the alpha4beta1 integrin. Journal of Cell Science, 111(Pt 9), 1165–1174.
Clements, J. M., Newham, P., Shepherd, M., Gilbert, R., Dudgeon, T. J., Needham, L. A., & Humphries, M. J. (1994). Identification of a key integrin-binding sequence in VCAM-1 homologous to the LDV active site in fibronectin. Journal of Cell Science, 107(Pt 8), 2127–2135.
Hemler, M. E., Huang, C., & Schwarz, L. (1987). The VLA protein family. Characterization of five distinct cell surface heterodimers each with a common 130,000 molecular weight beta subunit. The Journal of Biological Chemistry, 262(7), 3300–3309.
Hemler, M. E., Huang, C., Takada, Y., Schwarz, L., Strominger, J. L., & Clabby, M. L. (1987). Characterization of the cell surface heterodimer VLA-4 and related peptides. The Journal of Biological Chemistry, 262(24), 11478–11485.
Takada, Y., Elices, M. J., Crouse, C., & Hemler, M. E. (1989). The primary structure of the alpha 4 subunit of VLA-4: homology to other integrins and a possible cell-cell adhesion function. The EMBO Journal, 8(5), 1361–1368.
Chigaev, A., & Sklar, L. A. (2012). Aspects of VLA-4 and LFA-1 regulation that may contribute to rolling and firm adhesion. Frontiers in Immunology, 3, 242.
Elices, M. J., Osborn, L., Takada, Y., Crouse, C., Luhowskyj, S., Hemler, M. E., & Lobb, R. R. (1990). VCAM-1 on activated endothelium interacts with the leukocyte integrin VLA-4 at a site distinct from the VLA-4/fibronectin binding site. Cell, 60(4), 577–584.
Shimizu, Y., van Seventer, G. A., Horgan, K. J., & Shaw, S. (1990). Costimulation of proliferative responses of resting CD4+ T cells by the interaction of VLA-4 and VLA-5 with fibronectin or VLA-6 with laminin. Journal of Immunology, 145(1), 59–67.
Bridges, L. C., Sheppard, D., & Bowditch, R. D. (2005). ADAM disintegrin-like domain recognition by the lymphocyte integrins alpha4beta1 and alpha4beta7. The Biochemical Journal, 387(Pt 1), 101–108.
Humphries, J. D., & Humphries, M. J. (2007). CD14 is a ligand for the integrin alpha4beta1. FEBS Letters, 581(4), 757–763.
El Nemer, W., Wautier, M.-P., Rahuel, C., Gane, P., Hermand, P., Galactéros, F., & Le Van Kim, C. (2007). Endothelial Lu/BCAM glycoproteins are novel ligands for red blood cell alpha4beta1 integrin: role in adhesion of sickle red blood cells to endothelial cells. Blood, 109(8), 3544–3551.
Huang, J., Filipe, A., Rahuel, C., Bonnin, P., Mesnard, L., Guérin, C., & Tharaux, P.-L. (2014). Lutheran/basal cell adhesion molecule accelerates progression of crescentic glomerulonephritis in mice. Kidney International, 85(5), 1123–1136.
Yang, Y., Harrison, J. E., Print, C. G., Lehnert, K., Sammar, M., Lazarovits, A., & Krissansen, G. W. (1996). Interaction of monocytoid cells with the mucosal addressin MAdCAM-1 via the integrins VLA-4 and LPAM-1. Immunology and Cell Biology, 74(5), 383–393.
Lehnert, K., Print, C. G., Yang, Y., & Krissansen, G. W. (1998). MAdCAM-1 costimulates T cell proliferation exclusively through integrin alpha4beta7, whereas VCAM-1 and CS-1 peptide use alpha4beta1: evidence for “remote” costimulation and induction of hyperresponsiveness to B7 molecules. European Journal of Immunology, 28(11), 3605–3615.
Berlin, C., Berg, E. L., Briskin, M. J., Andrew, D. P., Kilshaw, P. J., Holzmann, B., & Butcher, E. C. (1993). Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell, 74(1), 185–195.
Alon, R., Kassner, P. D., Carr, M. W., Finger, E. B., Hemler, M. E., & Springer, T. A. (1995). The integrin VLA-4 supports tethering and rolling in flow on VCAM-1. The Journal of Cell Biology, 128(6), 1243–1253.
Berlin, C., Bargatze, R. F., Campbell, J. J., von Andrian, U. H., Szabo, M. C., Hasslen, S. R., & Butcher, E. C. (1995). Alpha 4 integrins mediate lymphocyte attachment and rolling under physiologic flow. Cell, 80(3), 413–422.
Chen, C., Mobley, J. L., Dwir, O., Shimron, F., Grabovsky, V., Lobb, R. R., & Alon, R. (1999). High affinity very late antigen-4 subsets expressed on T cells are mandatory for spontaneous adhesion strengthening but not for rolling on VCAM-1 in shear flow. Journal of immunology (Baltimore, Md.: 1950), 162(2), 1084–1095.
Grabovsky, V., Feigelson, S., Chen, C., Bleijs, D. A., Peled, A., Cinamon, G., & Alon, R. (2000). Subsecond induction of alpha4 integrin clustering by immobilized chemokines stimulates leukocyte tethering and rolling on endothelial vascular cell adhesion molecule 1 under flow conditions. The Journal of Experimental Medicine, 192(4), 495–506.
Ding, Z., Xiong, K., & Issekutz, T. B. (2001). Chemokines stimulate human T lymphocyte transendothelial migration to utilize VLA-4 in addition to LFA-1. Journal of Leukocyte Biology, 69(3), 458–466.
Nguyen, K., Sylvain, N. R., & Bunnell, S. C. (2008). T cell costimulation via the integrin VLA-4 inhibits the actin-dependent centralization of signaling microclusters containing the adaptor SLP-76. Immunity, 28(6), 810–821.
Burkhardt, J. K. (2008). Integrins put the brakes on microcluster dynamics at the immunological synapse. Immunity, 28(6), 732–734.
Carrasco, Y. R., & Batista, F. D. (2006). B-cell activation by membrane-bound antigens is facilitated by the interaction of VLA-4 with VCAM-1. The EMBO Journal, 25(4), 889–899.
Wang, Q.-Q., Li, H., Oliver, T., Glogauer, M., Guo, J., & He, Y.-W. (2008). Integrin beta 1 regulates phagosome maturation in macrophages through Rac expression. Journal of Immunology (Baltimore, Md.: 1950), 180(4), 2419–2428.
Milner, R., & Campbell, I. L. (2002). Developmental regulation of beta1 integrins during angiogenesis in the central nervous system. Molecular and Cellular Neurosciences, 20(4), 616–626.
Lawrence, M. B., & Springer, T. A. (1991). Leukocytes roll on a selectin at physiologic flow rates: distinction from and prerequisite for adhesion through integrins. Cell, 65(5), 859–873.
Von Andrian, U. H., Chambers, J. D., McEvoy, L. M., Bargatze, R. F., Arfors, K. E., & Butcher, E. C. (1991). Two-step model of leukocyte-endothelial cell interaction in inflammation: distinct roles for LECAM-1 and the leukocyte beta 2 integrins in vivo. Proceedings of the National Academy of Sciences of the United States of America, 88(17), 7538–7542.
Masumoto, A., & Hemler, M. E. (1993). Multiple activation states of VLA-4. Mechanistic differences between adhesion to CS1/fibronectin and to vascular cell adhesion molecule-1. The Journal of Biological Chemistry, 268(1), 228–234.
Chigaev, A., Waller, A., Zwartz, G. J., Buranda, T., & Sklar, L. A. (2007). Regulation of cell adhesion by affinity and conformational unbending of alpha4beta1 integrin. Journal of Immunology, 178(11), 6828–6839.
Chigaev, A., Waller, A., Amit, O., Halip, L., Bologa, C. G., & Sklar, L. A. (2009). Real-time analysis of conformation-sensitive antibody binding provides new insights into integrin conformational regulation. The Journal of Biological Chemistry, 284(21), 14337–14346.
Zwartz, G. J., Chigaev, A., Dwyer, D. C., Foutz, T. D., Edwards, B. S., & Sklar, L. A. (2004). Real-time analysis of very late antigen-4 affinity modulation by shear. The Journal of Biological Chemistry, 279(37), 38277–38286.
Chigaev, A., Waller, A., Amit, O., & Sklar, L. A. (2008). Galphas-coupled receptor signaling actively down-regulates alpha4beta1-integrin affinity: a possible mechanism for cell de-adhesion. BMC Immunology, 9, 26.
Chigaev, A., Smagley, Y., & Sklar, L. A. (2011). Nitric oxide/cGMP pathway signaling actively down-regulates α4β1-integrin affinity: an unexpected mechanism for inducing cell de-adhesion. BMC Immunology, 12, 28.
Fujita, M., Takada, Y. K., Izumiya, Y., & Takada, Y. (2014). The binding of monomeric C-reactive protein (mCRP) to Integrins αvβ3 and α4β1 is related to its pro-inflammatory action. PLoS ONE, 9(4), e93738.
Schmitz, P., Gerber, U., Schütze, N., Jüngel, E., Blaheta, R., Naggi, A., & Bendas, G. (2013). Cyr61 is a target for heparin in reducing MV3 melanoma cell adhesion and migration via the integrin VLA-4. Thrombosis and Haemostasis, 110(5), 1046–1054.
Fujita, M., Takada, Y. K., & Takada, Y. (2014). The chemokine fractalkine can activate integrins without CX3CR1 through direct binding to a ligand-binding site distinct from the classical RGD-binding site. PLoS ONE, 9(5), e96372.
Fiorcari, S., Brown, W. S., McIntyre, B. W., Estrov, Z., Maffei, R., O’Brien, S., & Burger, J. A. (2013). The PI3-kinase delta inhibitor idelalisib (GS-1101) targets integrin-mediated adhesion of chronic lymphocytic leukemia (CLL) cell to endothelial and marrow stromal cells. PLoS ONE, 8(12), e83830.
Schmid, M. C., Franco, I., Kang, S. W., Hirsch, E., Quilliam, L. A., & Varner, J. A. (2013). PI3-kinase γ promotes Rap1a-mediated activation of myeloid cell integrin α4β1, leading to tumor inflammation and growth. PLoS ONE, 8(4), e60226.
Feigelson, S. W., Grabovsky, V., Winter, E., Chen, L. L., Pepinsky, R. B., Yednock, T., & Alon, R. (2001). The Src kinase p56(lck) up-regulates VLA-4 integrin affinity. Implications for rapid spontaneous and chemokine-triggered T cell adhesion to VCAM-1 and fibronectin. The Journal of Biological Chemistry, 276(17), 13891–13901.
Brown, W. S., Khalili, J. S., Rodriguez-Cruz, T. G., Lizee, G., & McIntyre, B. W. (2014). B-Raf regulation of integrin α4β1-mediated resistance to shear stress through changes in cell spreading and cytoskeletal association in T cells. The Journal of Biological Chemistry. doi:10.1074/jbc.M114.562918.
De Bruyn, K. M. T., Rangarajan, S., Reedquist, K. A., Figdor, C. G., & Bos, J. L. (2002). The small GTPase Rap1 is required for Mn(2+)- and antibody-induced LFA-1- and VLA-4-mediated cell adhesion. The Journal of Biological Chemistry, 277(33), 29468–29476.
Sánchez-Mateos, P., Campanero, M. R., Balboa, M. A., & Sánchez-Madrid, F. (1993). Co-clustering of beta 1 integrins, cytoskeletal proteins, and tyrosine-phosphorylated substrates during integrin-mediated leukocyte aggregation. Journal of Immunology, 151(7), 3817–3828.
Alon, R., Feigelson, S. W., Manevich, E., Rose, D. M., Schmitz, J., Overby, D. R., & Ginsberg, M. H. (2005). Alpha4beta1-dependent adhesion strengthening under mechanical strain is regulated by paxillin association with the alpha4-cytoplasmic domain. The Journal of Cell Biology, 171(6), 1073–1084.
Manevich, E., Grabovsky, V., Feigelson, S. W., & Alon, R. (2007). Talin 1 and paxillin facilitate distinct steps in rapid VLA-4-mediated adhesion strengthening to vascular cell adhesion molecule 1. The Journal of Biological Chemistry, 282(35), 25338–25348.
Hyduk, S. J., Rullo, J., Cano, A. P., Xiao, H., Chen, M., Moser, M., & Cybulsky, M. I. (2011). Talin-1 and kindlin-3 regulate alpha4beta1 integrin-mediated adhesion stabilization, but not G protein-coupled receptor-induced affinity upregulation. Journal of Immunology, 187(8), 4360–4368.
Poste, G., & Fidler, I. J. (1980). The pathogenesis of cancer metastasis. Nature, 283(5743), 139–146.
Rice, G. E., Gimbrone, M. A., Jr., & Bevilacqua, M. P. (1988). Tumor cell-endothelial interactions. Increased adhesion of human melanoma cells to activated vascular endothelium. The American Journal of Pathology, 133(2), 204–210.
Rice, G. E., & Bevilacqua, M. P. (1989). An inducible endothelial cell surface glycoprotein mediates melanoma adhesion. Science, 246(4935), 1303–1306.
Taichman, D. B., Cybulsky, M. I., Djaffar, I., Longenecker, B. M., Teixidó, J., Rice, G. E., & Bevilacqua, M. P. (1991). Tumor cell surface alpha 4 beta 1 integrin mediates adhesion to vascular endothelium: demonstration of an interaction with the N-terminal domains of INCAM-110/VCAM-1. Cell Regulation, 2(5), 347–355.
Juneja, H. S., Schmalsteig, F. C., Lee, S., & Chen, J. (1993). Vascular cell adhesion molecule-1 and VLA-4 are obligatory adhesion proteins in the heterotypic adherence between human leukemia/lymphoma cells and marrow stromal cells. Experimental Hematology, 21(3), 444–450.
Csanaky, G., Matutes, E., Vass, J. A., Morilla, R., & Catovsky, D. (1997). Adhesion receptors on peripheral blood leukemic B cells. A comparative study on B cell chronic lymphocytic leukemia and related lymphoma/leukemias. Leukemia, 11(3), 408–415.
Martìn-Padura, I., Mortarini, R., Lauri, D., Bernasconi, S., Sanchez-Madrid, F., Parmiani, G., & Dejana, E. (1991). Heterogeneity in human melanoma cell adhesion to cytokine activated endothelial cells correlates with VLA-4 expression. Cancer Research, 51(8), 2239–2241.
Zhu, N. W., Perks, C. M., Burd, A. R., & Holly, J. M. (1999). Changes in the levels of integrin and focal adhesion kinase (FAK) in human melanoma cells following 532 nm laser treatment. International Journal of Cancer. Journal International Du Cancer, 82(3), 353–358.
Zhu, N., Eves, P. C., Katerinaki, E., Szabo, M., Morandini, R., Ghanem, G., & Haycock, J. W. (2002). Melanoma cell attachment, invasion, and integrin expression is upregulated by tumor necrosis factor alpha and suppressed by alpha melanocyte stimulating hormone. The Journal of Investigative Dermatology, 119(5), 1165–1171.
Klemke, M., Weschenfelder, T., Konstandin, M. H., & Samstag, Y. (2007). High affinity interaction of integrin alpha4beta1 (VLA-4) and vascular cell adhesion molecule 1 (VCAM-1) enhances migration of human melanoma cells across activated endothelial cell layers. Journal of Cellular Physiology, 212(2), 368–374.
Okahara, H., Yagita, H., Miyake, K., & Okumura, K. (1994). Involvement of very late activation antigen 4 (VLA-4) and vascular cell adhesion molecule 1 (VCAM-1) in tumor necrosis factor alpha enhancement of experimental metastasis. Cancer Research, 54(12), 3233–3236.
Garofalo, A., Chirivi, R. G., Foglieni, C., Pigott, R., Mortarini, R., Martin-Padura, I., & Dejana, E. (1995). Involvement of the very late antigen 4 integrin on melanoma in interleukin 1-augmented experimental metastases. Cancer Research, 55(2), 414–419.
Higashiyama, A., Watanabe, H., Okumura, K., & Yagita, H. (1996). Involvement of tumor necrosis factor alpha and very late activation antigen 4/vascular cell adhesion molecule 1 interaction in surgical-stress-enhanced experimental metastasis. Cancer Immunology, Immunotherapy: CII, 42(4), 231–236.
Cardones, A. R., Murakami, T., & Hwang, S. T. (2003). CXCR4 enhances adhesion of B16 tumor cells to endothelial cells in vitro and in vivo via beta(1) integrin. Cancer Research, 63(20), 6751–6757.
Rebhun, R. B., Cheng, H., Gershenwald, J. E., Fan, D., Fidler, I. J., & Langley, R. R. (2010). Constitutive expression of the alpha4 integrin correlates with tumorigenicity and lymph node metastasis of the B16 murine melanoma. Neoplasia, 12(2), 173–182.
Khatib, A.-M., Auguste, P., Fallavollita, L., Wang, N., Samani, A., Kontogiannea, M., & Brodt, P. (2005). Characterization of the host proinflammatory response to tumor cells during the initial stages of liver metastasis. The American Journal of Pathology, 167(3), 749–759.
Sipos, E., Chen, L., András, I. E., Wrobel, J., Zhang, B., Pu, H., & Toborek, M. (2012). Proinflammatory adhesion molecules facilitate polychlorinated biphenyl-mediated enhancement of brain metastasis formation. Toxicological Sciences, 126(2), 362–371.
Langley, R. R., Carlisle, R., Ma, L., Specian, R. D., Gerritsen, M. E., & Granger, D. N. (2001). Endothelial expression of vascular cell adhesion molecule-1 correlates with metastatic pattern in spontaneous melanoma. Microcirculation, 8(5), 335–345.
Haddad, O., Chotard-Ghodsnia, R., Verdier, C., & Duperray, A. (2010). Tumor cell/endothelial cell tight contact upregulates endothelial adhesion molecule expression mediated by NFkappaB: differential role of the shear stress. Experimental Cell Research, 316(4), 615–626.
Chiu, J.-J., Chen, L.-J., Lee, P.-L., Lee, C.-I., Lo, L.-W., Usami, S., & Chien, S. (2003). Shear stress inhibits adhesion molecule expression in vascular endothelial cells induced by coculture with smooth muscle cells. Blood, 101(7), 2667–2674.
Chiu, J.-J., Lee, P.-L., Chen, C.-N., Lee, C.-I., Chang, S.-F., Chen, L.-J., & Chien, S. (2004). Shear stress increases ICAM-1 and decreases VCAM-1 and E-selectin expressions induced by tumor necrosis factor-[alpha] in endothelial cells. Arteriosclerosis, Thrombosis, and Vascular Biology, 24(1), 73–79.
Partridge, J., Carlsen, H., Enesa, K., Chaudhury, H., Zakkar, M., Luong, L., & Evans, P. C. (2007). Laminar shear stress acts as a switch to regulate divergent functions of NF-kappaB in endothelial cells. FASEB Journal, 21(13), 3553–3561.
Qian, F., Vaux, D. L., & Weissman, I. L. (1994). Expression of the integrin alpha 4 beta 1 on melanoma cells can inhibit the invasive stage of metastasis formation. Cell, 77(3), 335–347.
Johnson, J. P. (1999). Cell adhesion molecules in the development and progression of malignant melanoma. Cancer Metastasis Reviews, 18(3), 345–357.
Huhtala, P., Humphries, M. J., McCarthy, J. B., Tremble, P. M., Werb, Z., & Damsky, C. H. (1995). Cooperative signaling by alpha 5 beta 1 and alpha 4 beta 1 integrins regulates metalloproteinase gene expression in fibroblasts adhering to fibronectin. The Journal of Cell Biology, 129(3), 867–879.
Altevogt, P., Hubbe, M., Ruppert, M., Lohr, J., von Hoegen, P., Sammar, M., & Butcher, E. C. (1995). The alpha 4 integrin chain is a ligand for alpha 4 beta 7 and alpha 4 beta 1. The Journal of Experimental Medicine, 182(2), 345–355.
Campanero, M. R., Arroyo, A. G., Pulido, R., Ursa, A., de Matías, M. S., Sánchez-Mateos, P., & Corbí, A. L. (1992). Functional role of alpha 2/beta 1 and alpha 4/beta 1 integrins in leukocyte intercellular adhesion induced through the common beta 1 subunit. European Journal of Immunology, 22(12), 3111–3119.
Bednarczyk, J. L., & McIntyre, B. W. (1990). A monoclonal antibody to VLA-4 alpha-chain (CDw49d) induces homotypic lymphocyte aggregation. Journal of Immunology, 144(3), 777–784.
Bednarczyk, J. L., Wygant, J. N., Szabo, M. C., Molinari-Storey, L., Renz, M., Fong, S., & McIntyre, B. W. (1993). Homotypic leukocyte aggregation triggered by a monoclonal antibody specific for a novel epitope expressed by the integrin beta 1 subunit: conversion of nonresponsive cells by transfecting human integrin alpha 4 subunit cDNA. Journal of Cellular Biochemistry, 51(4), 465–478.
Pulido, R., Elices, M. J., Campanero, M. R., Osborn, L., Schiffer, S., García-Pardo, A., & Sánchez-Madrid, F. (1991). Functional evidence for three distinct and independently inhibitable adhesion activities mediated by the human integrin VLA-4. Correlation with distinct alpha 4 epitopes. The Journal of Biological Chemistry, 266(16), 10241–10245.
Hart, I. R., Birch, M., & Marshall, J. F. (1991). Cell adhesion receptor expression during melanoma progression and metastasis. Cancer Metastasis Reviews, 10(2), 115–128.
Moretti, S., Martini, L., Berti, E., Pinzi, C., & Giannotti, B. (1993). Adhesion molecule profile and malignancy of melanocytic lesions. Melanoma Research, 3(4), 235–239.
Schadendorf, D., Gawlik, C., Haney, U., Ostmeier, H., Suter, L., & Czarnetzki, B. M. (1993). Tumour progression and metastatic behaviour in vivo correlates with integrin expression on melanocytic tumours. The Journal of Pathology, 170(4), 429–434.
Schadendorf, D., Heidel, J., Gawlik, C., Suter, L., & Czarnetzki, B. M. (1995). Association with clinical outcome of expression of VLA-4 in primary cutaneous malignant melanoma as well as P-selectin and E-selectin on intratumoral vessels. Journal of the National Cancer Institute, 87(5), 366–371.
Chen, Q., Zhang, X. H.-F., & Massagué, J. (2011). Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell, 20(4), 538–549.
Lin, K.-Y., Lu, D., Hung, C.-F., Peng, S., Huang, L., Jie, C., & Wu, T.-C. (2007). Ectopic expression of vascular cell adhesion molecule-1 as a new mechanism for tumor immune evasion. Cancer Research, 67(4), 1832–1841.
Kuai, W.-X., Wang, Q., Yang, X.-Z., Zhao, Y., Yu, R., & Tang, X.-J. (2012). Interleukin-8 associates with adhesion, migration, invasion and chemosensitivity of human gastric cancer cells. World Journal of Gastroenterology: WJG, 18(9), 979–985.
Ruco, L. P., de Laat, P. A., Matteucci, C., Bernasconi, S., Sciacca, F. M., van der Kwast, T. H., & Versnel, M. A. (1996). Expression of ICAM-1 and VCAM-1 in human malignant mesothelioma. The Journal of Pathology, 179(3), 266–271.
Ding, Y.-B., Chen, G.-Y., Xia, J.-G., Zang, X.-W., Yang, H.-Y., & Yang, L. (2003). Association of VCAM-1 overexpression with oncogenesis, tumor angiogenesis and metastasis of gastric carcinoma. World Journal of Gastroenterology: WJG, 9(7), 1409–1414.
Gupta, G. P., Minn, A. J., Kang, Y., Siegel, P. M., Serganova, I., Cordón-Cardo, C., & Massagué, J. (2005). Identifying site-specific metastasis genes and functions. Cold Spring Harbor Symposia on Quantitative Biology, 70, 149–158.
Minn, A. J., Gupta, G. P., Siegel, P. M., Bos, P. D., Shu, W., Giri, D. D., & Massagué, J. (2005). Genes that mediate breast cancer metastasis to lung. Nature, 436(7050), 518–524.
Vanharanta, S., & Massagué, J. (2013). Origins of metastatic traits. Cancer Cell, 24(4), 410–421.
Chen, Q., & Massagué, J. (2012). Molecular pathways: VCAM-1 as a potential therapeutic target in metastasis. Clinical Cancer Research, 18(20), 5520–5525.
Lu, X., Mu, E., Wei, Y., Riethdorf, S., Yang, Q., Yuan, M., & Kang, Y. (2011). VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging α4β1-positive osteoclast progenitors. Cancer Cell, 20(6), 701–714.
Hynes, R. O. (2011). Metastatic cells will take any help they can get. Cancer Cell, 20(6), 689–690.
Lin, K. Y., Guarnieri, F. G., Staveley-O’Carroll, K. F., Levitsky, H. I., August, J. T., Pardoll, D. M., & Wu, T. C. (1996). Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Research, 56(1), 21–26.
Wu, T. C., Guarnieri, F. G., Staveley-O’Carroll, K. F., Viscidi, R. P., Levitsky, H. I., Hedrick, L., & Pardoll, D. M. (1995). Engineering an intracellular pathway for major histocompatibility complex class II presentation of antigens. Proceedings of the National Academy of Sciences of the United States of America, 92(25), 11671–11675.
Wu, T.-C. (2007). The role of vascular cell adhesion molecule-1 in tumor immune evasion. Cancer Research, 67(13), 6003–6006.
Rose, D. M., Grabovsky, V., Alon, R., & Ginsberg, M. H. (2001). The affinity of integrin alpha(4)beta(1) governs lymphocyte migration. Journal of Immunology, 167(5), 2824–2830.
Rose, D. M., Han, J., & Ginsberg, M. H. (2002). Alpha4 integrins and the immune response. Immunological Reviews, 186, 118–124.
Liu, S., Thomas, S. M., Woodside, D. G., Rose, D. M., Kiosses, W. B., Pfaff, M., & Ginsberg, M. H. (1999). Binding of paxillin to alpha4 integrins modifies integrin-dependent biological responses. Nature, 402(6762), 676–681.
Rose, D. M., Liu, S., Woodside, D. G., Han, J., Schlaepfer, D. D., & Ginsberg, M. H. (2003). Paxillin binding to the alpha 4 integrin subunit stimulates LFA-1 (integrin alpha L beta 2)-dependent T cell migration by augmenting the activation of focal adhesion kinase/proline-rich tyrosine kinase-2. Journal of Immunology, 170(12), 5912–5918.
Kaplan, R. N., Riba, R. D., Zacharoulis, S., Bramley, A. H., Vincent, L., Costa, C., & Lyden, D. (2005). VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature, 438(7069), 820–827.
Müller, A., Homey, B., Soto, H., Ge, N., Catron, D., Buchanan, M. E., & Zlotnik, A. (2001). Involvement of chemokine receptors in breast cancer metastasis. Nature, 410(6824), 50–56.
Zlotnik, A., Burkhardt, A. M., & Homey, B. (2011). Homeostatic chemokine receptors and organ-specific metastasis. Nature Reviews Immunology, 11(9), 597–606.
Williams, S. A., Harata-Lee, Y., Comerford, I., Anderson, R. L., Smyth, M. J., & McColl, S. R. (2010). Multiple functions of CXCL12 in a syngeneic model of breast cancer. Molecular Cancer, 9, 250.
Koizumi, K., Kozawa, Y., Ohashi, Y., Nakamura, E. S., Aozuka, Y., Sakurai, H., & Saiki, I. (2007). CCL21 promotes the migration and adhesion of highly lymph node metastatic human non-small cell lung cancer Lu-99 in vitro. Oncology Reports, 17(6), 1511–1516.
Mantovani, A., Bottazzi, B., Colotta, F., Sozzani, S., & Ruco, L. (1992). The origin and function of tumor-associated macrophages. Immunology Today, 13(7), 265–270.
Conti, I., & Rollins, B. J. (2004). CCL2 (monocyte chemoattractant protein-1) and cancer. Seminars in Cancer Biology, 14(3), 149–154.
Zhao, L., Lim, S. Y., Gordon-Weeks, A. N., Tapmeier, T. T., Im, J. H., Cao, Y., & Muschel, R. J. (2013). Recruitment of a myeloid cell subset (CD11b/Gr1 mid) via CCL2/CCR2 promotes the development of colorectal cancer liver metastasis. Hepatology, 57(2), 829–839.
Lim, S. Y., Gordon-Weeks, A. N., Zhao, L., Tapmeier, T. T., Im, J. H., Cao, Y., & Muschel, R. J. (2013). Recruitment of myeloid cells to the tumor microenvironment supports liver metastasis. Oncoimmunology, 2(3), e23187.
Qian, B.-Z., Li, J., Zhang, H., Kitamura, T., Zhang, J., Campion, L. R., & Pollard, J. W. (2011). CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature, 475(7355), 222–225.
Qian, B., Deng, Y., Im, J. H., Muschel, R. J., Zou, Y., Li, J., & Pollard, J. W. (2009). A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PloS One, 4(8), e6562.
Gil-Bernabé, A. M., Ferjancic, S., Tlalka, M., Zhao, L., Allen, P. D., Im, J. H., & Muschel, R. J. (2012). Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood, 119(13), 3164–3175.
Ferjancic, S., Gil-Bernabé, A. M., Hill, S. A., Allen, P. D., Richardson, P., Sparey, T., & Muschel, R. J. (2013). VCAM-1 and VAP-1 recruit myeloid cells that promote pulmonary metastasis in mice. Blood, 121(16), 3289–3297.
Murdoch, C., Muthana, M., Coffelt, S. B., & Lewis, C. E. (2008). The role of myeloid cells in the promotion of tumour angiogenesis. Nature Reviews Cancer, 8(8), 618–631.
Tazzyman, S., Lewis, C. E., & Murdoch, C. (2009). Neutrophils: key mediators of tumour angiogenesis. International Journal of Experimental Pathology, 90(3), 222–231.
Lindau, D., Gielen, P., Kroesen, M., Wesseling, P., & Adema, G. J. (2013). The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology, 138(2), 105–115.
Yang, L., DeBusk, L. M., Fukuda, K., Fingleton, B., Green-Jarvis, B., Shyr, Y., & Lin, P. C. (2004). Expansion of myeloid immune suppressor Gr+CD11b+cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell, 6(4), 409–421.
Jin, H., Su, J., Garmy-Susini, B., Kleeman, J., & Varner, J. (2006). Integrin alpha4beta1 promotes monocyte trafficking and angiogenesis in tumors. Cancer Research, 66(4), 2146–2152.
Joshi, S., Singh, A. R., Zulcic, M., Bao, L., Messer, K., Ideker, T., & Durden, D. L. (2014). Rac2 controls tumor growth, metastasis and M1-M2 macrophage differentiation in vivo. PLoS ONE, 9(4), e95893.
Folkman, J. (1971). Tumor angiogenesis: therapeutic implications. The New England Journal of Medicine, 285(21), 1182–1186.
Ide, A. G., Baker, N. H., & Warren, S. L. (1939). Vascularization of the Brown-Pearce rabbit epithelioma transplant as seen in the transparent ear chamber. American Journal of Roentgenology, 42, 891–899.
Algire, G. H., Chalkley, H. W., Legallais, F. Y., & Park, H. D. (1945). Vascular reactions of normal and malignant tissues in vivo. I. Vascular reactions of mice to wounds and to normal and neoplastic transplants. Journal of the National Cancer Institute, 6(1), 73–85.
Folkman, J., Merler, E., Abernathy, C., & Williams, G. (1971). Isolation of a tumor factor responsible for angiogenesis. The Journal of Experimental Medicine, 133(2), 275–288.
Gimbrone, M. A., Jr., Leapman, S. B., Cotran, R. S., & Folkman, J. (1972). Tumor dormancy in vivo by prevention of neovascularization. The Journal of Experimental Medicine, 136(2), 261–276.
Avraamides, C. J., Garmy-Susini, B., & Varner, J. A. (2008). Integrins in angiogenesis and lymphangiogenesis. Nature Reviews Cancer, 8(8), 604–617.
Garmy-Susini, B., & Varner, J. A. (2008). Roles of integrins in tumor angiogenesis and lymphangiogenesis. Lymphatic Research and Biology, 6(3–4), 155–163.
Grant, M. B., May, W. S., Caballero, S., Brown, G. A. J., Guthrie, S. M., Mames, R. N., & Scott, E. W. (2002). Adult hematopoietic stem cells provide functional hemangioblast activity during retinal neovascularization. Nature Medicine, 8(6), 607–612.
Patterson, L. J., Gering, M., & Patient, R. (2005). Scl is required for dorsal aorta as well as blood formation in zebrafish embryos. Blood, 105(9), 3502–3511.
Wang, L., Li, L., Shojaei, F., Levac, K., Cerdan, C., Menendez, P., & Bhatia, M. (2004). Endothelial and hematopoietic cell fate of human embryonic stem cells originates from primitive endothelium with hemangioblastic properties. Immunity, 21(1), 31–41.
Lyden, D., Hattori, K., Dias, S., Costa, C., Blaikie, P., Butros, L., & Rafii, S. (2001). Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nature Medicine, 7(11), 1194–1201.
Chen, L., Ackerman, R., Saleh, M., Gotlinger, K. H., Kessler, M., Mendelowitz, L. G., & Guo, A. M. (2014). 20-HETE regulates the angiogenic functions of human endothelial progenitor cells and contributes to angiogenesis in vivo. The Journal of Pharmacology and Experimental Therapeutics, 348(3), 442–451.
Jin, H., Aiyer, A., Su, J., Borgstrom, P., Stupack, D., Friedlander, M., & Varner, J. (2006). A homing mechanism for bone marrow-derived progenitor cell recruitment to the neovasculature. The Journal of Clinical Investigation, 116(3), 652–662.
Schmid, M. C., & Varner, J. A. (2009). Circulating endothelial progenitor cells. Methods in Molecular Biology, 467, 139–155.
Ruzinova, M. B., Schoer, R. A., Gerald, W., Egan, J. E., Pandolfi, P. P., Rafii, S., & Benezra, R. (2003). Effect of angiogenesis inhibition by Id loss and the contribution of bone-marrow-derived endothelial cells in spontaneous murine tumors. Cancer Cell, 4(4), 277–289.
Ono, M., Torisu, H., Fukushi, J., Nishie, A., & Kuwano, M. (1999). Biological implications of macrophage infiltration in human tumor angiogenesis. Cancer Chemotherapy and Pharmacology, 43(Suppl), S69–71.
Jin, D. K., Shido, K., Kopp, H.-G., Petit, I., Shmelkov, S. V., Young, L. M., & Rafii, S. (2006). Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nature Medicine, 12(5), 557–567.
Mantovani, A., Sozzani, S., Locati, M., Allavena, P., & Sica, A. (2002). Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends in Immunology, 23(11), 549–555.
De Palma, M., Murdoch, C., Venneri, M. A., Naldini, L., & Lewis, C. E. (2007). Tie2-expressing monocytes: regulation of tumor angiogenesis and therapeutic implications. Trends in Immunology, 28(12), 519–524.
Venneri, M. A., De Palma, M., Ponzoni, M., Pucci, F., Scielzo, C., Zonari, E., & Naldini, L. (2007). Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood, 109(12), 5276–5285.
Ghiringhelli, F., Puig, P. E., Roux, S., Parcellier, A., Schmitt, E., Solary, E., & Zitvogel, L. (2005). Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. The Journal of Experimental Medicine, 202(7), 919–929.
Nagaraj, S., & Gabrilovich, D. I. (2010). Myeloid-derived suppressor cells in human cancer. Cancer Journal, 16(4), 348–353.
Tazzyman, S., Niaz, H., & Murdoch, C. (2013). Neutrophil-mediated tumour angiogenesis: subversion of immune responses to promote tumour growth. Seminars in Cancer Biology, 23(3), 149–158.
Looi, L. M. (1987). Tumor-associated tissue eosinophilia in nasopharyngeal carcinoma. A pathologic study of 422 primary and 138 metastatic tumors. Cancer, 59(3), 466–470.
Reed, J. A., McNutt, N. S., Bogdany, J. K., & Albino, A. P. (1996). Expression of the mast cell growth factor interleukin-3 in melanocytic lesions correlates with an increased number of mast cells in the perilesional stroma: implications for melanoma progression. Journal of Cutaneous Pathology, 23(6), 495–505.
Griffioen, A. W., Damen, C. A., Blijham, G. H., & Groenewegen, G. (1996). Tumor angiogenesis is accompanied by a decreased inflammatory response of tumor-associated endothelium. Blood, 88(2), 667–673.
Griffioen, A. W., Tromp, S. C., & Hillen, H. F. (1998). Angiogenesis modulates the tumour immune response. International Journal of Experimental Pathology, 79(6), 363–368.
Tromp, S. C., oude Egbrink, M. G., Dings, R. P., van Velzen, S., Slaaf, D. W., Hillen, H. F., & Griffioen, A. W. ((2000). Tumor angiogenesis factors reduce leukocyte adhesion in vivo. International Immunology, 12(5), 671–676.
Dirkx, A. E. M., Oude Egbrink, M. G. A., Kuijpers, M. J. E., van der Niet, S. T., Heijnen, V. V. T., Bouma-ter Steege, J. C. A., & Griffioen, A. W. (2003). Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Research, 63(9), 2322–2329.
Dirkx, A. E. M., Oude Egbrink, M. G. A., Wagstaff, J., & Griffioen, A. W. (2006). Monocyte/macrophage infiltration in tumors: modulators of angiogenesis. Journal of Leukocyte Biology, 80(6), 1183–1196.
Jain, R. K., & Booth, M. F. (2003). What brings pericytes to tumor vessels? The Journal of Clinical Investigation, 112(8), 1134–1136.
Abramsson, A., Lindblom, P., & Betsholtz, C. (2003). Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. The Journal of Clinical Investigation, 112(8), 1142–1151.
Koch, A. E., Halloran, M. M., Haskell, C. J., Shah, M. R., & Polverini, P. J. (1995). Angiogenesis mediated by soluble forms of E-selectin and vascular cell adhesion molecule-1. Nature, 376(6540), 517–519.
Fukushi, J., Ono, M., Morikawa, W., Iwamoto, Y., & Kuwano, M. (2000). The activity of soluble VCAM-1 in angiogenesis stimulated by IL-4 and IL-13. Journal of Immunology, 165(5), 2818–2823.
Nakao, S., Kuwano, T., Ishibashi, T., Kuwano, M., & Ono, M. (2003). Synergistic effect of TNF-alpha in soluble VCAM-1-induced angiogenesis through alpha 4 integrins. Journal of Immunology, 170(11), 5704–5711.
Calzada, M. J., Zhou, L., Sipes, J. M., Zhang, J., Krutzsch, H. C., Iruela-Arispe, M. L., & Roberts, D. D. (2004). Alpha4beta1 integrin mediates selective endothelial cell responses to thrombospondins 1 and 2 in vitro and modulates angiogenesis in vivo. Circulation Research, 94(4), 462–470.
Garmy-Susini, B., Jin, H., Zhu, Y., Sung, R.-J., Hwang, R., & Varner, J. (2005). Integrin alpha4beta1-VCAM-1-mediated adhesion between endothelial and mural cells is required for blood vessel maturation. The Journal of Clinical Investigation, 115(6), 1542–1551.
Banerji, S., Ni, J., Wang, S. X., Clasper, S., Su, J., Tammi, R., & Jackson, D. G. (1999). LYVE-1, a new homologue of the CD44 glycoprotein, is a lymph-specific receptor for hyaluronan. The Journal of Cell Biology, 144(4), 789–801.
Wigle, J. T., & Oliver, G. (1999). Prox1 function is required for the development of the murine lymphatic system. Cell, 98(6), 769–778.
Breiteneder-Geleff, S., Soleiman, A., Kowalski, H., Horvat, R., Amann, G., Kriehuber, E., & Kerjaschki, D. (1999). Angiosarcomas express mixed endothelial phenotypes of blood and lymphatic capillaries: podoplanin as a specific marker for lymphatic endothelium. The American Journal of Pathology, 154(2), 385–394.
Baluk, P., & McDonald, D. M. (2008). Markers for microscopic imaging of lymphangiogenesis and angiogenesis. Annals of the New York Academy of Sciences, 1131, 1–12.
Huang, X. Z., Wu, J. F., Ferrando, R., Lee, J. H., Wang, Y. L., Farese, R. V., Jr., & Sheppard, D. (2000). Fatal bilateral chylothorax in mice lacking the integrin alpha9beta1. Molecular and Cellular Biology, 20(14), 5208–5215.
Mishima, K., Watabe, T., Saito, A., Yoshimatsu, Y., Imaizumi, N., Masui, S., & Miyazono, K. (2007). Prox1 induces lymphatic endothelial differentiation via integrin alpha9 and other signaling cascades. Molecular Biology of the Cell, 18(4), 1421–1429.
Bazigou, E., Xie, S., Chen, C., Weston, A., Miura, N., Sorokin, L., & Makinen, T. (2009). Integrin-alpha9 is required for fibronectin matrix assembly during lymphatic valve morphogenesis. Developmental Cell, 17(2), 175–186.
Hong, Y.-K., Lange-Asschenfeldt, B., Velasco, P., Hirakawa, S., Kunstfeld, R., Brown, L. F., & Detmar, M. (2004). VEGF-A promotes tissue repair-associated lymphatic vessel formation via VEGFR-2 and the alpha1beta1 and alpha2beta1 integrins. FASEB Journal, 18(10), 1111–1113.
Grimaldo, S., Yuen, D., Ecoiffier, T., & Chen, L. (2011). Very late antigen-1 mediates corneal lymphangiogenesis. Investigative Ophthalmology & Visual Science, 52(7), 4808–4812.
Dietrich, T., Onderka, J., Bock, F., Kruse, F. E., Vossmeyer, D., Stragies, R., & Cursiefen, C. (2007). Inhibition of inflammatory lymphangiogenesis by integrin alpha5 blockade. The American Journal of Pathology, 171(1), 361–372.
Garmy-Susini, B., Avraamides, C. J., Schmid, M. C., Foubert, P., Ellies, L. G., Barnes, L., & Varner, J. A. (2010). Integrin alpha4beta1 signaling is required for lymphangiogenesis and tumor metastasis. Cancer Research, 70(8), 3042–3051.
Garmy-Susini, B., Makale, M., Fuster, M., & Varner, J. A. (2007). Methods to study lymphatic vessel integrins. Methods in Enzymology, 426, 415–438.
Zhou, F., Chang, Z., Zhang, L., Hong, Y.-K., Shen, B., Wang, B., & He, Y. (2010). Akt/Protein kinase B is required for lymphatic network formation, remodeling, and valve development. The American Journal of Pathology, 177(4), 2124–2133.
Garmy-Susini, B., Avraamides, C. J., Desgrosellier, J. S., Schmid, M. C., Foubert, P., Ellies, L. G., & Varner, J. (2013). PI3Kα activates integrin α4β1 to establish a metastatic niche in lymph nodes. Proceedings of the National Academy of Sciences of the United States of America, 110(22), 9042–9047.
Testaz, S., & Duband, J. L. (2001). Central role of the alpha4beta1 integrin in the coordination of avian truncal neural crest cell adhesion, migration, and survival. Developmental Dynamics, 222(2), 127–140.
Alvarez-Dolado, M. (2007). Cell fusion: biological perspectives and potential for regenerative medicine. Frontiers in Bioscience: a Journal and Virtual Library, 12, 1–12.
Nygren, J. M., Liuba, K., Breitbach, M., Stott, S., Thorén, L., Roell, W., & Jacobsen, S. E. W. (2008). Myeloid and lymphoid contribution to non-haematopoietic lineages through irradiation-induced heterotypic cell fusion. Nature Cell Biology, 10(5), 584–592.
Nygren, J. M., Jovinge, S., Breitbach, M., Säwén, P., Röll, W., Hescheler, J., & Jacobsen, S. E. W. (2004). Bone marrow-derived hematopoietic cells generate cardiomyocytes at a low frequency through cell fusion, but not transdifferentiation. Nature Medicine, 10(5), 494–501.
Singec, I., & Snyder, E. Y. (2008). Inflammation as a matchmaker: revisiting cell fusion. Nature Cell Biology, 10(5), 503–505.
Mortensen, K., Lichtenberg, J., Thomsen, P. D., & Larsson, L.-I. (2004). Spontaneous fusion between cancer cells and endothelial cells. Cellular and Molecular life Sciences: CMLS, 61(16), 2125–2131.
Song, K., Zhu, F., Zhang, H., & Shang, Z. (2012). Tumor necrosis factor-α enhanced fusions between oral squamous cell carcinoma cells and endothelial cells via VCAM-1/VLA-4 pathway. Experimental Cell Research, 318(14), 1707–1715.
Durand, R. E., & Sutherland, R. M. (1972). Effects of intercellular contact on repair of radiation damage. Experimental Cell Research, 71(1), 75–80.
Fridman, R., Giaccone, G., Kanemoto, T., Martin, G. R., Gazdar, A. F., & Mulshine, J. L. (1990). Reconstituted basement membrane (Matrigel) and laminin can enhance the tumorigenicity and the drug resistance of small cell lung cancer cell lines. Proceedings of the National Academy of Sciences of the United States of America, 87(17), 6698–6702.
Meredith, J. E., Jr., Fazeli, B., & Schwartz, M. A. (1993). The extracellular matrix as a cell survival factor. Molecular Biology of the Cell, 4(9), 953–961.
Schuetz, J. D., & Schuetz, E. G. (1993). Extracellular matrix regulation of multidrug resistance in primary monolayer cultures of adult rat hepatocytes. Cell growth & differentiation: the molecular biology journal of the American Association for Cancer Research, 4(1), 31–40.
Bates, R. C., Buret, A., van Helden, D. F., Horton, M. A., & Burns, G. F. (1994). Apoptosis induced by inhibition of intercellular contact. The Journal of Cell Biology, 125(2), 403–415.
Scott, G., Cassidy, L., & Busacco, A. (1997). Fibronectin suppresses apoptosis in normal human melanocytes through an integrin-dependent mechanism. The Journal of Investigative Dermatology, 108(2), 147–153.
Bradstock, K. F., & Gottlieb, D. J. (1995). Interaction of acute leukemia cells with the bone marrow microenvironment: implications for control of minimal residual disease. Leukemia & Lymphoma, 18(1–2), 1–16.
St Croix, B., & Kerbel, R. S. (1997). Cell adhesion and drug resistance in cancer. Current Opinion in Oncology, 9(6), 549–556.
Matsunaga, T., Takemoto, N., Sato, T., Takimoto, R., Tanaka, I., Fujimi, A., & Niitsu, Y. (2003). Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nature Medicine, 9(9), 1158–1165.
Damiano, J. S., Cress, A. E., Hazlehurst, L. A., Shtil, A. A., & Dalton, W. S. (1999). Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood, 93(5), 1658–1667.
Mori, Y., Shimizu, N., Dallas, M., Niewolna, M., Story, B., Williams, P. J., & Yoneda, T. (2004). Anti-alpha4 integrin antibody suppresses the development of multiple myeloma and associated osteoclastic osteolysis. Blood, 104(7), 2149–2154.
Park, C. C., Zhang, H., Pallavicini, M., Gray, J. W., Baehner, F., Park, C. J., & Bissell, M. J. (2006). Beta1 integrin inhibitory antibody induces apoptosis of breast cancer cells, inhibits growth, and distinguishes malignant from normal phenotype in three dimensional cultures and in vivo. Cancer Research, 66(3), 1526–1535.
Meads, M. B., Gatenby, R. A., & Dalton, W. S. (2009). Environment-mediated drug resistance: a major contributor to minimal residual disease. Nature Reviews Cancer, 9(9), 665–674.
Chen, Y.-X., Wang, Y., Fu, C.-C., Diao, F., Song, L.-N., Li, Z.-B., & Lu, J. (2010). Dexamethasone enhances cell resistance to chemotherapy by increasing adhesion to extracellular matrix in human ovarian cancer cells. Endocrine-Related Cancer, 17(1), 39–50.
Scalici, J. M., Harrer, C., Allen, A., Jazaeri, A., Atkins, K. A., McLachlan, K. R., & Slack-Davis, J. K. (2014). Inhibition of α4β1 integrin increases ovarian cancer response to carboplatin. Gynecologic Oncology, 132(2), 455–461.
Weekes, C. D., Pirruccello, S. J., Vose, J. M., Kuszynski, C., & Sharp, J. G. (1998). Lymphoma cells associated with bone marrow stromal cells in culture exhibit altered growth and survival. Leukemia & Lymphoma, 31(1–2), 151–165.
Weekes, C. D., Kuszynski, C. A., & Sharp, J. G. (2001). VLA-4 mediated adhesion to bone marrow stromal cells confers chemoresistance to adherent lymphoma cells. Leukemia & Lymphoma, 40(5–6), 631–645.
St Croix, B., Flørenes, V. A., Rak, J. W., Flanagan, M., Bhattacharya, N., Slingerland, J. M., & Kerbel, R. S. (1996). Impact of the cyclin-dependent kinase inhibitor p27Kip1 on resistance of tumor cells to anticancer agents. Nature Medicine, 2(11), 1204–1210.
Fukai, F., Mashimo, M., Akiyama, K., Goto, T., Tanuma, S., & Katayama, T. (1998). Modulation of apoptotic cell death by extracellular matrix proteins and a fibronectin-derived antiadhesive peptide. Experimental Cell Research, 242(1), 92–99.
Ilić, D., Almeida, E. A., Schlaepfer, D. D., Dazin, P., Aizawa, S., & Damsky, C. H. (1998). Extracellular matrix survival signals transduced by focal adhesion kinase suppress p53-mediated apoptosis. The Journal of Cell Biology, 143(2), 547–560.
Damiano, J. S., & Dalton, W. S. (2000). Integrin-mediated drug resistance in multiple myeloma. Leukemia & Lymphoma, 38(1–2), 71–81.
Higashimoto, I., Chihara, J., Kakazu, T., Kawabata, M., Nakajima, S., & Osame, M. (1996). Regulation of eosinophil cell death by adhesion to fibronectin. International Archives of Allergy and Immunology, 111(Suppl 1), 66–69.
De la Fuente, M. T., Casanova, B., Garcia-Gila, M., Silva, A., & Garcia-Pardo, A. (1999). Fibronectin interaction with alpha4beta1 integrin prevents apoptosis in B cell chronic lymphocytic leukemia: correlation with Bcl-2 and Bax. Leukemia, 13(2), 266–274.
Konopleva, M., Konoplev, S., Hu, W., Zaritskey, A. Y., Afanasiev, B. V., & Andreeff, M. (2002). Stromal cells prevent apoptosis of AML cells by up-regulation of anti-apoptotic proteins. Leukemia, 16(9), 1713–1724.
Liu, C.-C., Leclair, P., Yap, S. Q., & Lim, C. J. (2013). The membrane-proximal KXGFFKR motif of α-integrin mediates chemoresistance. Molecular and Cellular Biology, 33(21), 4334–4345.
Layani-Bazar, A., Skornik, I., Berrebi, A., Pauker, M. H., Noy, E., Silberman, A., & Sredni, B. (2014). Redox modulation of adjacent thiols in VLA-4 by AS101 converts myeloid leukemia cells from a drug-resistant to drug-sensitive state. Cancer Research. doi:10.1158/0008-5472.CAN-13-2159.
Jacamo, R., Chen, Y., Wang, Z., Ma, W., Zhang, M., Spaeth, E. L., & Andreeff, M. (2014). Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood, 123(17), 2691–2702.
Brachtl, G., Piñón Hofbauer, J., Greil, R., & Hartmann, T. N. (2014). The pathogenic relevance of the prognostic markers CD38 and CD49d in chronic lymphocytic leukemia. Annals of Hematology, 93(3), 361–374.
Gattei, V., Bulian, P., Del Principe, M. I., Zucchetto, A., Maurillo, L., Buccisano, F., & Del Poeta, G. (2008). Relevance of CD49d protein expression as overall survival and progressive disease prognosticator in chronic lymphocytic leukemia. Blood, 111(2), 865–873.
Nückel, H., Switala, M., Collins, C. H., Sellmann, L., Grosse-Wilde, H., Dührsen, U., & Rebmann, V. (2009). High CD49d protein and mRNA expression predicts poor outcome in chronic lymphocytic leukemia. Clinical Immunology, 131(3), 472–480.
Balcer, L. J., Galetta, S. L., Calabresi, P. A., Confavreux, C., Giovannoni, G., Havrdova, E., & Panzara, M. A. (2007). Natalizumab reduces visual loss in patients with relapsing multiple sclerosis. Neurology, 68(16), 1299–1304.
Havrdova, E., Galetta, S., Hutchinson, M., Stefoski, D., Bates, D., Polman, C. H., & Hyde, R. (2009). Effect of natalizumab on clinical and radiological disease activity in multiple sclerosis: a retrospective analysis of the Natalizumab Safety and Efficacy in Relapsing-Remitting Multiple Sclerosis (AFFIRM) study. The Lancet. Neurology, 8(3), 254–260.
Armuzzi, A., & Felice, C. (2013). Natalizumab in Crohn’s disease: past and future areas of applicability. Annals of Gastroenterology: Quarterly Publication of the Hellenic Society of Gastroenterology, 26(3), 189–190.
Lanzarotto, F., Carpani, M., Chaudhary, R., & Ghosh, S. (2006). Novel treatment options for inflammatory bowel disease: targeting alpha 4 integrin. Drugs, 66(9), 1179–1189.
Polman, C. H., O’Connor, P. W., Havrdova, E., Hutchinson, M., Kappos, L., Miller, D. H., & Investigators, A. F. F. I. R. M. (2006). A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. The New England Journal of Medicine, 354(9), 899–910.
Rudick, R. A., Stuart, W. H., Calabresi, P. A., Confavreux, C., Galetta, S. L., Radue, E.-W., & Investigators, S. E. N. T. I. N. E. L. (2006). Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. The New England Journal of Medicine, 354(9), 911–923.
Berger, J. R., & Houff, S. (2006). Progressive multifocal leukoencephalopathy: lessons from AIDS and natalizumab. Neurological Research, 28(3), 299–305.
Tur, C., & Montalban, X. (2014). Natalizumab: risk stratification of individual patients with multiple sclerosis. CNS Drugs, 28(7), 641–648.
Cutter, G. R., & Stüve, O. (2014). Does risk stratification decrease the risk of natalizumab-associated PML? Where is the evidence? Multiple Sclerosis, 20(10), 1304–1305.
Vavricka, B. M. P., Baumberger, P., Russmann, S., & Kullak-Ublick, G. A. (2011). Diagnosis of melanoma under concomitant natalizumab therapy. Multiple Sclerosis, 17(2), 255–256.
Mullen, J. T., Vartanian, T. K., & Atkins, M. B. (2008). Melanoma complicating treatment with natalizumab for multiple sclerosis. The New England Journal of Medicine, 358(6), 647–648.
Olson, D. L., Burkly, L. C., Leone, D. R., Dolinski, B. M., & Lobb, R. R. (2005). Anti-alpha4 integrin monoclonal antibody inhibits multiple myeloma growth in a murine model. Molecular Cancer Therapeutics, 4(1), 91–99.
Podar, K., Zimmerhackl, A., Fulciniti, M., Tonon, G., Hainz, U., Tai, Y.-T., & Anderson, K. C. (2011). The selective adhesion molecule inhibitor Natalizumab decreases multiple myeloma cell growth in the bone marrow microenvironment: therapeutic implications. British Journal of Haematology, 155(4), 438–448.
Study of Natalizumab in Relapsed/Refractory Multiple Myeloma - ClinicalTrials.gov. (n.d.). Retrieved August 28, 2014, from https://clinicaltrials.gov/ct2/show/NCT00675428
Ramirez, P., Rettig, M. P., Uy, G. L., Deych, E., Holt, M. S., Ritchey, J. K., & DiPersio, J. F. (2009). BIO5192, a small molecule inhibitor of VLA-4, mobilizes hematopoietic stem and progenitor cells. Blood, 114(7), 1340–1343.
Rettig, M. P., Ansstas, G., & DiPersio, J. F. (2012). Mobilization of hematopoietic stem and progenitor cells using inhibitors of CXCR4 and VLA-4. Leukemia, 26(1), 34–53.
Davenport, R. J., & Munday, J. R. (2007). Alpha4-integrin antagonism—an effective approach for the treatment of inflammatory diseases? Drug Discovery Today, 12(13–14), 569–576.
Davenport, R. J., & Munday, J. R. (2008). Blocking alpha4-integrins—a small molecule approach to treatment of multiple sclerosis. Journal of the Neurological Sciences, 274(1–2), 27–30.
Schmid, M. C., Avraamides, C. J., Foubert, P., Shaked, Y., Kang, S. W., Kerbel, R. S., & Varner, J. A. (2011). Combined blockade of integrin-α4β1 plus cytokines SDF-1α or IL-1β potently inhibits tumor inflammation and growth. Cancer Research, 71(22), 6965–6975.
Schlesinger, M., Roblek, M., Ortmann, K., Naggi, A., Torri, G., Borsig, L., & Bendas, G. (2014). The role of VLA-4 binding for experimental melanoma metastasis and its inhibition by heparin. Thrombosis Research, 133(5), 855–862.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Schlesinger, M., Bendas, G. Contribution of very late antigen-4 (VLA-4) integrin to cancer progression and metastasis. Cancer Metastasis Rev 34, 575–591 (2015). https://doi.org/10.1007/s10555-014-9545-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-014-9545-x