
Abstract
The metastasis of cancer is a complex and life-threatening process that is only partially understood. Immune suppressive cells are recognized as important contributors to tumour progression and may also promote the development and growth of tumour metastases. Specifically, regulatory T cells (Tregs) have been found to promote primary tumour progression, and emerging pre-clinical data suggests that Tregs may promote metastasis and metastatic tumour growth. While the precise role that Tregs play in metastatic progression is understudied, recent findings have indicated that by suppressing innate and adaptive anti-tumour immunity, Tregs may shield tumour cells from immune detection, and thereby allow tumour cells to survive, proliferate and acquire characteristics that facilitate dissemination. This review will highlight our current understanding of Tregs in metastasis, including an overview of pre-clinical findings and discussion of clinical data regarding Tregs and therapeutic outcome. Evolving strategies to directly ablate Tregs or to inhibit their function will also be discussed. Improving our understanding of how Tregs may influence tumour metastasis may lead to novel treatments for metastatic cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Tregs are a functionally diverse subset of immune suppressive T cells necessary for the maintenance of immune homeostasis. Under normal physiological conditions, Tregs function to prevent the reaction of the immune system to self-antigens, thereby limiting autoimmunity. However, Tregs may also aberrantly suppress the activity of lymphoid and myeloid lineage immune cells, contributing to the pathogenesis of many diseases, including cancer [1] and chronic infection [2]. Despite their active role in disease pathogenesis, Tregs are systemically necessary for the suppression of self-reactive immune cells in peripheral tissues, termed ‘peripheral tolerance’. The existence of a regulatory immune cell population was first recognized during organ transplantation in 1970, when Gershon and Kondo noted that thymus-derived lymphocytes were capable of inducing tolerance when adoptively transferred to thymectomized, lethally irradiated, bone marrow-reconstituted recipient mice treated with sheep red blood cells [3]. This work denoted the presence of ‘suppressor cells’ for the first time, and these suppressor cell populations were later implicated in host immunological tolerance to tumour antigens [4]. The phenotype of these cells remained ambiguous until Sakagucki and colleagues identified CD4+CD25+ ‘regulatory’ T cells as the mediators of tolerance following transplantation of splenocytes into thymectomized mice [5]. Sustained expression of the master regulator and transcription factor forkhead box P3 (Foxp3), or scurfin, is required for the maintenance and immune suppressive function of most Treg subsets [6, 7].
In general, Tregs can be segregated into two subsets based on their ontogeny, including natural or thymus-derived tTregs and peripherally generated pTregs [8–10]. Thymic and peripheral subsets of Tregs are phenotypically characterized by the expression of CD4+CD25highFoxp3+ and based on the methylation status of their Treg-specific demethylated region (TSDR) at the Foxp3 promoter [11]. Thymic Tregs are generated in the thymic medulla from immature CD4+ T cells in response to high-affinity interaction with a self-peptide:major histocompatibility (MHC) complex presented by medullary thymic epithelial cells (mTECs) or dendritic cells (DCs) and function predominantly to regulate autoimmunity [10, 8]. Both subsets require engagement of their T cell receptor (TCR) to become activated. Peripheral Tregs are generated extrathymically from CD4+ T cells (Tconvs) in response to sub-optimal engagement of their TCR co-stimulatory receptors in a tolerogenic environment that induces Foxp3 expression. For example, pTregs may be generated through exposure of naive CD4+ T cells to transforming growth factor-β (TGF-β), interleukin-2 (IL-2) and immature dendritic cells presenting antigen. Tregs may also be generated from Tconvs exposed to prostaglandin E2 (PGE2) and IL-10. Peripheral Tregs are thought to be principally generated at sites of inflammation and have been described to have a diverse recognition of antigens. Additional subsets of T cells defined to have regulatory activity include Tr1 and Th3 helper T cells, double-negative T cells, γδ T cells, NK T cells [12].
While the role of FoxP3 in mediating transcription of the suppressive gene signature in murine Tregs is well established, determining the function of Foxp3 in human Tregs is complicated by the presence of two isoforms, Foxp3A and Foxp3B, for which the cellular distribution and immune suppressive function of each is unknown [13]. However, research has shown that the level of Foxp3 expression in humans is associated with the function of Tregs, where Foxp3high cells display immune suppressive regulatory function [14]. Indeed, individuals born with immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome resulting from a mutation in the Foxp3 gene develop a severe, often fatal, lymphoproliferative disease [6], suggesting an important role for Foxp3 in mediating the immune suppressive function of human Tregs.
Through a variety of mechanisms, Tregs can suppress both adaptive and innate immune responses in a contact-dependant or contact-independent manner. Prior to mediating their suppressive activity, Tregs must be activated by recognition of antigen complexed with MHC II on an antigen presenting cell (APC). Research investigating the antigen specificity of Tregs is still in its infancy, especially with regard to the existence of intratumoural neoantigen-specific Tregs. It is known that a large fraction of Tregs, especially those originating in the thymus, are specific to self-antigens expressed on APCs (as discussed by Gratz and Campbell [15]). This autoreactive nature of Tregs is required for the maintenance of peripheral tolerance and prevention of autoimmune disease. However, within inflammatory environments, antigen recognition by Tregs appears to be more diverse, with Tregs characterized to have reactivity against bacterial, viral, parasitic and alloantigens [8, 9]. Once activated, Tregs may suppress the immune response in an antigen non-specific manner, termed bystander suppression [16, 17]. The method of suppression used by Tregs is a point of controversy; however, it is thought that under homeostatic conditions, thymic Tregs may suppress auto-reactive T cell activation through the deprivation of co-stimulatory signals and IL-2 (reviewed in detail by Yamaguchi et al. [18]). In contrast, during an inflammatory reaction, thymic or peripheral Tregs may inactivate or kill effector T cells and APCs via the release of the suppressive cytokines IL-10, IL-35 and TGF-β or through the release of granzymes and perforin [18].
Regardless of the mechanism of Treg-mediated immune suppression, it is clear that Tregs are relevant to the pathogenesis of many diseases including autoimmune conditions, such as type 1 diabetes, and chronic inflammatory conditions including asthma and inflammatory bowel disease (IBD) [19]. Based on pre-clinical tumour models, a role for Tregs in the progression of many types of cancer has become apparent, especially with regard to the ability of Tregs to promote the development and growth of murine primary tumours. The influence of Tregs on tumour progression in the clinic is less clear, with intratumoural Treg levels correlating with better or worse outcomes depending on the tumour type. There is some pre-clinical evidence that Tregs can accumulate in metastatic tumours, and whether Tregs are involved in promoting the development and growth of tumour metastases is an area of active investigation. Knowledge of the phenotype of Tregs in primary tumours, the mechanisms by which Tregs may promote tumour cell invasion and spread and the potential role of Tregs in the development of metastases may help to facilitate the design of therapies to block the tumour-promoting functions of Tregs and to bypass anti-tumour tolerance. In this review, we will discuss the relationship between Treg levels and therapeutic outcome in the clinic, the phenotypic and functional heterogeneity of Tregs in tumour-bearing hosts and potential mechanisms by which Tregs may influence the development and growth of primary tumours and metastatic tumour foci. We will also discuss recent approaches designed to specifically target Tregs in cancer and examine the effects of these therapies on tumour progression.
2 Accumulation and clinical significance of Tregs in patient tumours
Tregs are observed in many types of solid primary tumours (as reviewed in Facciabene et al. [20]), and Tregs may represent important contributors to tumour progression. Elevated levels of Tregs in solid tumours may be due to recruitment of Tregs from the peripheral circulation and/or expansion of Tregs within the solid tumour microenvironment. There are various proposed mechanisms for the recruitment of Tregs into primary tumours, including attraction to the tumour site by chemotactic gradients. Examples of chemokine interactions resulting in Treg recruitment to the tumour have been reviewed extensively by Mailloux et al. [21] and include CCL22/CCR4 [22, 23], CCL5/CCR5 [24], hypoxia-mediated CCL28/CCR10 [25] and CXCL12/CXCR4 [26]. Other lines of experimentation have suggested the de novo generation of pTregs from conventional CD4+ cells within the primary tumour [27, 28], although the local expansion of infiltrating tTregs may also occur [29]. Clonal expansion of tTregs and generation of pTregs in situ due to production of IL-10, TGF-β and adenosine by tumour cells and myeloid-derived suppressor cells (MDSCs) have been also reported [30]. Finally, it has been found that effector T cells are more vulnerable to reactive oxygen species (ROS) in the tumour microenvironment than Tregs, resulting in an increased abundance of Tregs relative to effector T cells in the ROS-rich tumour milieu [31].
The relationship between intra-tumoural Tregs (or elevated numbers of Tregs in the peripheral circulation) and cancer progression in the clinic is unclear. Systemic accumulation of Tregs, assessed by the immunohistochemical detection of Foxp3+ lymphocytes, has been associated with improved outcome in many haematological malignancies [32–34]. In solid tumours, the prognostic significance of tumour-infiltrating Foxp3+ lymphocytes remains controversial and varies with the tumour type and location. On one hand, a high density of tumour-infiltrating Foxp3+ cells has been associated with poor outcome in ovarian [22, 35, 36], hepatocellular [37, 38], renal [39, 40], pancreatic [41], melanoma [42], colorectal [43] and lung [44] cancers. On the other hand, there is evidence that Treg accumulation is associated with good prognosis in colorectal [45–47], gastric [48] and head and neck carcinomas [49–51]. Various factors, including the functional heterogeneity of tumour-infiltrating Tregs, their accumulation in peritumoural versus intratumoural sites, the levels of CD8+ cytotoxic T cells in the tumour or the immunogenicity of the tumour cells may explain these seemingly contradictory results (reviewed by Elkord et al. 2014 [52]). In addition, the conflicting prognostic significance of Tregs in cancer may reflect methodological differences in the identification and assessment of Tregs in biopsy samples and/or the changing cancer-immune response relationship over the different stages of the proposed immunoediting process [53]. While one of the important functions of the immune system is to detect and destroy aberrant, transformed cells by a process called immunosurveillance, the immune system can also shape and/or promote cancer development by eliminating highly immune-reactive tumour cell clones in the process of immunoediting (reviewed in Dunn et al. [54]). The host-protective and tumour-sculpting actions of the immune system during cancer development are composed of three phases: elimination (i.e. immunosurveillance), equilibrium (between tumour cells and the immune system) and escape of the tumour from immune-mediated equilibrium (extensively reviewed in Dunn et al. [54]). Tregs are crucial players in the escape phase of the cancer immunoediting process. Depending on the immunoediting phase of a particular tumour at the time of biopsy, it is plausible that tumour-infiltrating Tregs found in the biopsy may have growth-inhibiting or growth-promoting effects.
The specificity of Foxp3 to identify Tregs is emerging as an important consideration in the immunohistochemical evaluation of Tregs. Although Foxp3 expression has been considered the most specific and accurate marker to define Tregs in cancer patients [55, 56], naïve CD4+ T cells can transiently express low levels of Foxp3 upon stimulation while they are non-suppressive [13]. Moreover, small numbers of CD8+ cells can exhibit reactivity with anti-Foxp3 antibody [57], and Foxp3 can be expressed by tumour cells in some cases. Foxp3 expression in non-haematopoietic cells and tissues was first published in 2007, when Hinz et al. reported the expression of Foxp3 in pancreatic adenocarcinoma tissue and pancreatic carcinoma cell lines [58]. Similarly, Foxp3 protein expression has been reported in normal and cancerous human breast tissues [59] and, later, in other types of cancer cells (reviewed by Triulzi et al. [60]). Although non-haematopoietic Foxp3 expression has been proposed as a tumour suppressor [61, 62], Foxp3 expression in cancer cells has also been shown to predict poor clinical outcome and metastases in breast, lung, esophageal and urinary bladder cancers [63–66]. This discrepancy could be explained by the recent finding that Foxp3 can induce several genes implicated in migration, EMT and metastasis, in addition to its anti-proliferative action [60]. Foxp3 expression by cancer cells can therefore be viewed as a double-edged sword, and whether one or both roles will take effect in a specific tumour type requires further investigation.
Other markers in addition to Foxp3 have been used to identify Tregs in a range of clinical studies, with some of these markers being related to the immune suppressive function of the cells. Indeed, Foxp3+ tumour-infiltrating Tregs may be divisible into suppressive and non-suppressive populations [67–69], with a decrease in CD39 expression postulated as a major impairment to the suppressive capacity of Tregs. Non-suppressive Foxp3+ Tregs can secrete pro-inflammatory cytokines [67] which may confer better prognosis to some colon cancer patients [70]. A substantial number of IL-17+ Tregs have been identified in patients with colorectal cancer [71]. These cells induced proinflammatory cytokine production and suppressed local T cell immunity simultaneously and, therefore, were linked to active inflammation and tumour development [71]. Another subset of human pTreg found in healthy volunteers co-express the ectonucleotidases CD39 and CD73 and have been found to mediate suppression of effector immune cells ex vivo via the adenosine pathway [72]. In head and neck squamous cell carcinoma (HNSCC), two different CD4+CD39+ T cell subsets (CD25+Foxp3+ and CD25negFoxp3neg) were significantly elevated in the tumour site with the CD4+CD39+CD25+Foxp3+ subset having remarkably higher immunosuppressive function [73]. Melanoma-infiltrating Foxp3+ Tregs predominantly contain a Foxp3hi CD45RAneg aTreg immunosuppressive sub-population [74], while Foxp3+ Tregs infiltrating colon cancers contain higher frequencies of non-suppressive Foxp3low CD45RAneg Treg populations (reviewed by Nishikawa and Sakaguchi [70]). Treg function in primary tumours may also be influenced by different tumour phenotypes, molecular subtypes or microenvironmental parameters. For example, the favourable prognosis associated with the high density of Foxp3+ T cells infiltrating colorectal carcinomas could be attributed to their ability to suppress tumour-promoting inflammatory immune responses generated by infectious stimuli from bacteria translocated through the mucosal barrier [46]. Furthermore, Foxp3+ Tregs may suppress the pro-inflammatory and tumour-promoting capacities of Th17 cells infiltrating colorectal cancer [75, 46]. Regardless of the mechanisms involved, it is clear that regulatory T cell populations can have a variable impact on clinical outcome depending on the tumour type, and researchers are cautioned to consider whether Tregs (alone) can be considered robust biomarkers of the tumour-associated immune response and/or therapeutic outcome in a particular patient population. In the following section, we will discuss the prognostic value of Tregs infiltrating breast carcinomas as a prototype for the heterogeneous prognostic role of tumour-infiltrating Tregs within the same cancer type.
3 Prognostic significance of Tregs in primary breast tumours
Breast cancer is a remarkably heterogeneous disease, embracing a wide range of clinical patterns, stages of presentation, biological behaviour, prognostic characteristics and response to different types of treatment. The number of immunosuppressive CD4+CD25+ Tregs has been shown to significantly increase in the peripheral blood and the primary tumour of patients with invasive breast cancer [76, 77]. Table 1 summarizes several prognostic studies that investigated Foxp3+ Tregs infiltrating breast carcinomas. The first study to report the relationship between tumour-infiltrating Treg numbers and clinical outcome was performed by Bates et al. [76]. They demonstrated that Foxp3+ infiltrating cells were an independent factor for poor prognosis in patients with oestrogen receptor (ER) positive tumours, but not in patients with ER-negative tumours [76]. Similarly, a high number of Foxp3+ Tregs infiltrating breast tumours were significantly associated with a high risk of relapse and death [76, 78, 79, 24, 80], especially in ER-positive [24, 80] and basal-like tumours [80]. Interestingly, and opposite to the previously reported results, high Foxp3+ tumour-infiltrating leukocyte (TIL) levels were significantly associated with a favourable outcome in 143 ER-negative breast tumours, independent of standard prognostic factors [81]. The same results have been confirmed for triple-negative (n = 82) and basal-like (n = 65) tumours. Intriguingly, the association of Foxp3+ TIL with good outcome was dependent on the presence of large numbers of CD8+ TIL [81]. In patients who received chemotherapy, the presence of Tregs resulted in significantly less relapses independent of other factors such as nodal stage and HLA class I expression level [82]. In a larger (n = 1445), well-characterized series of invasive breast cancer cases with long-term follow-up, Mahmoud et al. investigated the prognostic value of tumour-infiltrating Foxp3+ cell density and tumour microenvironmental localization [83]. Although higher numbers of Foxp3+ infiltrating cells were associated with a worse prognosis on univariate analysis, the number of Foxp3+ cells was not identified as an independent prognostic factor in multivariate analysis [83]. The Mahmoud et al. data set [83] was recently integrated with a larger study to evaluate the prognostic value of T lymphocytes in breast cancer [84]. Foxp3+ T lymphocytes infiltrating breast cancer tissues from 5239 patients did not have any significant association with clinical outcome after adjustment for known prognostic factors [84]. Moreover, the interaction between different immune cells infiltrating 1113 breast carcinomas, assessed by the CLOPE clustering algorithm (for clustering large transactional databases with high dimensions) [85], did not include Foxp3+ Tregs as a leading discriminative marker in the prognostic cluster [86].
Foxp3 expression in breast tumour cells has been identified as an independent prognostic factor for improved clinical outcome in 103 patients with HER2 over-expressing tumours [87]. These data are in apparent contradiction with Merlo et al., who reported an independent association between high expression of Foxp3 by breast tumour cells and poor prognosis [64]. However, it was shown that Foxp3 acts as a transcriptional repressor for the breast cancer oncogenes SKP2 and HER2, suppressing breast tumour cell growth in mice and humans [88, 59]. These data suggest that the function of Foxp3 in breast cancer cells may closely relate to the HER2 oncogenic pathway. Consistent with Foxp3 functioning as an important tumour suppressor in breast cancer, loss of Foxp3 nuclear expression in mammary epithelial cells has been recently shown to augment CXCR4 expression and, consequently, metastasis via the chemotactic response of CXCR4+ tumour cells to CXCL12 expressed in peripheral tissues [89].
It is important to consider the clinical significance of Treg accumulation in breast tumours prior to and after administration of neoadjuvant therapies, which are commonly employed in breast cancer patients to decrease tumour size prior to surgery. Pathologic complete response (pCR), as recently refined by von Minckwitz et al. [90], has been used to predict long-term outcome after neo-adjuvant chemotherapy of patients with breast cancer based on the presence or absence of residual disease. Intratumoural Treg accumulation in patients who have received neo-adjuvant therapy has been shown to affect the pCR rate of these patients. For example, a study of 101 locally advanced breast cancer patients found that a decrease in intratumoural Treg levels after neo-adjuvant chemotherapy was associated with a positive pCR [91]. Futhermore, the density of intratumoural Tregs prior to neo-adjuvant therapy was a strong predictive factor for patient survival, with higher intratumoural Treg levels prior to chemotherapy associated with decreased overall survival [91]. Several additional studies have highlighted a correlation between Treg levels and response to neo-adjuvant therapy [78, 92, 93]. Specifically, Verma et al. found that the percent of systemic Foxp3+ Tregs predicted the response of tumours to neo-adjuvant chemotherapy, with lower systemic Treg levels prior to neo-adjuvant therapy conferring improved response to treatment in women with large and locally advanced breast tumours [93]. Interestingly, neoadjuvant therapy and surgery were unable to restore Tregs to the levels observed in healthy women, which may suggest the presence of residual metastatic disease [93]. Indeed, the potential influence of Tregs on the development and growth of tumours is an important area of research, and both clinical and pre-clinical data are emerging to support the concept that Tregs may play important roles in metastasis.
4 Association of tumour-infiltrating Tregs with metastatic disease
The recruitment of Tregs to primary lesions has been associated with the development of metastases in patients with breast [94–97, 81], gastric [98], prostate [99], colorectal [43], renal [100], thyroid [101] or lung cancers [102]. In breast cancer, intratumoural Foxp3+ lymphocytes were found to increase with progression from normal breast tissue to ductal carcinoma in situ (DCIS) to invasive ductal carcinoma (IDC) and were highly correlated with the invasive characteristics of the tumour [95, 97]. The accumulation of Tregs has also been observed in colorectal cancer, where patients with liver metastases have elevated levels of functionally immune suppressive CD4+CD25+CD127dim/- Tregs in their peripheral blood prior to surgery [43]. Similarly, elevated levels of CD4+CD25+CD127low/- Tregs were identified in the peripheral blood of patients with metastatic castration-resistant prostate cancer (mCRPC) versus healthy donors [99]. Populations of Tregs from the peripheral blood with similar suppressive activity were analyzed for differential gene expression, revealing that Tregs from mCRPC patients over-expressed factors involving T cell proliferation (C-FOS, C-JUN, DUSP1), cell cycle progression and inhibition of cellular migration (RGS-1) [99]. Interestingly, the upregulation of proliferation and differentiation factors suggested that regulatory cells from mCRPC were capable of bypassing anergy, potentially in response to activation signals such as tumour secreted factors, IL-2 and exposure to their cognate antigen. It was hypothesized that the increased expression of the anti-migratory factor, RGS-1, was related to Treg accumulation and arrest within mCRPC tumours [99]. Furthermore, in clear cell renal cell carcinoma (CCRCC) the intratumoural infiltration of Foxp3+ and programmed cell death-1+ (PD-1+) lymphocytes correlated with risk of distant metastatic relapse [100]. Thus, the accumulation of Tregs systemically and within primary lesions has been associated with metastatic disease in several tumour models, as outlined in Tables 2 and 3, and Tregs exhibit a variety of functions that may promote dissemination of tumour cells from the primary tumour mass.
5 Functions of Tregs in promoting metastatic tumour cell dissemination
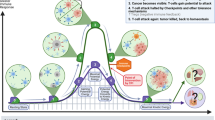
Regulatory T cells may promote tumour metastasis by suppressing anti-tumour immune responses and/or by directly facilitating the invasion and migration of tumour cells (Fig. 1). Tregs may suppress cytotoxic immune responses against tumour cells by releasing soluble immune suppressive molecules including granzymes [103], galectin-1 [104], adenosine [105], PGE2 [43] or indolamine dioxygenase (IDO) [106] (Fig. 1a). Mechanisms of Treg contact-dependent suppression include cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) [107], programmed death-ligand 1 (PD-L1) [108, 106], lymphocyte-activation gene 3 (LAG-3) [109], neuropilin 1 (Nrp1) [110] and CD39/73 [105] expression. The mechanisms used by Tregs to promote primary tumour growth vary with the type of tumour, its location and the surrounding tissue microenvironment (reviewed in detail by Facciabene et al. [20]). Protumourigenic functions of Tregs identified in pre-clinical tumour models include roles in immunosuppression and angiogenesis [111], and Tregs may influence the function of other immune cells as well as malignant and stromal cells [112]. However, mechanisms by which Tregs may influence metastatic tumour cell dissemination or metastatic tumour growth are relatively poorly understood and represent an area of increasingly active investigation.

Tregs promote metastasis of the primary tumour through a direct mechanisms such as RANKL expression and maspin suppression in RANK+ tumour cells or through secretion of the angiogenic factor VEGF-A. Tregs may indirectly promote tumour metastasis by suppressing intratumoural immunity using contact-dependant or contact-independent mechanisms. Depending on the type of cancer, tumour cells may metastasize via the bloodstream or lymphatics to a variety of secondary organs such as the lungs or lymph nodes. b In the metastatic lungs, tumour cells can induce tolerance by secreting factors chemotactic for Tregs, including CCL17 and CCL22 in the 4T1 model of murine mammary carcinoma. Once recruited to the metastatic lungs, Tregs may inhibit the function of cytotoxic T lymphocytes (CTLs) or initiate the apoptosis of CTLs and/or NK cells. c In the metastatic lymph nodes, DCs loaded with tumour antigen prime CTLs for the recognition and destruction of tumour cells. DCs may also induce Treg generation through the secretion of soluble factors such as TGF-β, IL-10, retinoic acid, IDO and vitamin D. Furthermore, DCs can activate tumour-antigen specific Tregs which then suppress anti-tumor cytotoxicity in an antigen specific or non-specific manner
In patients with colorectal cancer, cyclooxygenase-2 (COX2) and PGE2 expression are associated with recurrent metastatic disease of the liver. CD4+CD25+CD127dim/- Tregs were found to actively suppress tumour-specific effector and cytotoxic responses against the colorectal antigen CEA61–69 as evident by a decrease in interferon-γ (IFNγ) and tumour necrosis factor-α (TNFα) expression, and this suppression could be inhibited with the COX antagonist indomethacin [43]. Correspondingly, patients with recurrent metastatic disease accumulated intratumoural Tregs expressing high levels of COX2 and had increased plasma levels of the tumour proliferation, angiogenesis and immune suppressive factor PGE2 [43]. The expression of COX2 by Tregs suggests that Tregs facilitate immune suppression and tumour development via PGE2 expression within colorectal tumours [43]. A study of murine breast cancer found that COX2 expressing tumour cells produced PGE2 and recruited Tregs expressing the cognate receptors EP2 or EP4 to the primary tumour to promote bone metastasis [113]. Additionally, lipocalin 2 (LCN2) expressing tumour cells were found to promote the expansion of Tregs and thereby facilitate tumour cell metastasis in a murine model of colorectal cancer [114]. For instance, CCL2 secreted by Snail+ tumour cells had a dual function in promoting tumour cell epithelial to mesenchymal transition (EMT) while inducing LCN2 production. LCN2 produced by Snail+ tumour cells promoted the generation of CD4+Foxp3+ Tregs that promoted tumour metastasis by suppressing cytotoxic T lymphocytes, thereby generating a suppressive microenvironment that shielded tumour cells from immune attack and indirectly facilitated their EMT [114].
Neuropilin-1 (Nrp1) expression by intratumoural Tregs is thought to potentiate anti-tumour immune suppression by stabilizing the Treg phenotype through an increase in survival and quiescence factors while repressing Treg differentiation [110]. Expression of the Nrp1 ligand Sema4a was enriched on tumour-infiltrating plasmacytoid dendritic cells, which are thought to aid Tregs in promoting tolerance induction [110]. Cre/Lox-based Foxp3-specific ablation of Nrp1 reduced tumour growth in colon carcinoma (MC38), melanoma (B16.F10) and thymoma (EL4) [110]. Furthermore, Tregs from B16 tumour-bearing mice lacking Nrp1 had fewer lung metastases and increased intratumoural CD8+ T cells expressing IFNγ and TNFα, suggesting that Tregs promoted metastasis of melanoma cells through the induction of intratumoural tolerance [110]. Nrp1 expressing Tregs are recruited by intratumoural expression of their cognate ligand vascular endothelial growth factor (VEGF) [115]. Blockade of this signalling axis through ablation of Nrp1 or VEGF significantly reduced intratumoural Treg accumulation and resulted in attenuated tumour growth and increased activated CD8+ T cell infiltrates in murine models of melanoma and fibrosarcoma [115]. These findings suggest that Nrp-1 expression stabilizes the Treg phenotype and permits Treg migration toward a gradient of VEGF. Once in the tumour, Nrp-1+ Tregs contribute to tolerance and tumour dissemination [110, 115]. Additionally, these studies provide mechanistic insight into previous observations of the correlation between VEGF levels and Treg infiltration in ovarian [116] and breast cancer [117].
Aside from suppressing innate and adaptive immunity against tumour cells, Tregs may facilitate cancer progression by directly promoting tumour cell angiogenesis and invasion (Fig. 1a). CD4+CD25+ cells purified from the peripheral blood mononuclear cells (PBMCs) of patients with stage III or IV epithelial ovarian cancer, as well as CD4+CD25+Foxp3+ Tregs from murine spleens, secrete VEGF-A and induce endothelial cell accumulation and expansion in vitro [25]. Additionally, in pre-clinical models, CD4+CD25+Foxp3+ Tregs were found to produce receptor activator of nuclear factor kappa-B ligand (RANKL), a factor important for osteoclast differentiation and activation, as well as mammary gland lactational hyperplasia [118]. RANKL production in metastatic mammary carcinomas overexpressing the proto-oncogene Erbb2 directly stimulated the invasion of RANK+ breast tumour cells by suppression of the metastasis inhibitor maspin [24]. These studies highlight that Tregs may promote metastasis by mechanisms other than immune suppression, although the direct impact of Tregs on tumour cells to facilitate metastasis warrants further study.
Tregs play an active role in promoting tumour growth and may directly promote metastasis by interacting with tumour cells to suppress metastasis inhibiting factors, such as maspin, or by releasing angiogenic factors like VEGF-A. Tregs can also indirectly promote tumour spread by suppressing tumour-specific cytotoxic responses, and these findings highlight the role of tumour-infiltrating Tregs in promoting the growth of primary tumours and dissemination of metastatic tumour cells. Interestingly, emerging pre-clinical evidence suggests that Tregs may also influence the development and growth of metastatic tumour foci by accumulating in tissues distant from the primary tumour.
6 Accumulation of Tregs in metastases
Tregs are enriched within the metastatic lymph nodes [119, 101, 120, 121] and peritoneal metastases [122] of some cancer patients; however, the association of Tregs with the incidence of recurrent disease varies with the type of cancer. For instance, CD4+CD25+Foxp3+ Tregs were increased in metastatic lymph nodes of patients with recurrent papillary thyroid cancer (PTC) versus metastases-free nodes [101]. Correspondingly, metastatic lymph nodes with evidence of extranodal invasion had a high frequency of PD-1+ T cells, and the expression of PD-1 has been associated with T cell exhaustion and as a mechanism of immune suppression utilized by Tregs [101]. Concurrent Foxp3 and PD-1 expression was therefore suggested as a biomarker of aggressive or recurrent disease in patients with PTC [101]. CD4+CD25+CD127low/- Tregs have also been shown to accumulate in the axillary tumour draining lymph nodes (TDLNs) of previously untreated breast cancer patients and to decrease the expression of pro-inflammatory factors such as IL-17 and IFNγ [119]. It is thought that through this mechanism, these regulatory cells thereby function to skew the immune response toward an anti-inflammatory, pro-tumorigenic phenotype within TDLNs to facilitate tumour metastasis [119] (Fig. 1c). Additionally, sentinel lymph nodes (SLN) of breast cancer patients positive for Foxp3+ lymphocytes and IDO were highly associated with metastatic disease [121]. Patients with gastric cancer have also displayed elevated levels of Foxp3+ Tregs in primary lesions as well as metastatic lymph nodes [120]. The accumulation of Foxp3+ lymphocytes in gastric tumours was inversely correlated with CD83+ dendritic cell density and was associated with tumour progression and poor prognosis [120]. Unfortunately, the study of Tregs in humans is largely restricted to the primary lesion, blood or lymphatics (due to the relative ease of obtaining diagnostic tissue biopsies and blood samples), and there is a paucity of clinical information regarding the presence of Tregs in other organs that harbour tumour metastases (Table 3).
Tregs have been isolated from the peritoneal metastases of ovarian cancer patients [122], although CD4+CD25high cells present in peritoneal metastases were associated with improved overall and progression-free survival. While these findings may be attributed to the inclusion of activated conventional T cells in the analyzed population of CD4+CD25high cells [122], it remains possible that Treg infiltration into tumour metastases may result in improved outcome for some tumour types. Thus, establishing how Tregs may influence metastatic tumour growth in patients must be addressed before therapeutic strategies to target Tregs can be implemented in the clinic. Interestingly, Tregs have also been identified and isolated from the lungs of mice bearing metastatic primary tumours [123–131] as indicated in Table 2, where they promote metastatic tumour growth in lung tissue.
Immune suppressive Tregs have been observed in the lungs of mice bearing metastatic breast tumours (4T1, 4T1.2) [123, 124, 126–128, 130, 131], melanoma (B16F10) [125], colorectal cancer [129, 132] and acute lymphoblastic leukaemia (CCRF-CEM) [124]. Importantly, ablation or inhibition of the regulatory T cell population using a variety of methods (outlined further below) hindered the development of pulmonary metastases [108, 126, 130, 131], providing evidence that Tregs may promote metastatic growth in these model systems.
7 Functions of Tregs in promoting the growth of metastatic foci
It is known that within the primary tumour, Tregs may suppress tumour-specific cytotoxic immune responses or directly interact with tumour cells to facilitate proliferation and metastasis of the malignancy. However, both the recruitment of Tregs to metastatic sites and the potential activity of Tregs in metastatic target organs are less clear. A study by Biragyn et al. [124] found that tumour-bearing lungs secreted CCL22 and CCL17 to recruit CCR4+ Tregs [130] (Fig. 1b). Once in the metastatic lungs, Tregs initiated the apoptosis of natural killer cells via the production of beta-galactoside-binding protein (β-GBP) (Fig. 1b) [130]. Tregs have also been observed to accumulate in the metastatic lungs of a murine model of melanoma (B16F10), where they were associated with the generation of anti-tumour tolerance and metastatic foci development [125]. In this study, administration of recombinant TNF was found to promote tumour metastasis and was associated with enhanced recruitment of Tregs to metastatic lungs. Pulmonary-infiltrating Tregs were preferentially enriched for TNF-R2 expression and promoted a tolerogenic environment that fostered metastatic tumour growth, evident by a reduction in the number of CD8+ T cells in the metastatic lungs of mice given TNF [125]. Furthermore, ablation of Tregs by administration of diphtheria toxin to B6.Foxp3.Luci.DTR-4 mice significantly reduced pulmonary metastasis, even in the presence of recombinant TNF, suggesting that Tregs were necessary to facilitate melanoma metastasis [125].
In a subsequent study, Biragyn et al. (2011) found that tumour-evoked CD25+CD19+B220+ regulatory B cells (tBregs) constitutively expressing signal transducer and activator of transcription 3 (STAT3), a key transcriptional regulator of cytokine and growth factor signalling, promoted the pulmonary metastasis of tumour cells by facilitating the TGF-β-dependant generation of Tregs that suppressed T cells in vitro [131]. Importantly, the adoptive transfer of tBregs from Balb/c mice in conjunction with CD4+CD25- non-Tregs was sufficient to facilitate the lung metastasis of 4T1 cells in NOD/SCID mice [131]. Immunodeficient NOD/SCID mice lack functional T cells and B cells and, therefore, the use of this model removes the confounding affect of host Tregs in metastasis. This finding was also supported in a study using resveratrol (RSV) to inactivate STAT3. RSV treatment prevented tBreg formation, thereby dampening TGF-β production and Foxp3+ Treg generation to hinder pulmonary metastasis [133]. In the pre-metastatic lungs of 4T1 tumour-bearing mice, CD4+ and CD8+ T cells were suppressed by a signalling axis that involved complement anaphylatoxin C5a receptor (C5aR) [134]. C5a expression in the lungs preceded tumour cell arrival and recruited C5aR+ MDSCs that favoured Treg development via the production of TGF-β and IL-10 [134]. Importantly, by generating an immune suppressive microenvironment, C5aR facilitated the development of lung metastases [134].
Tregs have been shown to participate in the metastasis of primary tumours as well as in the initiation and development of metastatic foci in pre-clinical tumour models. Additional study is needed to determine if there are preferential mechanisms of immune suppression utilized by Tregs within metastatic foci or if Tregs display non-immune-related functions in distant tissues that may promote metastatic tumour development and growth. It also remains to be determined if mechanisms of Treg accumulation and immune suppression observed in pre-clinical tumour models occur in cancer patients. Knowledge of Treg function at primary and secondary tumour sites may facilitate the design of therapies that help to prevent tumour cell dissemination as well as the development of metastatic tumour foci. Treatments of this nature could be advantageous in combination with current immunotherapies to stimulate anti-tumour immune responses while depleting tumour-specific immune suppressive Tregs in patients with potential or established metastases.
8 Methods to target Tregs
The presence of Tregs in the tumour microenvironment can create tolerance toward tumour antigens [135], and Tregs may promote the growth and spread of many malignancies by suppressing the function of tumour-antigen-specific effector T cells and APCs [136]. By suppressing anti-tumour cytotoxicity, Tregs limit the efficacy of immune-stimulating therapies in eradicating tumours. Targeting Tregs either as a monotherapy or as an adjuvant to vaccination has emerged as a viable immunotherapeutic strategy to break tolerance against tumour antigens [137]. Several lines of therapy have been developed to systemically deplete Tregs or hinder their expansion or immune suppressive function. Taxanes, as well as CD25 blocking antibodies or antibody-conjugated immunotoxins, such as denileukin diftitox, have been shown to deplete Tregs [138, 139], whereas tyrosine kinase (TK) inhibitors such as imatinib have been shown to reduce Treg expansion [138]. The immune suppressive function of Tregs can be attenuated by TK inhibitors, low-dose cyclophosphamide, IDO inhibition or checkpoint blockade of PD-L1 and/or CTLA-4 [137]. Depending on the therapeutic utilized, the global modification of Tregs has been associated with off-target elimination of effector T cells, increased conversion of effector T cells to Tregs or, in contrast, the development of an autoimmune phenotype [138]. Attempts to target global populations of Tregs are limited by the systemic requirement for these cells to maintain peripheral tolerance, and therefore therapeutic strategies to specifically target Tregs that accumulate in solid tumours would represent a significant advancement in the treatment of many malignancies.
9 Impact of Treg ablation on metastasis and metastatic foci development
Tregs have been targeted in several pre-clinical tumour models including osteosarcoma [140], breast cancer [141, 124, 142], melanoma [143], hepatocellular carcinoma [144], colorectal [129] and renal cancers [145]. In many cases, the ablation or functional inactivation of the regulatory cell population is associated with primary tumour regression and the prevention or inhibition of metastases development [141, 124, 142–144, 129]. However, additional research is necessary to clarify whether the observed reduction in metastases development upon ablation or functional inhibition of Tregs is attributed to a localized and specific effect on metastatic foci or to a regression of the primary tumour and concomitant decrease in tumour cell dissemination. For example, treatment of tumour-bearing mice with the adenosine A2B receptor antagonist PSB603 reduced splenic Treg accumulation, primary tumour growth and pulmonary nodule formation in a B16F10 model of melanoma without directly affecting tumour cell viability [143]. This effect was attributed to reduced generation of pTregs from CD4+ T cells and increased systemic populations of helper and cytotoxic T cells leading to an increased anti-tumour cytotoxicity response [143]. In a subsequent study, treatment of HER2+ tumours with the MVA-BN®-HER2, a Vaccinia Ankara-based recombinant vaccine, was associated with regression of pulmonary tumour cell foci generated by i.v. injection of CT26-HER2+ tumour cells and a decrease in intrapulmonary Tregs [129]. Following administration of MVA-BN®-HER2, the frequency of Tregs within the pulmonary lesions of a murine lung metastasis model (CT26-HER2+) corresponded with increased infiltration of functional HER-2 specific CD8+CD11c+ T cells [129]. Depletion of CD25+ Tregs with an anti-CD25 monoclonal antibody (PC61) reduced pulmonary tumour burden; however, its combination with MVA-BN®-HER2 was most efficacious in prolonging survival [129]. Thus, while a few studies have suggested that Tregs may impact metastatic tumour growth independently from decreasing primary tumour growth, additional Treg inhibitory studies are necessary using pre-clinical models of spontaneous and experimental metastasis to further define Treg function at metastatic sites.
Methods of Treg specific inactivation have been developed in murine systems using targeted anti-sense oligonucleotide delivery systems such as TARC-arp and RANTES-arp. The ‘arp’ domains represent a single DNA/RNA-binding domain (RBD) from the capsid of hepatitis B virus (HBV) fused to TARC (CCL17) or RANTES (CCL5) to permit Foxp3 siRNA binding by the associated chemokine. TARC and RANTES are chemotactic for CCR4- and CCR5-expressing cells, respectively [124]. These chemokines are internalized into the cytosol of cells expressing the appropriate receptors to transiently silence Foxp3 expression and function with antisense oligonucleotide/RNAi [124]. In the 4T1.2 model of breast cancer, treatment of mice with TARC-arp conjugated to siRNA reduced Foxp3 expression up to 50 % in CCR4+ Tregs. Inactivation of CCR4+ Tregs resulted in a substantial reduction in pulmonary metastasis [124]. A reduction in pulmonary metastatic foci was also observed when IL-10 was knocked out or silenced with TARC-arp siIL-10, affirming an important role for IL-10 in mediating the immune regulatory functions of Tregs [124]. Chemo-arp systems, such as those discussed above, have not yet been applied in therapeutic settings, but could potentially be adapted for use in cancer patients for the selective silencing of genes in Tregs. A reduction in metastasis and metastatic foci development with the disruption of Tregs has also been observed in osteosarcoma, where administration of anti-CD25 monoclonal antibody (PC61) systemically depleted Tregs and corresponded with regression of the primary tumour and a decrease in the number and size of lung and liver metastases [140]. Overall, the reduction in metastasis associated with Treg ablation seems to be attributed to a decrease in Treg differentiation or accumulation in the primary tumour coupled with an increase in effector cells with anti-tumour activity.
While several pre-clinical studies have investigated the therapeutic potential of targeting Tregs to reduce murine primary tumour growth and metastasis, there are several drugs in clinical trial as immune-based therapeutics for cancer patients that may also target Tregs. For the treatment of HER2-positive breast cancer, novel vaccination approaches to break immune tolerance toward HER2 have been employed using attenuated bacteria (Listeria monocytogenes) designed to secrete adjuvant fusion proteins to stimulate an immune response against HER2 [146]. One such vaccine, ADXS31–164, stimulated an effector response against HER2, increased the CD8+/Treg ratio and delayed tumour progression [146]. Live-attenuated L. monocytogenes vaccines (Lm-LLO) have been used in phase I clinical trials for advanced cervix carcinoma [147]. Interestingly, the administration of Lm-LLO vaccines in pre-clinical models decreased the suppressive activity of MDSCs and Tregs, associated with a reduction in IL-10 and arginase I expression [148]. Additional approaches to target HER2 involve the use of MVA-BN®-HER2, which is currently in phase I clinical trial for breast cancer (NCT0048277) [129].
Cyclophosphamide (CTX), a nitrogen mustard alkylating agent, is frequently used in a clinical setting for the treatment of a variety of solid tumours and has also been shown to deplete Tregs. In one study, repeated low-dose administration of CTX as a monotherapeutic agent in treatment-refractory patients with metastatic (stage IV) breast cancer initially reduced circulating Tregs by 40 % [142]. However, the level of functionally immune suppressive Tregs rebounded 42 days after initial drug administration to reach pre-treatment levels by day 84 [142]. This transient Treg depletion corresponded with an increase in the generation of tumour reactive effector cells that were stable in frequency throughout the treatment period [142]. In contrast, a single dose of cyclophosphamide administered in combination with the non-specific immune-stimulating agent bacille Calmette–Guérin (BCG) failed to alter Treg number or function in patients with metastatic cancers of the breast, lung, kidney, stomach, colon, bladder or prostate [149]. These results suggest that a single treatment with CTX is insufficient to deplete circulating Treg numbers and that perhaps continuous low-dose (i.e., metronomic [150]) treatment with CTX may be necessary to increase the overall survival of patients with metastatic cancer [149]. The mechanism for CTX depletion of Tregs is unclear; however, the effect of this drug is thought to be related to an increase in the availability of tumour antigens and/or a transient decrease in functional Tregs with the preservation of conventional T cells [149, 142].
Many drugs that are capable of depleting Tregs have been investigated as a combination therapy with immune-stimulating agents. A recent study by Quezada et al. investigated the efficacy of administration of anti-CTLA-4 in combination with an irradiated tumour cell vaccine actively secreting GM-CSF (GVAX+α) to protect against B16-BL6 melanoma progression [151]. When administered separately, these agents were unable to prolong survival; however, when given in combination, they significantly reduced tumour progression and increased the Teff/Treg ratio [151]. A separate study examined the role of combination therapy for the depletion of Tregs using an orthotopic, polyomavirus middle-T antigen-driven model of breast tumour progression in Foxp3DTR mice [108]. Whereas αCTLA-4 or αPD-1 + αPDL1 alone did not cause a significant reduction in tumour burden, administration of diphtheria toxin to ablate Tregs in conjunction with either αCTLA-4 or αPD-1 + αPDL1 significantly decreased tumour volume [108]. Additionally, diphtheria toxin administration alone or in conjunction with αCTLA-4 or αPD-1 + αPDL1 significantly reduced the number of metastatic lung foci [108]. Consistent with previous findings, αCTLA-4 alone did not decrease the number of lung metastases, whereas αPD-1 + αPDL1 treatment significantly decreased the number of lung nodules. This study also found that a single-dose administration of ionizing radiation (12 Gy) to the tumour in combination with diphtheria toxin administration synergistically reduced tumour growth, decreased the number of metastatic lung nodules and increased the survival of tumour-bearing mice [108]. These results highlight the synergistic effect of Treg depletion with immune-stimulating agents or radiation. However, it should be noted that the systemic ablation of Tregs has been associated with a myriad of autoimmune pathologies as well as lymphoproliferative syndrome, indicating that transient Treg inhibition may be more ideal for use in the clinic in combination with other treatments. By inhibiting multiple avenues of Treg function and recruitment, combination therapies may offer enhanced specificity for targeting tumour-infiltrating Tregs.
10 Conclusions
The accumulation of Tregs in solid tumours has been associated with unfavourable prognosis in many pre-clinical tumour models and cancer patients. However, some clinical studies have found that intratumoural Treg accumulation can correlate with improved patient prognosis, especially when associated with an increase in intratumoural CD8+ TIL. Collectively, it is important to consider the net result of immune cell interactions in the primary tumour microenvironment and observed differences between tumour types when considering how Tregs may influence patient prognosis. Ultimately, the clinical relevance of tumour-infiltrating Tregs may be best studied by unifying the markers used to define and isolate Tregs and by better defining the functional role of Tregs within different types of cancer. In view of the emerging pivotal role of the host immune system in controlling tumour progression, an international task force has been initiated to promote the utilization of a “cancer immunoscore” into routine clinical practice [152]. This new approach for cancer classification provides recommendations for the worldwide harmonization and implementation of the immunoscore (I) as a new component of the tumour-node-metastasis (TNM) cancer classification system (TNM-I) [152, 153]. This initiative will provide important information about the influence of tumour-infiltrating Tregs (and other immune cells) on therapeutic outcome, and will help to identify patients that may benefit from novel therapeutic strategies designed to target Tregs.
The mechanism(s) used by Tregs to promote primary tumour growth seem to vary with the tumour model, and for this reason, it is important to validate the functions of Tregs most relevant to tumour progression in cancer patients. Analysis of Treg immune suppressive activity in patient-derived xenografts using mice with a humanized immune system [154] may help to better define the Treg subsets and suppressive mechanisms most relevant to clinical tumour progression. Within the primary tumour, Tregs may indirectly facilitate tumour progression by suppressing immune-mediated cytotoxicity, thereby masking tumour cells from eradication by the immune system and allowing the cells to metastasize. Tregs may also directly facilitate tumour progression by releasing VEGF or by reducing the expression of the metastasis-inhibiting factor maspin. Overall, the protumorigenic functions of Tregs have been linked to tumour cell dissemination from the primary tumour to secondary sites. Tregs have also been identified within metastatic lymph nodes and peritoneal metastases of cancer patients and in the lungs of mice bearing metastatic primary tumours. Relatively few mechanisms have been established describing the recruitment of Tregs to metastatic target organs or the role of Tregs within metastatic foci; however, evidence suggests that the induction of tumour-antigen tolerance at secondary sites is likely important for metastatic outgrowth. It remains to be determined if similar immune suppressive mechanisms are used by Tregs at the primary tumour and within metastatic foci. If so, targeting conserved avenues of Treg function at multiple tumour sites may enhance anti-tumour immunity and the eradication of disseminated tumours.
While the ablation of Tregs has been shown to increase anti-tumour immunity in some tumour models, especially when administered as a combination therapy with immune stimulating agents, therapies to ablate Tregs are complicated by the systemic requirement of tTregs for the maintenance of peripheral tolerance. In the absence of Tregs, chronic inflammation and autoimmune conditions may develop, suggesting that targeting specific tumour-promoting functions of Tregs may represent an attractive alternative to directly targeting Tregs themselves. Regardless, the ablation or functional inactivation of Tregs has been shown to induce primary tumour regression and the prevention or inhibition of metastases development in several tumour models, providing support for the concept that targeting Tregs to reduce tumour burden may represent a viable therapeutic strategy. As tumour-infiltrating Tregs may be of thymic or peripheral origin, the identification of a single marker or set of markers that could be used to specifically target the intratumoural Treg population remains an open question. Additional studies to determine the context in which Tregs promote or inhibit tumour progression is also critical to the development of therapeutic strategies to target Tregs in patients. Ultimately, the study of Treg phenotype and function within the primary tumour and metastatic foci should aid in the design of therapies with increased specificity for the elimination of intratumoural Tregs while minimizing the disruption of peripheral tolerance.
Abbreviations
- Tregs:
-
Regulatory T cells
- pTregs:
-
Peripheral regulatory T cells
- tTregs:
-
Thymic regulatory T cells
- Foxp3:
-
Forkhead box P3
References
Beyer, M., & Schultze, J. L. (2006). Regulatory T cells in cancer. Blood, 108(3), 804–811.
Maizels, R. M., & Smith, K. A. (2011). Regulatory T cells in infection. Advances in Immunology, 112, 73–136.
Gershon, R. K., & Kondo, K. (1970). Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology, 18(5), 723–737.
Berendt, M. J., & North, R. J. (1980). T-cell-mediated suppression of anti-tumor immunity. An explanation for progressive growth of an immunogenic tumor. Journal of Experimental Medicine, 151(1), 69–80.
Sakaguchi, S., Sakaguchi, N., Asano, M., Itoh, M., & Toda, M. (1995). Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. Journal of Immunology, 155(3), 1151–1164.
Bennett, C. L., Christie, J., Ramsdell, F., Brunkow, M. E., Ferguson, P. J., Whitesell, L., et al. (2001). The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nature Genetics, 27(1), 20–21.
Williams, L. M., & Rudensky, A. Y. (2007). Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nature Immunology, 8(3), 277–284.
McMurchy, A. N., Bushell, A., Levings, M. K., & Wood, K. J. (2011). Moving to tolerance: clinical application of T regulatory cells. Seminars in Immunology, 23(4), 304–313.
Pacholczyk, R., & Kern, J. (2008). The T-cell receptor repertoire of regulatory T cells. Immunology, 125(4), 450–458.
Bilate, A. M., & Lafaille, J. J. (2012). Induced CD4+Foxp3+ regulatory T cells in immune tolerance. Annual Review of Immunology, 30, 733–758.
Toker, A., Engelbert, D., Garg, G., Polansky, J. K., Floess, S., Miyao, T., et al. (2013). Active demethylation of the Foxp3 locus leads to the generation of stable regulatory T cells within the thymus. Journal of Immunology, 190(7), 3180–3188.
Shalev, I., Schmelzle, M., Robson, S. C., & Levy, G. (2011). Making sense of regulatory T cell suppressive function. Seminars in Immunology, 23(4), 282–292.
Sakaguchi, S., Miyara, M., Costantino, C. M., & Hafler, D. A. (2010). Foxp3+ regulatory T cells in the human immune system. Nature Reviews Immunology, 10(7), 490–500.
Allan, S. E., Song-Zhao, G. X., Abraham, T., McMurchy, A. N., & Levings, M. K. (2008). Inducible reprogramming of human T cells into Treg cells by a conditionally active form of Foxp3. European Journal of Immunology, 38(12), 3282–3289.
Gratz, I. K., & Campbell, D. J. (2014). Organ-specific and memory treg cells: specificity, development, function, and maintenance. Frontiers in Immunology, 5, 333.
Corthay, A. (2009). How do regulatory T cells work? Scandinavian Journal of Immunology, 70(4), 326–336.
Morlacchi, S., Dal Secco, V., Soldani, C., Glaichenhaus, N., Viola, A., & Sarukhan, A. (2011). Regulatory T cells target chemokine secretion by dendritic cells independently of their capacity to regulate T cell proliferation. Journal of Immunology, 186(12), 6807–6814.
Yamaguchi, T., Wing, J. B., & Sakaguchi, S. (2011). Two modes of immune suppression by Foxp3(+) regulatory T cells under inflammatory or non-inflammatory conditions. Seminars in Immunology, 23(6), 424–430.
Wright, G. P., Notley, C. A., Xue, S. A., Bendle, G. M., Holler, A., Schumacher, T. N., et al. (2009). Adoptive therapy with redirected primary regulatory T cells results in antigen-specific suppression of arthritis. Proceedings of the National Academy of Sciences of the United States of America, 106(45), 19078–19083.
Facciabene, A., Motz, G. T., & Coukos, G. (2012). T-regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Research, 72(9), 2162–2171.
Mailloux, A. W., & Young, M. R. (2010). Regulatory T-cell trafficking: from thymic development to tumor-induced immune suppression. Critical Reviews in Immunology, 30(5), 435–447.
Curiel, T. J., Coukos, G., Zou, L., Alvarez, X., Cheng, P., Mottram, P., et al. (2004). Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nature Medicine, 10(9), 942–949.
Gobert, M., Treilleux, I., Bendriss-Vermare, N., Bachelot, T., Goddard-Leon, S., Arfi, V., et al. (2009). Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Research, 69(5), 2000–2009.
Tan, W., Zhang, W., Strasner, A., Grivennikov, S., Cheng, J. Q., Hoffman, R. M., et al. (2011). Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature, 470(7335), 548–553.
Facciabene, A., Peng, X., Hagemann, I. S., Balint, K., Barchetti, A., Wang, L. P., et al. (2011). Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature, 475(7355), 226–230.
Wei, S., Kryczek, I., Edwards, R. P., Zou, L., Szeliga, W., Banerjee, M., et al. (2007). Interleukin-2 administration alters the CD4+Foxp3+ T-cell pool and tumor trafficking in patients with ovarian carcinoma. Cancer Research, 67(15), 7487–7494.
Liu, V. C., Wong, L. Y., Jang, T., Shah, A. H., Park, I., Yang, X., et al. (2007). Tumor evasion of the immune system by converting CD4+CD25− T cells into CD4+CD25+ T regulatory cells: role of tumor-derived TGF-beta. Journal of Immunology, 178(5), 2883–2892.
Valzasina, B., Piconese, S., Guiducci, C., & Colombo, M. P. (2006). Tumor-induced expansion of regulatory T cells by conversion of CD4+CD25− lymphocytes is thymus and proliferation independent. Cancer Research, 66(8), 4488–4495.
Elkord, E., Sharma, S., Burt, D. J., & Hawkins, R. E. (2011). Expanded subpopulation of FoxP3+ T regulatory cells in renal cell carcinoma co-express Helios, indicating they could be derived from natural but not induced Tregs. Clinical Immunology, 140(3), 218–222.
Nizar, S., Meyer, B., Galustian, C., Kumar, D., & Dalgleish, A. (2010). T regulatory cells, the evolution of targeted immunotherapy. Biochimica et Biophysica Acta, 1806(1), 7–17.
Mougiakakos, D., Johansson, C. C., & Kiessling, R. (2009). Naturally occurring regulatory T cells show reduced sensitivity toward oxidative stress-induced cell death. Blood, 113(15), 3542–3545.
Carreras, J., Lopez-Guillermo, A., Fox, B. C., Colomo, L., Martinez, A., Roncador, G., et al. (2006). High numbers of tumor-infiltrating Foxp3-positive regulatory T cells are associated with improved overall survival in follicular lymphoma. Blood, 108(9), 2957–2964.
Lee, A. M., Clear, A. J., Calaminici, M., Davies, A. J., Jordan, S., MacDougall, F., et al. (2006). Number of CD4+ cells and location of forkhead box protein P3-positive cells in diagnostic follicular lymphoma tissue microarrays correlates with outcome. Journal of Clinical Oncology, 24(31), 5052–5059.
Tzankov, A., Meier, C., Hirschmann, P., Went, P., Pileri, S. A., & Dirnhofer, S. (2008). Correlation of high numbers of intratumoral Foxp3+ regulatory T cells with improved survival in germinal center-like diffuse large B-cell lymphoma, follicular lymphoma and classical Hodgkin’s lymphoma. Haematologica, 93(2), 193–200.
Polcher, M., Braun, M., Friedrichs, N., Rudlowski, C., Bercht, E., Fimmers, R., et al. (2010). Foxp3(+) cell infiltration and granzyme B(+)/Foxp3(+) cell ratio are associated with outcome in neoadjuvant chemotherapy-treated ovarian carcinoma. Cancer Immunology, Immunotherapy, 59(6), 909–919.
Wolf, D., Wolf, A. M., Rumpold, H., Fiegl, H., Zeimet, A. G., Muller-Holzner, E., et al. (2005). The expression of the regulatory T cell-specific forkhead box transcription factor Foxp3 is associated with poor prognosis in ovarian cancer. Clinical Cancer Research, 11(23), 8326–8331.
Chen, K. J., Zhou, L., Xie, H. Y., Ahmed, T. E., Feng, X. W., & Zheng, S. S. (2012). Intratumoral regulatory T cells alone or in combination with cytotoxic T cells predict prognosis of hepatocellular carcinoma after resection. Medical Oncology, 29(3), 1817–1826.
Gao, Q., Qiu, S. J., Fan, J., Zhou, J., Wang, X. Y., Xiao, Y. S., et al. (2007). Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. Journal of Clinical Oncology, 25(18), 2586–2593.
Li, J. F., Chu, Y. W., Wang, G. M., Zhu, T. Y., Rong, R. M., Hou, J., et al. (2009). The prognostic value of peritumoral regulatory T cells and its correlation with intratumoral cyclooxygenase-2 expression in clear cell renal cell carcinoma. BJU International, 103(3), 399–405.
Siddiqui, S. A., Frigola, X., Bonne-Annee, S., Mercader, M., Kuntz, S. M., Krambeck, A. E., et al. (2007). Tumor-infiltrating Foxp3-CD4+CD25+ T cells predict poor survival in renal cell carcinoma. Clinical Cancer Research, 13(7), 2075–2081.
Hiraoka, N., Onozato, K., Kosuge, T., & Hirohashi, S. (2006). Prevalence of Foxp3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clinical Cancer Research, 12(18), 5423–5434.
Mougiakakos, D., Johansson, C. C., Trocme, E., All-Ericsson, C., Economou, M. A., Larsson, O., et al. (2010). Intratumoral forkhead box P3-positive regulatory T cells predict poor survival in cyclooxygenase-2-positive uveal melanoma. Cancer, 116(9), 2224–2233.
Brudvik, K. W., Henjum, K., Aandahl, E. M., Bjornbeth, B. A., & Tasken, K. (2012). Regulatory T-cell-mediated inhibition of antitumor immune responses is associated with clinical outcome in patients with liver metastasis from colorectal cancer. Cancer Immunology, Immunotherapy, 61(7), 1045–1053.
Shimizu, K., Nakata, M., Hirami, Y., Yukawa, T., Maeda, A., & Tanemoto, K. (2010). Tumor-infiltrating Foxp3+ regulatory T cells are correlated with cyclooxygenase-2 expression and are associated with recurrence in resected non-small cell lung cancer. Journal of Thoracic Oncology, 5(5), 585–590.
Correale, P., Rotundo, M. S., Del Vecchio, M. T., Remondo, C., Migali, C., Ginanneschi, C., et al. (2010). Regulatory (Foxp3+) T-cell tumor infiltration is a favorable prognostic factor in advanced colon cancer patients undergoing chemo or chemoimmunotherapy. Journal of Immunotherapy, 33(4), 435–441.
Ladoire, S., Martin, F., & Ghiringhelli, F. (2011). Prognostic role of Foxp3+ regulatory T cells infiltrating human carcinomas: the paradox of colorectal cancer. Cancer Immunology, Immunotherapy, 60(7), 909–918.
Salama, P., Phillips, M., Grieu, F., Morris, M., Zeps, N., Joseph, D., et al. (2009). Tumor-infiltrating Foxp3+ T regulatory cells show strong prognostic significance in colorectal cancer. Journal of Clinical Oncology, 27(2), 186–192.
Haas, M., Dimmler, A., Hohenberger, W., Grabenbauer, G. G., Niedobitek, G., & Distel, L. V. (2009). Stromal regulatory T-cells are associated with a favourable prognosis in gastric cancer of the cardia. BMC Gastroenterology, 9, 65.
Badoual, C., Hans, S., Rodriguez, J., Peyrard, S., Klein, C., Agueznay Nel, H., et al. (2006). Prognostic value of tumor-infiltrating CD4+ T-cell subpopulations in head and neck cancers. Clinical Cancer Research, 12(2), 465–472.
Bron, L., Jandus, C., Andrejevic-Blant, S., Speiser, D. E., Monnier, P., Romero, P., et al. (2013). Prognostic value of arginase-II expression and regulatory T-cell infiltration in head and neck squamous cell carcinoma. International Journal of Cancer, 132(3), E85–E93.
Strauss, L., Bergmann, C., Gooding, W., Johnson, J. T., & Whiteside, T. L. (2007). The frequency and suppressor function of CD4+CD25highFoxp3+ T cells in the circulation of patients with squamous cell carcinoma of the head and neck. Clinical Cancer Research, 13(21), 6301–6311.
Chaudhary, B., Abd Al Samid, M., al-Ramadi, B. K., & Elkord, E. (2014). Phenotypic alterations, clinical impact and therapeutic potential of regulatory T cells in cancer. Expert Opinion on Biological Therapy, 14(7), 931–945.
Zitvogel, L., Kepp, O., & Kroemer, G. (2011). Immune parameters affecting the efficacy of chemotherapeutic regimens. Nature Reviews. Clinical Oncology, 8(3), 151–160.
Dunn, G. P., Old, L. J., & Schreiber, R. D. (2004). The three Es of cancer immunoediting. Annual Review of Immunology, 22, 329–360.
Hori, S., Nomura, T., & Sakaguchi, S. (2003). Control of regulatory T cell development by the transcription factor Foxp3. Science, 299(5609), 1057–1061.
Kryczek, I., Liu, R., Wang, G., Wu, K., Shu, X., Szeliga, W., et al. (2009). Foxp3 defines regulatory T cells in human tumor and autoimmune disease. Cancer Research, 69(9), 3995–4000.
Roncador, G., Brown, P. J., Maestre, L., Hue, S., Martinez-Torrecuadrada, J. L., Ling, K. L., et al. (2005). Analysis of Foxp3 protein expression in human CD4+CD25+ regulatory T cells at the single-cell level. European Journal of Immunology, 35(6), 1681–1691.
Hinz, S., Pagerols-Raluy, L., Oberg, H. H., Ammerpohl, O., Grussel, S., Sipos, B., et al. (2007). Foxp3 expression in pancreatic carcinoma cells as a novel mechanism of immune evasion in cancer. Cancer Research, 67(17), 8344–8350.
Zuo, T., Wang, L., Morrison, C., Chang, X., Zhang, H., Li, W., et al. (2007). Foxp3 is an X-linked breast cancer suppressor gene and an important repressor of the HER-2/ErbB2 oncogene. Cell, 129(7), 1275–1286.
Triulzi, T., Tagliabue, E., Balsari, A., & Casalini, P. (2013). Foxp3 expression in tumor cells and implications for cancer progression. Journal of Cellular Physiology, 228(1), 30–35.
Li, W., Wang, L., Katoh, H., Liu, R., Zheng, P., & Liu, Y. (2011). Identification of a tumor suppressor relay between the Foxp3 and the Hippo pathways in breast and prostate cancers. Cancer Research, 71(6), 2162–2171.
McInnes, N., Sadlon, T. J., Brown, C. Y., Pederson, S., Beyer, M., Schultze, J. L., et al. (2012). Foxp3 and Foxp3-regulated microRNAs suppress SATB1 in breast cancer cells. Oncogene, 31(8), 1045–1054.
Dimitrakopoulos, F. I., Papadaki, H., Antonacopoulou, A. G., Kottorou, A., Gotsis, A. D., Scopa, C., et al. (2011). Association of Foxp3 expression with non-small cell lung cancer. Anticancer Research, 31(5), 1677–1683.
Merlo, A., Casalini, P., Carcangiu, M. L., Malventano, C., Triulzi, T., Menard, S., et al. (2009). Foxp3 expression and overall survival in breast cancer. Journal of Clinical Oncology, 27(11), 1746–1752.
Winerdal, M. E., Marits, P., Winerdal, M., Hasan, M., Rosenblatt, R., Tolf, A., et al. (2011). Foxp3 and survival in urinary bladder cancer. BJU International, 108(10), 1672–1678.
Xue, L., Lu, H. Q., He, J., Zhao, X. W., Zhong, L., Zhang, Z. Z., et al. (2010). Expression of Foxp3 in esophageal squamous cell carcinoma relating to the clinical data. Diseases of the Esophagus, 23(4), 340–346.
Miyara, M., Yoshioka, Y., Kitoh, A., Shima, T., Wing, K., Niwa, A., et al. (2009). Functional delineation and differentiation dynamics of human CD4+ T cells expressing the Foxp3 transcription factor. Immunity, 30(6), 899–911.
Rech, A. J., Mick, R., Kaplan, D. E., Chang, K. M., Domchek, S. M., & Vonderheide, R. H. (2010). Homeostasis of peripheral Foxp3(+) CD4(+) regulatory T cells in patients with early and late stage breast cancer. Cancer Immunology, Immunotherapy, 59(4), 599–607.
Xu, L., Xu, W., Qiu, S., & Xiong, S. (2010). Enrichment of CCR6+Foxp3+ regulatory T cells in the tumor mass correlates with impaired CD8+ T cell function and poor prognosis of breast cancer. Clinical Immunology, 135(3), 466–475.
Nishikawa, H., & Sakaguchi, S. (2014). Regulatory T cells in cancer immunotherapy. Current Opinion in Immunology, 27, 1–7.
Kryczek, I., Wu, K., Zhao, E., Wei, S., Vatan, L., Szeliga, W., et al. (2011). IL-17+ regulatory T cells in the microenvironments of chronic inflammation and cancer. Journal of Immunology, 186(7), 4388–4395.
Mandapathil, M., Hilldorfer, B., Szczepanski, M. J., Czystowska, M., Szajnik, M., Ren, J., et al. (2010). Generation and accumulation of immunosuppressive adenosine by human CD4+CD25highFoxp3+ regulatory T cells. Journal of Biological Chemistry, 285(10), 7176–7186.
Schuler, P. J., Schilling, B., Harasymczuk, M., Hoffmann, T. K., Johnson, J., Lang, S., et al. (2012). Phenotypic and functional characteristics of CD4+ CD39+ Foxp3+ and CD4+ CD39+ Foxp3neg T-cell subsets in cancer patients. European Journal of Immunology, 42(7), 1876–1885.
Sugiyama, D., Nishikawa, H., Maeda, Y., Nishioka, M., Tanemura, A., Katayama, I., et al. (2013). Anti-CCR4 mAb selectively depletes effector-type Foxp3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proceedings of the National Academy of Sciences of the United States of America, 110(44), 17945–17950.
Crome, S. Q., Clive, B., Wang, A. Y., Kang, C. Y., Chow, V., Yu, J., et al. (2010). Inflammatory effects of ex vivo human Th17 cells are suppressed by regulatory T cells. Journal of Immunology, 185(6), 3199–3208.
Bates, G. J., Fox, S. B., Han, C., Leek, R. D., Garcia, J. F., Harris, A. L., et al. (2006). Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. Journal of Clinical Oncology, 24(34), 5373–5380.
Liyanage, U. K., Moore, T. T., Joo, H. G., Tanaka, Y., Herrmann, V., Doherty, G., et al. (2002). Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. Journal of Immunology, 169(5), 2756–2761.
Aruga, T., Suzuki, E., Saji, S., Horiguchi, S., Horiguchi, K., Sekine, S., et al. (2009). A low number of tumor-infiltrating Foxp3-positive cells during primary systemic chemotherapy correlates with favorable anti-tumor response in patients with breast cancer. Oncology Reports, 22(2), 273–278.
Liu, F., Lang, R., Zhao, J., Zhang, X., Pringle, G. A., Fan, Y., et al. (2011). CD8(+) cytotoxic T cell and Foxp3(+) regulatory T cell infiltration in relation to breast cancer survival and molecular subtypes. Breast Cancer Research and Treatment, 130(2), 645–655.
Yan, M., Jene, N., Byrne, D., Millar, E. K., O’Toole, S. A., McNeil, C. M., et al. (2011). Recruitment of regulatory T cells is correlated with hypoxia-induced CXCR4 expression, and is associated with poor prognosis in basal-like breast cancers. Breast Cancer Research, 13(2), R47.
West, N. R., Kost, S. E., Martin, S. D., Milne, K., Deleeuw, R. J., Nelson, B. H., et al. (2013). Tumour-infiltrating Foxp3(+) lymphocytes are associated with cytotoxic immune responses and good clinical outcome in oestrogen receptor-negative breast cancer. British Journal of Cancer, 108(1), 155–162.
de Kruijf, E. M., van Nes, J. G., Sajet, A., Tummers, Q. R., Putter, H., Osanto, S., et al. (2010). The predictive value of HLA class I tumor cell expression and presence of intratumoral Tregs for chemotherapy in patients with early breast cancer. Clinical Cancer Research, 16(4), 1272–1280.
Mahmoud, S. M., Paish, E. C., Powe, D. G., Macmillan, R. D., Lee, A. H., Ellis, I. O., et al. (2011). An evaluation of the clinical significance of Foxp3+ infiltrating cells in human breast cancer. Breast Cancer Research and Treatment, 127(1), 99–108.
Ali, H. R., Provenzano, E., Dawson, S. J., Blows, F. M., Liu, B., Shah, M., et al. (2014). Association between CD8+ T-cell infiltration and breast cancer survival in 12,439 patients. Annals of Oncology, 25(8), 1536–1543.
Yang, Y., Guan, X., You, J. CLOPE: a fast and effective clustering algorithm for transactional data. In Proceedings of the eighth ACM SIGKDD international conference on knowledge discovery and data mining, Edmonton, Alberta, Canada, 2002 (pp. 682–687): ACM
Mahmoud, S. M. (2011). Inflammation and immunosurveillance in breast Cancer [PhD thesis]. Nottingham, UK: The University of Nottingham. http://etheses.nottingham.ac.uk/1827/
Ladoire, S., Arnould, L., Mignot, G., Coudert, B., Rebe, C., Chalmin, F., et al. (2011). Presence of Foxp3 expression in tumor cells predicts better survival in HER2-overexpressing breast cancer patients treated with neoadjuvant chemotherapy. Breast Cancer Research and Treatment, 125(1), 65–72.
Zuo, T., Liu, R., Zhang, H., Chang, X., Liu, Y., Wang, L., et al. (2007). Foxp3 is a novel transcriptional repressor for the breast cancer oncogene SKP2. Journal of Clinical Investigation, 117(12), 3765–3773.
Douglass, S., Meeson, A. P., Overbeck-Zubrzycka, D., Brain, J. G., Bennett, M. R., Lamb, C. A., et al. (2014). Breast cancer metastasis: demonstration that Foxp3 regulates CXCR4 expression and the response to CXCL12. Journal of Pathology, 234 (1), 74–85.
von Minckwitz, G., Untch, M., Blohmer, J. U., Costa, S. D., Eidtmann, H., Fasching, P. A., et al. (2012). Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. Journal of Clinical Oncology, 30(15), 1796–1804.
Demir, L., Yigit, S., Ellidokuz, H., Erten, C., Somali, I., Kucukzeybek, Y., et al. (2013). Predictive and prognostic factors in locally advanced breast cancer: effect of intratumoral Foxp3+ Tregs. Clinical and Experimental Metastasis, 30(8), 1047–1062.
Ladoire, S., Arnould, L., Apetoh, L., Coudert, B., Martin, F., Chauffert, B., et al. (2008). Pathologic complete response to neoadjuvant chemotherapy of breast carcinoma is associated with the disappearance of tumor-infiltrating Foxp3+ regulatory T cells. Clinical Cancer Research, 14(8), 2413–2420.
Verma, C., Eremin, J. M., Robins, A., Bennett, A. J., Cowley, G. P., El-Sheemy, M. A., et al. (2013). Abnormal T regulatory cells (Tregs: Foxp3+, CTLA-4+), myeloid-derived suppressor cells (MDSCs: monocytic, granulocytic) and polarised T helper cell profiles (Th1, Th2, Th17) in women with large and locally advanced breast cancers undergoing neoadjuvant chemotherapy (NAC) and surgery: failure of abolition of abnormal Treg profile with treatment and correlation of Treg levels with pathological response to NAC. Journal of Translational Medicine, 11, 16.
Decker, T., Fischer, G., Bucke, W., Bucke, P., Stotz, F., Gruneberger, A., et al. (2012). Increased number of regulatory T cells (T-regs) in the peripheral blood of patients with HER-2/neu-positive early breast cancer. Journal of Cancer Research and Clinical Oncology, 138(11), 1945–1950.
Lal, A., Chan, L., Devries, S., Chin, K., Scott, G. K., Benz, C. C., et al. (2013). Foxp3-positive regulatory T lymphocytes and epithelial Foxp3 expression in synchronous normal, ductal carcinoma in situ, and invasive cancer of the breast. Breast Cancer Research and Treatment, 139(2), 381–390.
Recchia, F., Candeloro, G., Necozione, S., Desideri, G., Cesta, A., Recchia, L., et al. (2013). Vascular endothelial growth factor expression and T-regulatory cells in premenopausal breast cancer. Oncology Letters, 5(4), 1117–1122.
Takenaka, M., Seki, N., Toh, U., Hattori, S., Kawahara, A., Yamaguchi, T., et al. (2013). Foxp3 expression in tumor cells and tumor-infiltrating lymphocytes is associated with breast cancer prognosis. Molecular and Clinical Oncology, 1(4), 625–632.
Zhou, S., Xu, S., Tao, H., Zhen, Z., Chen, G., Zhang, Z., et al. (2013). CCR7 expression and intratumoral Foxp3+ regulatory T cells are correlated with overall survival and lymph node metastasis in gastric cancer. PloS One, 8(9), e74430.
Huen, N. Y., Pang, A. L., Tucker, J. A., Lee, T. L., Vergati, M., Jochems, C., et al. (2013). Up-regulation of proliferative and migratory genes in regulatory T cells from patients with metastatic castration-resistant prostate cancer. International Journal of Cancer, 133(2), 373–382.
Kang, M. J., Kim, K. M., Bae, J. S., Park, H. S., Lee, H., Chung, M. J., et al. (2013). Tumor-infiltrating PD1-positive lymphocytes and Foxp3-positive regulatory T cells predict distant metastatic relapse and survival of clear cell renal cell carcinoma. Translational Oncology, 6(3), 282–289.
French, J. D., Kotnis, G. R., Said, S., Raeburn, C. D., McIntyre, R. C., Jr., Klopper, J. P., et al. (2012). Programmed death-1+ T cells and regulatory T cells are enriched in tumor-involved lymph nodes and associated with aggressive features in papillary thyroid cancer. Journal of Clinical Endocrinology and Metabolism, 97(6), E934–E943.
Fu, H. Y., Li, C., Yang, W., Gai, X. D., Jia, T., Lei, Y. M., et al. (2013). Foxp3 and TLR4 protein expression are correlated in non-small cell lung cancer: implications for tumor progression and escape. Acta Histochemica, 115(2), 151–157.
Efimova, O. V., & Kelley, T. W. (2009). Induction of granzyme B expression in T-cell receptor/CD28-stimulated human regulatory T cells is suppressed by inhibitors of the PI3K-mTOR pathway. BMC Immunology, 10, 59.
Garin, M. I., Chu, C. C., Golshayan, D., Cernuda-Morollon, E., Wait, R., & Lechler, R. I. (2007). Galectin-1: a key effector of regulation mediated by CD4+CD25+ T cells. Blood, 109(5), 2058–2065.
Borsellino, G., Kleinewietfeld, M., Di Mitri, D., Sternjak, A., Diamantini, A., Giometto, R., et al. (2007). Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood, 110(4), 1225–1232.
Spranger, S., Spaapen, R. M., Zha, Y., Williams, J., Meng, Y., Ha, T. T., et al. (2013). Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Science Translational Medicine, 5(200), 200ra116.
Wing, K., Onishi, Y., Prieto-Martin, P., Yamaguchi, T., Miyara, M., Fehervari, Z., et al. (2008). CTLA-4 control over Foxp3+ regulatory T cell function. Science, 322(5899), 271–275.
Bos, P. D., Plitas, G., Rudra, D., Lee, S. Y., & Rudensky, A. Y. (2013). Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. Journal of Experimental Medicine, 210(11), 2435–2466.
Camisaschi, C., Casati, C., Rini, F., Perego, M., De Filippo, A., Triebel, F., et al. (2010). LAG-3 expression defines a subset of CD4(+)CD25(high)Foxp3(+) regulatory T cells that are expanded at tumor sites. Journal of Immunology, 184(11), 6545–6551.
Delgoffe, G. M., Woo, S. R., Turnis, M. E., Gravano, D. M., Guy, C., Overacre, A. E., et al. (2013). Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature, 501(7466), 252–256.
Facciabene, A., Santoro, S., & Coukos, G. (2012). Know thy enemy: why are tumor-infiltrating regulatory T cells so deleterious? Oncoimmunology, 1(4), 575–577.
Whiteside, T. L. (2008). The tumor microenvironment and its role in promoting tumor growth. Oncogene, 27(45), 5904–5912.
Karavitis, J., Hix, L. M., Shi, Y. H., Schultz, R. F., Khazaie, K., & Zhang, M. (2012). Regulation of COX2 expression in mouse mammary tumor cells controls bone metastasis and PGE2-induction of regulatory T cell migration. PloS One, 7(9), e46342.
Kudo-Saito, C., Shirako, H., Ohike, M., Tsukamoto, N., & Kawakami, Y. (2013). CCL2 is critical for immunosuppression to promote cancer metastasis. Clinical and Experimental Metastasis, 30(4), 393–405.
Hansen, W., Hutzler, M., Abel, S., Alter, C., Stockmann, C., Kliche, S., et al. (2012). Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth. Journal of Experimental Medicine, 209(11), 2001–2016.
Wada, J., Suzuki, H., Fuchino, R., Yamasaki, A., Nagai, S., Yanai, K., et al. (2009). The contribution of vascular endothelial growth factor to the induction of regulatory T-cells in malignant effusions. Anticancer Research, 29(3), 881–888.
Gupta, S., Joshi, K., Wig, J. D., & Arora, S. K. (2007). Intratumoral Foxp3 expression in infiltrating breast carcinoma: its association with clinicopathologic parameters and angiogenesis. Acta Oncologica, 46(6), 792–797.
Boyce, B. F., & Xing, L. (2007). Biology of RANK, RANKL, and osteoprotegerin. Arthritis Research and Therapy, 9(Suppl 1), S1.
Faghih, Z., Erfani, N., Haghshenas, M. R., Safaei, A., Talei, A. R., & Ghaderi, A. (2014). Immune profiles of CD4+ lymphocyte subsets in breast cancer tumor draining lymph nodes. Immunology Letters, 158(1–2), 57–65.
Kashimura, S., Saze, Z., Terashima, M., Soeta, N., Ohtani, S., Osuka, F., et al. (2012). CD83(+) dendritic cells and Foxp3(+) regulatory T cells in primary lesions and regional lymph nodes are inversely correlated with prognosis of gastric cancer. Gastric Cancer, 15(2), 144–153.
Mansfield, A. S., Heikkila, P. S., Vaara, A. T., von Smitten, K. A., Vakkila, J. M., & Leidenius, M. H. (2009). Simultaneous Foxp3 and IDO expression is associated with sentinel lymph node metastases in breast cancer. BMC Cancer, 9, 231.
Tsiatas, M. L., Gyftaki, R., Liacos, C., Politi, E., Rodolakis, A., Dimopoulos, M. A., et al. (2009). Study of T lymphocytes infiltrating peritoneal metastases in advanced ovarian cancer: associations with vascular endothelial growth factor levels and prognosis in patients receiving platinum-based chemotherapy. International Journal of Gynecological Cancer, 19(8), 1329–1334.
Abadi, Y. M., Jeon, H., Ohaegbulam, K. C., Scandiuzzi, L., Ghosh, K., Hofmeyer, K. A., et al. (2013). Host b7x promotes pulmonary metastasis of breast cancer. Journal of Immunology, 190(7), 3806–3814.
Biragyn, A., Bodogai, M., Olkhanud, P. B., Denny-Brown, S. R., Puri, N., Ayukawa, K., et al. (2013). Inhibition of lung metastasis by chemokine CCL17-mediated in vivo silencing of genes in CCR4+ Tregs. Journal of Immunotherapy, 36(4), 258–267.
Chopra, M., Riedel, S. S., Biehl, M., Krieger, S., von Krosigk, V., Bauerlein, C. A., et al. (2013). Tumor necrosis factor receptor 2-dependent homeostasis of regulatory T cells as a player in TNF-induced experimental metastasis. Carcinogenesis, 34(6), 1296–1303.
Dalotto-Moreno, T., Croci, D. O., Cerliani, J. P., Martinez-Allo, V. C., Dergan-Dylon, S., Mendez-Huergo, S. P., et al. (2013). Targeting galectin-1 overcomes breast cancer-associated immunosuppression and prevents metastatic disease. Cancer Research, 73(3), 1107–1117.
Ghochikyan, A., Davtyan, A., Hovakimyan, A., Davtyan, H., Poghosyan, A., Bagaev, A., et al. (2014). Primary 4T1 tumor resection provides critical “window of opportunity” for immunotherapy. Clinical and Experimental Metastasis, 31(2), 185–198.
Kim, P. S., Jochems, C., Grenga, I., Donahue, R. N., Tsang, K. Y., Gulley, J. L., et al. (2014). Pan-Bcl-2 inhibitor, GX15-070 (obatoclax), decreases human T regulatory lymphocytes while preserving effector T lymphocytes: a rationale for its use in combination immunotherapy. Journal of Immunology, 192(6), 2622–2633.
Mandl, S. J., Rountree, R. B., Dalpozzo, K., Do, L., Lombardo, J. R., Schoonmaker, P. L., et al. (2012). Immunotherapy with MVA-BN(R)-HER2 induces HER-2-specific Th1 immunity and alters the intratumoral balance of effector and regulatory T cells. Cancer Immunology, Immunotherapy, 61(1), 19–29.
Olkhanud, P. B., Baatar, D., Bodogai, M., Hakim, F., Gress, R., Anderson, R. L., et al. (2009). Breast cancer lung metastasis requires expression of chemokine receptor CCR4 and regulatory T cells. Cancer Research, 69(14), 5996–6004.
Olkhanud, P. B., Damdinsuren, B., Bodogai, M., Gress, R. E., Sen, R., Wejksza, K., et al. (2011). Tumor-evoked regulatory B cells promote breast cancer metastasis by converting resting CD4(+) T cells to T-regulatory cells. Cancer Research, 71(10), 3505–3515.
Tanikawa, T., Wilke, C. M., Kryczek, I., Chen, G. Y., Kao, J., Nunez, G., et al. (2012). Interleukin-10 ablation promotes tumor development, growth, and metastasis. Cancer Research, 72(2), 420–429.
Lee-Chang, C., Bodogai, M., Martin-Montalvo, A., Wejksza, K., Sanghvi, M., Moaddel, R., et al. (2013). Inhibition of breast cancer metastasis by resveratrol-mediated inactivation of tumor-evoked regulatory B cells. Journal of Immunology, 191(8), 4141–4151.
Vadrevu, S. K., Chintala, N. K., Sharma, S. K., Sharma, P., Cleveland, C., Riediger, L., et al. (2014). Complement C5a receptor facilitates cancer metastasis by altering T-cell responses in the metastatic niche. Cancer Research, 74(13), 3454–3465.
Weiss, J. M., Subleski, J. J., Back, T., Chen, X., Watkins, S. K., Yagita, H., et al. (2014). Regulatory T cells and myeloid-derived suppressor cells in the tumor microenvironment undergo Fas-dependent cell death during IL-2/alphaCD40 therapy. Journal of Immunology, 192(12), 5821–5829.
Kim, R., Emi, M., & Tanabe, K. (2007). Cancer immunoediting from immune surveillance to immune escape. Immunology, 121(1), 1–14.
Spranger, S., Koblish, H. K., Horton, B., Scherle, P. A., Newton, R., & Gajewski, T. F. (2014). Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. Journal of Immunotherapy of Cancer, 2, 3.
Byrne, W. L., Mills, K. H., Lederer, J. A., & O’Sullivan, G. C. (2011). Targeting regulatory T cells in cancer. Cancer Research, 71(22), 6915–6920.
von Boehmer, H., & Daniel, C. (2013). Therapeutic opportunities for manipulating T(Reg) cells in autoimmunity and cancer. Nature Reviews Drug Discovery, 12(1), 51–63.
Kozawa, E., Sugiura, H., Wasa, J., Kohyama, K., Yamada, K., Nishioka, A., et al. (2010). Suppression of tumour metastasis in a murine osteosarcoma model with anti-CD25 monoclonal antibody treatment. Anticancer Research, 30(12), 5019–5022.
Allard, B., Pommey, S., Smyth, M. J., & Stagg, J. (2013). Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clinical Cancer Research, 19(20), 5626–5635.
Ge, Y., Domschke, C., Stoiber, N., Schott, S., Heil, J., Rom, J., et al. (2012). Metronomic cyclophosphamide treatment in metastasized breast cancer patients: immunological effects and clinical outcome. Cancer Immunology, Immunotherapy, 61(3), 353–362.
Kaji, W., Tanaka, S., Tsukimoto, M., & Kojima, S. (2014). Adenosine A(2B) receptor antagonist PSB603 suppresses tumor growth and metastasis by inhibiting induction of regulatory T cells. Journal of Toxicological Sciences, 39(2), 191–198.
Li, C. X., Wong, B. L., Ling, C. C., Ma, Y. Y., Shao, Y., Geng, W., et al. (2014). A novel oxygen carrier “YQ23” suppresses the liver tumor metastasis by decreasing circulating endothelial progenitor cells and regulatory T cells. BMC Cancer, 14(1), 293.
Pohla, H., Buchner, A., Stadlbauer, B., Frankenberger, B., Stevanovic, S., Walter, S., et al. (2012). High immune response rates and decreased frequencies of regulatory T cells in metastatic renal cell carcinoma patients after tumor cell vaccination. Molecular Medicine, 18, 1499–1508.
Shahabi, V., Seavey, M. M., Maciag, P. C., Rivera, S., & Wallecha, A. (2011). Development of a live and highly attenuated Listeria monocytogenes-based vaccine for the treatment of HER2/neu-overexpressing cancers in human. Cancer Gene Therapy, 18(1), 53–62.
Maciag, P. C., Radulovic, S., & Rothman, J. (2009). The first clinical use of a live-attenuated Listeria monocytogenes vaccine: a phase I safety study of Lm-LLO-E7 in patients with advanced carcinoma of the cervix. Vaccine, 27(30), 3975–3983.
Wallecha, A., Singh, R., & Malinina, I. (2013). Listeria monocytogenes (Lm)-LLO immunotherapies reduce the immunosuppressive activity of myeloid-derived suppressor cells and regulatory T cells in the tumor microenvironment. Journal of Immunotherapy, 36(9), 468–476.
Audia, S., Nicolas, A., Cathelin, D., Larmonier, N., Ferrand, C., Foucher, P., et al. (2007). Increase of CD4+CD25+ regulatory T cells in the peripheral blood of patients with metastatic carcinoma: a phase I clinical trial using cyclophosphamide and immunotherapy to eliminate CD4+CD25+ T lymphocytes. Clinical and Experimental Immunology, 150(3), 523–530.
Emmenegger, U., Francia, G., Chow, A., Shaked, Y., Kouri, A., Man, S., et al. (2011). Tumors that acquire resistance to low-dose metronomic cyclophosphamide retain sensitivity to maximum tolerated dose cyclophosphamide. Neoplasia, 13(1), 40–48.
Simpson, T. R., Li, F., Montalvo-Ortiz, W., Sepulveda, M. A., Bergerhoff, K., Arce, F., et al. (2013). Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. Journal of Experimental Medicine, 210(9), 1695–1710.
Galon, J., Pages, F., Marincola, F. M., Angell, H. K., Thurin, M., Lugli, A., et al. (2012). Cancer classification using the immunoscore: a worldwide task force. Journal of Translational Medicine, 10, 205.
Galon, J., Mlecnik, B., Bindea, G., Angell, H. K., Berger, A., Lagorce, C., et al. (2014). Towards the introduction of the ‘immunoscore’ in the classification of malignant tumours. Journal of Pathology, 232(2), 199–209.
Gonzalez, L., Strbo, N., & Podack, E. R. (2013). Humanized mice: novel model for studying mechanisms of human immune-based therapies. Immunologic Research, 57(1–3), 326–334.
Budiu, R. A., Elishaev, E., Brozick, J., Lee, M., Edwards, R. P., Kalinski, P., et al. (2013). Immunobiology of human mucin 1 in a preclinical ovarian tumor model. Oncogene, 32(32), 3664–3675.
Drennan, S., Stafford, N. D., Greenman, J., & Green, V. L. (2013). Increased frequency and suppressive activity of CD127(low/-) regulatory T cells in the peripheral circulation of patients with head and neck squamous cell carcinoma are associated with advanced stage and nodal involvement. Immunology, 140(3), 335–343.
Schneider, T., Kimpfler, S., Warth, A., Schnabel, P. A., Dienemann, H., Schadendorf, D., et al. (2011). Foxp3(+) regulatory T cells and natural killer cells distinctly infiltrate primary tumors and draining lymph nodes in pulmonary adenocarcinoma. Journal of Thoracic Oncology, 6(3), 432–438.
Acknowledgments
This work was supported by the Canadian Institutes of Health Research (Grant MOP-126138) and the BC Cancer Foundation. E.C.H. is supported by a Frederick Banting and Charles Best Canada Graduate Scholarship from the Canadian Institutes of Health Research and by a 4-year doctoral fellowship from the University of British Columbia. K.L.B. is a Michael Smith Foundation for Health Research Biomedical Research scholar.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Halvorsen, E.C., Mahmoud, S.M. & Bennewith, K.L. Emerging roles of regulatory T cells in tumour progression and metastasis. Cancer Metastasis Rev 33, 1025–1041 (2014). https://doi.org/10.1007/s10555-014-9529-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-014-9529-x