
Abstract
Purpose
The transcription factors ZEB1 and ZEB2 mediate epithelial-to-mesenchymal transition (EMT) and metastatic progression in numerous malignancies including breast cancer. ZEB1 and ZEB2 drive EMT through transcriptional repression of cell–cell junction proteins and members of the tumor suppressive miR200 family. However, in estrogen receptor positive (ER +) breast cancer, the role of ZEB2 as an independent driver of metastasis has not been fully investigated.
Methods
In the current study, we induced exogenous expression of ZEB2 in ER + MCF-7 and ZR-75–1 breast cancer cell lines and examined EMT gene expression and metastasis using dose–response qRT-PCR, transwell migration assays, proliferation assays with immunofluorescence of Ki-67 staining. We used RNA sequencing to identify pathways and genes affected by ZEB2 overexpression. Finally, we treated ZEB2-overexpressing cells with 17β-estradiol (E2) or ICI 182,780 to evaluate how ZEB2 affects estrogen response.
Results
Contrary to expectation, we found that ZEB2 did not increase canonical epithelial nor decrease mesenchymal gene expressions. Furthermore, ZEB2 overexpression did not promote a mesenchymal cell morphology. However, ZEB1 and ZEB2 protein expression induced significant migration of MCF-7 and ZR-75-1 breast cancer cells in vitro and MCF-7 xenograft metastasis in vivo. Transcriptomic (RNA sequencing) pathway analysis revealed alterations in estrogen signaling regulators and pathways, suggesting a role for ZEB2 in endocrine sensitivity in luminal A breast cancer. Expression of ZEB2 was negatively correlated with estrogen receptor complex genes in luminal A patient tumors. Furthermore, treatment with 17β-estradiol (E2) or the estrogen receptor antagonist ICI 182,780 had no effect on growth of ZEB2-overexpressing cells.
Conclusion
ZEB2 is a multi-functional regulator of drug sensitivity, cell migration, and metastasis in ER + breast cancer and functions through non-canonical mechanisms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer remains the malignancy with the second highest rate of mortality in women [1], accounting for over 40,000 deaths a year [2]. While overall five-year survival rates for women diagnosed with localized breast cancer is 94.3%, overall survival rates for patients with metastatic disease was reported to be 54.2% [3]. Tumors with luminal A subtypes make up the largest proportion of diagnosed breast cancer cases [4]. This subtype is characterized primarily by low rates of proliferation and motility accompanied by a high dependence on estrogen signaling [5]. These tumors generally show a favorable response to endocrine-targeting therapies, including tamoxifen, fulvestrant, and aromatase inhibitors [6]. However, a significant number of women with luminal A breast cancer develop resistance to endocrine-targeting therapy [7, 8], and the acquisition of resistance to hormone targeting therapies is associated with increased invasive capacity in breast cancer cells and metastasis [9, 10].
Mechanisms associated with breast cancer metastasis include epithelial-mesenchymal transition [11,12,13], a complex program in which tumor cells of epithelial origin and phenotype undergo changes allowing them to progress to a mesenchymal, motile phenotype [14]. Hallmarks of EMT include loss of cellular adhesions, changes in cytoskeletal proteins, and increased cell migration [15]. This change in cellular molecular and morphologic phenotype facilitates cancer cells; ability to escape the primary tumor site and seed distant metastatic sites [11, 15]. EMT transcription factors ZEB1 and ZEB2 have been linked to poor outcome and metastatic progression in multiple malignancies [16]. ZEB1 and ZEB2 have been shown to promote this transition through direct transcriptional downregulation of epithelial cadherin 1 (CDH1) and miR200 family members [17,18,19,20]. Other studies have shown that ZEB2 represses expression of other cell junction proteins, including those encoded by claudins, desmosome, and plakophilin genes [21]. While this phenomenon has been well characterized in an array of cancer types, the ability of ZEB family member proteins to initiate cell motility and metastasis in a luminal A breast cancer cell is incompletely understood and the link between ZEB-induced metastasis and endocrine sensitivity remains unexplored.
In the present study, we demonstrate ZEB2 expression induces cell migration and metastasis, independent of their roles as EMT-inducing transcription factors. Furthermore, no changes in cell morphology and expression of CDH1 and miR200 family members were observed, indicating an EMT-independent effect. We further show that overexpression of ZEB2 downregulated ER, GATA3 and FOXA1, essential components of the ER binding complex. Collectively, the results strongly support a novel role for ZEB2 as a multifunctional driver of metastasis and endocrine resistance in luminal A breast cancer.
Methods
Cell lines and reagents
Human breast cancer MCF-7 and ZR-75–1 cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA). Liquid nitrogen stocks were made upon receipt and maintained until the start of each study. Cells were used for no more than six months after being thawed with periodic recording of morphology and doubling times to ensure maintenance of phenotype. Cells were maintained at 37 °C, 5% CO2 in 10% DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Hyclone, Salt Lake City, UT, USA) and 1% penicillin/streptomycin (Invitrogen). 17β-estradiol (E2, Sigma, St. Louis, MO) and ICI182,780 were dissolved in dimethyl sulfoxide (DMSO) (Invitrogen) as a 1 mM stock solution and kept at -20 °C. Each drug was diluted in culture media and used at various concentrations as indicated.
Transfection of cell lines
MCF-7 and ZR-75–1 cells were plated at approximately 50% confluency in 10cm2 culture dishes in 10% DMEM and allowed to adhere overnight in a 37 °C incubator. The following day cells were transfected with 5 μg of CMV-eGFP vector, CMV-eGFP-ZEB1, or CMV-eGFP-ZEB2 (GeneCopoeia, Rockville, MD) plasmids using Lipofectamine 2000 (Invitrogen, Grand Isles, NY) per manufacturer’s instructions. After 24 h, the transfection media was replaced with fresh cultured media and cells were treated with neomycin to select for transfected cells. Cells were also monitored for GFP expression. Upon stable selection, pooled populations were confirmed by qPCR and GFP visualization (Supplementary Figure 1a, b).
Transwell migration and invasion assays
Migration assays were performed following the manufacturer’s instructions (BD Biosciences) and as previously published [22]. Cells were treated at time of plating. After 48 h migrated cells were fixed to the membranes and stained with crystal violet (0.1% in 20% methanol), and migrated cells were visualized and quantified by microscopy. Data represented as number of migrated or invaded cells per field of view ± SEM for triplicate experiments.
RNA isolation
Cells were harvested by trypsinization and total RNA was isolated using the RNeasy kit, according to the manufacturer’s instructions (Qiagen, Valencia, CA, USA). The quantity and quality of the RNA were determined by absorbance at 260 and 280 nm using the NanoDrop ND-1000 (NanoDrop, Wilmington, DE, USA).
Quantitative reverse transcription real-time PCR
Total RNA was reverse-transcribed using the BioRad First Strand cDNA synthesis kit, following the manufacturer’s protocol (BioRad, Hercules, CA) and then assayed via quantitative real-time PCR (qPCR) to assess gene expression changes as previously published [23]. To obtain cDNA and run PCR studies for miRNA experiments, the miScript II RT kit and the miScript SYBR Green PCR kit were utilized (Qiagen, Valencia, CA, USA). Data is represented as normalized ΔΔCt (fold expression) compared to β-actin and vector control samples of biological triplicate samples ± S.E.M. Primer sequences for genes examined are included below. Primers for miR200 family and miR141 genes were obtained from a kit provided by Qiagen with the following catalog numbers: miR200a #MS00003738, miR200b, Cat. No. MS00009016; miR200c, Cat. No. MS00003752; miR141, Cat. No. MS00003507.
Gene | Sequence |
|---|---|
β-Actin | F: GGCACCCAGCACAATGAAGA |
R: ACTCCTGCTTGCTGATCCAC | |
BCL2 | F: CCGCAAGACAGGCTTTTCAG |
R: TAATAGTCGCCCCCTCTCGG | |
CDH1 | F: CCTGCCATTCTGGGGATTCT |
R: CCGAAGAAACAGCAAGAGCAG | |
CDH2 | F: GCCCCTCAAGTGTTACCTCAA |
R: AGCCGAGTGATGGTCCAATTT | |
ESR1 | F: GGCATGGTGGAGATCTTCGA |
R: CCTCTCCCTGCAGATTCATCA | |
FOXA1 | F: ACTCCAAGCCTCCTCAACTGCG |
R: GTGCCAAGCCGTGTGCCG | |
GATA3 | F: CGAAGGCCTTGTGAACAGAT |
R: CTGCAGCCCAGATTTCTCA | |
PLAUR | F: GGTGACGCCTTCAGCATGA |
R: CCCACTGCGGTACTGGACAT | |
ZEB2 | F: CGCTTGACATCACTGAAGGA |
R: CTTGCCACACTCTGTGCATT | |
SNAI1 | F: AGCCGTGCCTTCGCTGACC |
R: GGACTCTTGGTGCTTGTGGAGC | |
TWIST | F: TGTCCGCGTCCCACTAGC |
R: TGTCCATTTTCTCCTTCTCTGGA | |
NME1 | F: CTGCAGCCGGAGTTCAAAC |
R: GCAATGAAGGTACGCTCACAGT | |
CLDN1 | F: CACCTGCCACCCCTGAGCC |
R: GCGGTCACGATGTTGTCGCC | |
CLDN3 | F: GCTGCTCTGCTGCTCGTGT |
R: TTGCGGTCGTAGCCTGTGCC | |
CLDN4 | F: CGGGCAGAAGCGGGAGATGG |
R: CAGCAGAGCGGGCAGCAGAA | |
CLDN7 | F: TCCCGCAGTGGCAGATGAGC |
R: GGACAGGGCGAGCACCGAGT | |
DSP | F: GCTTTCCTCCCGCTCCTGCC |
R: GTCGCTGCTCGCTACCTGGG | |
PKP2 | F: AGGGCTCACTACACGCACAGC |
R: CCCGCCTGCTTTCTTGGTGGT | |
PKP3 | F: GACCGCCGACCACCACCAG |
R: CTTCTCACCCACGCTGCCCG | |
GJB2 | F: CCACGGCGGGAGACAGGTG |
R: TCTGGGCGGTTTGCTCTGCG | |
GJB3 | F: CTGTGCCCCTTCCCGCTGTG |
R: GGACCTTGACCGTGCGTGGG | |
ZO3 | F: CCACCTGCCCGCCACCAAAG |
R: TCCACCTCCTCCACCCGCTG | |
CDH3 | F: CTCACCTCCTCCGCCTCCGA |
R: TAGTCGTCCTCCCCGCCACC | |
PGR | F: TACCCGCCCTATCTCAACTACC |
R: TGCTTCATCCCCACAGATTAAACA | |
TFF1 | F: GAGGCCCAGACAGAGACGTG |
R: CCCTGCAGAAGTGTCTAAAATTCA |
Colony assays
MCF-7 VECTOR- or ZEB2-expressing cells were plated in 6-well plates at a density of 100 cells/well in 2 in phenol red-free DMEM with 5% FBS. Twenty-four hours later DMSO or E2 (10 nM) were added and cells were monitored for colony growth. Ten days later the cells were fixed with 3% glutaraldehyde for 20 min. Following fixation, the plates were washed and stained with a solution of 0.1% crystal violet in 20% methanol for 20 min. Colonies of ≥ 5 cells, visualized using brightfield microscopy, were counted as positive.
Crystal violet staining to examine cell morphology
Cells were plated at a density of 5000–10,000 cells, depending on average cell size, per well in a 96-well plate in 5% charcoal–dextran stripped media and allowed to adhere overnight at 37 °C in humidified 5% CO2. The following day (day 0), cells were treated with drug or vehicle. Plates were harvested on days 0, 3, and 5, fixed with glutaraldehyde, and stained with 1% crystal violet in 10% methanol solution. Morphological changes were observed under an inverted microscope with brightfield microscopy.
Fluorescence imaging for cell morphology and proliferation
MCF-7 and ZR75 VECTOR, or ZEB2 cells (5,000 cells per well) were seeded in wells of a 96-well plate for 24 h. Cells were fixed in formalin (Fischer Scientific, Hampton, NH) and permeabilized with Triton-X100 (MP Biomedicals, St. Ana CA). Confirmation of expression was performed through visualization of GFP tag in expression constructs. Cytoskeletal components were stained with AlexaFluor 555-conjugated phalloidin (Cell Signaling, Danvers MA). Ki-67 staining was performed using AlexaFluor 555-conjugated mouse anti Ki-67 (BD Biosciences, Rockville, MD). Cells were counterstained with DAPI (NucBlue Fixed Cell Stain ReadyProbe, Life Technologies, Carlsbad CA). Fluorescent images were acquired using a Carl Zeiss Cell Observer inverted microscope (Carl Zeiss, Thornwood, NY) 20 × or 40 × objectives.
Animal xenograft studies
Xenograft tumor studies were conducted as previously described [24]. All procedures using animal data was performed per guidelines outlined by the Tulane Institutional Animal Care & Use Committee. Immune-compromised SCID/beige female mice (4–6 weeks old) were obtained from Charles River Laboratories (Wilmington, MA). The animals were allowed a period of adaptation in a sterile and pathogen-free environment with food and water ad libitum. Breast cancer cells were collected and viable cells (5 million cells per injection) in suspension with 50 μL of sterile PBS mixed with 100 μL Matrigel™ (BD Biosciences, San Jose, CA). Injections were made bilaterally into the inguinal mammary fat pads on day 0 (n = 5 animals/group) using 27 1/2 gauge sterile syringes. All the procedures in animals were carried out under anesthesia using a mix of isoflurane and oxygen delivered by mask. Tumor size was measured biweekly for 30 days using a digital caliper. Tumor volume was calculated using the following formula: 4/3πLS2, where L is the larger radius and S is the smaller radius. All procedures involving these animals were conducted in compliance with State and Federal laws, standards of the U.S. Department of Health and Human Services, and guidelines established by the Tulane University Animal Care and Use Committee. The Tulane University Animal Care and Use Committee approved the use of animals in this specific study. The facilities and laboratory animal program of Tulane University are accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care. At necropsy, animals were euthanized by cervical dislocation following CO2 exposure. Lungs were removed and fixed in 10% formalin. Tumors were dissected; half of the tumor was preserved in – 20 °C freezer for genomic analyses and the other half was fixed in 10% formalin for immunohistochemical analyses.
Immunohistochemical staining
Lungs were fixed in 10% neutral buffered formalin for 24 to 36 h. Paraffin-embedded sections were prepared at 4 μm thickness followed by standard H&E staining. Section images were captured using the Aperio ScanScope and metastatic burden was calculated with Spectrum software (Aperio Technologies, Vista, CA, USA). One section was analyzed to quantify metastasis for each mouse.
RNA sequencing
Read preparation, repeat masking, and read mapping were conducted as we have previously published [25]. Gene expression changes are represented as fold change ZEB2 compared to vector. Fold change greater than 2.0 and p value of less than 0.05 were considered statistically significant. Pathway analysis and prediction of upstream regulators were performed using Gene Set Enrichment Analysis (GSEA). GSEA was run using the JAVA program (http://www.broadinstitute.org/gsea) using MSigDB C2 curated gene set collection. Gene sets with a false discovery rate (FDR) value < 0.05 after performing 1,000 permutations were considered to be significantly enriched. Pathways and upstream regulators with a z-score of > 1.5 or < − 1.5 with a p value of < 0.05 were considered significantly changed.
Breast cancer data mining
Targeted gene expression correlation analysis for ZEB2, ESR1, PGR, FOXA1, GATA3, TFF1, and BCL2 was derived from DNA microarrays of pooled breast cancer samples obtained from bc-GenExMiner-v3.0 [26]. Datasets represented gene correlation [26] and expression [27] analyses from DNA microarrays (n = 10,001 patients) and RNA sequencing (n = 4712 patients). Correlation maps were then generated based on estrogen receptor status. More detailed information regarding how analyses were performed are described by Jézéquel et al., 2012 [26] and Jézéquel et al. [27].
ERE-luciferase assay
Cells were seeded in 24-well plates at a density of 5 × 105 cells/well in media containing phenol red-free DMEM with 5% FBS and allowed to attach overnight. The cells were then transfected for 5 h with 300 μg pGL2-ERE2X-TK-luciferase plasmid (Panomics) using Effectene according to the manufacturer’s protocol (Qiagen). After 5 h DMSO or 10 nM E2 were added to the cells and cells were incubated at 37 °C overnight. The following morning, the media was removed, and 100 μl of lysis buffer was added per well and then incubated for 5 min at room temperature. Cell debris was pelleted by centrifugation at 15000×g for 5 min. Luciferase activity of 100 μL of cell extracts was determined from an equal volume of Bright Glo luciferase assay reagent (Promega Corp.) in an Autolumat Plus luminometer (Berthhold Technologies).
Statistical analyses
Statistical analyses were carried out with GraphPad Prism software (Graph-Pad Software, Inc., San Diego, CA, USA). Statistics were subjected to unpaired Student’s t test, with p < 0.05 considered statistically significant.
Results
ZEB2 does not act through their canonical role as EMT-inducing transcription factors
We sought to determine the ability of ZEB2 to induce a metastatic phenotype in our system. To accomplish this, we exogenously expressed ZEB2 in ER + MCF-7 and ZR-75–1 cell lines which are non-invasive and exhibit an epithelial phenotype. Expression was confirmed using qPCR analysis and nuclear localization of the GFP tagged protein (Supplementary Figure S1). As ZEB2 has been characterized as EMT-inducing transcription factors and are thought to function as transcriptional repressors [28, 29], we anticipated that exogenous ZEB2 expression would induce a transcriptional downregulation of CDH1 expression and of adhesion-related genes. We then evaluated how ZEB2 overexpression in the luminal cell lines affected genes that are associated with maintenance of an epithelial phenotype, tight junction, and cell adhesion genes including claudins and desmosomes (CLDN1, CLDN3, CLDN4, CLDN7, DSP, PKP2, PKP3, GJB2, GJB3, ZO-3, CDH3) (Fig. 1a, b). The non-significant p-values for CLDN1, CLDN3, CLDN4, CLDN7, DSP, PKP3, GJB2, GJB3, ZO-3, CDH3 in MCF-7-ZEB2 and CLDN3, CLDN4, CLDN7, DSP, PKP3, GJB2, GJB3, ZO-3, CDH3 in ZR-75-1-ZEB2 cells compared to vector controls were 0.7154, 0.2433, 0.2102, 0.8714, 0.9248, 0.9141, 0.7784, 0.4884, 0.8317, 0.8737 and 0.0634, 0.2539, 0.9460, 0.1903, 0.2608, 0.8286, 0.3919, 0.7946, 0.4855, respectively. While cells overexpressing ZEB2 significantly increased PKP2 in MCF-7 and ZR-75-1 cells (p = 0.0211 and p = 0.0145, respectively) and CLDN1 in ZR-75-1 cells (p = 0.0041), these findings did not appear to be meaningful especially since the other cell adhesion and tight junction genes were not significantly altered in response to ZEB2 overexpression. Next, we sought to assess ZEB2 overexpression effects on EMT-related genes; ZEB2 overexpression did not alter expression of EMT genes, and neither suppressed CDH1 expression nor increased mesenchymal gene expression (CDH2, VIM, SNAI1, TWIST, PLAUR, NME) (Fig. 1c, d). Due to prominent roles of the miR200 family in regulating EMT plasticity, we then sought to determine how ZEB2 transcriptionally regulates this family. We found transcription of miR200 family members, miR200a, miR200b, miR200c, and miR141 remained unchanged in ZEB2-overexpressing cells compared to vector controls. The non-significant p-values for miR200a, miR200b, miR200c and miR141 in MCF-7-ZEB2, and ZR-75-1-ZEB2 cells compared to vector controls were 0.6697, 0.2837, 0.5040, 0.4313 and 0.4321, 0.7498 0.2039, and 0.1617, respectively (Fig. 2a, b). Furthermore, analysis of cell morphology with crystal violet staining and brightfield imaging revealed that ZEB2-expressing cells maintained an epithelial like phenotype, as characterized by growth in colonies, cobblestone like shape, and maintenance of cell-to-cell contact (Fig. 2c). Follow-up morphology evaluation with phalloidin staining and fluorescence imaging confirmed these observations. As these results directly contradict our original hypothesis that ZEB2 induced the mesenchymal phenotype through suppression of epithelial genes and upregulation of mesenchymal genes, we further investigated potential mechanisms through which ZEB2 drives metastasis in luminal A breast cancers.

ZEB2 overexpression does not induce EMT in Luminal A breast cancer cells. a Cell–cell adhesion and junction genes were not changed in ZEB2-overexpressing a MCF-7 cells or b ZR-75-1 cells compared to vector controls. EMT genes including the epithelial gene CDH1 and mesenchymal genes CDH2, VIM, SNAI1, TWIST, PLAUR, NME were not affected in ZEB2-overexpressing c MCF-7 cells or d ZR-75-1 cells compared to vector controls. Data was acquired using qRT-PCR and normalized to β-actin and vector controls. Error bars represent mean ± S.E.M. ZR-75-1 is abbreviated to ZR-75 in the graphs within the figure

ZEB2 overexpression did not change expression of EMT-associated miRNAs and did not alter cell morphology. a, b Analysis of ZEB2-mediated miRNA gene expressions that regulate EMT (miR-200a, miR-200b, miR-200c, miR-141) using qRT-PCR. Data was normalized to β-actin and vector controls. Error bars represent mean ± S.E.M.. Cell morphology of MCF-7 and ZR-75–1 cells expressing empty vector or ZEB2 using (c) crystal violet staining and brightfield imaging and (d) fluorescence staining with phalloidin to highlight actin cytoskeleton proteins (red) and DAPI nuclear stain (blue). Representative images for brightfield imaging were captured at 10 × and scale bars for fluorescence images represent 0.25 mm. ZR-75-1 is abbreviated to ZR-75 in the graphs within the figure
ZEB2 promotes cell motility and metastasis in luminal A breast cancer cell lines.
ZEB family member proteins have been characterized primarily in the context of induction of EMT [17,18,19], However, recent work demonstrated that the loss of expression of cell–cell junction proteins is not a necessary step in metastasis [30, 31], exemplified by observations of collective cell migration and invasion [32, 33]. Therefore, we sought to examine the ability of ZEB2 to potentiate a metastatic phenotype independent of their function in EMT in a murine model, and we compared these data to mice inoculated with ZEB1-overexpressing cells. MCF-7-vector, ZEB1, or ZEB2-expressing cells were injected into the mammary fat pad SCID/beige mice (5 mice per group) and monitored for tumor growth over 40 days. When the tumors reached 1000 mm3 in size, they were removed and the mice were monitored until they began to show signs of morbidity around 40 days later. Lungs were harvested, sectioned, and H&E stained to examine metastasis. We found that tumor growth was not affected by ZEB1 nor ZEB2 expression (Fig. 3a). Furthermore, quantification of one section per mouse in each group revealed that mice with ZEB1- or ZEB2-overexpressing tumor cells had significantly greater number and overall area of metastatic lung lesions when compared to the control group (Fig. 3b–d).

ZEB1 and ZEB2 are implicated in cell migration and metastasis. Ovariectomized SCID/beige mice (n = 5/group) implanted with a 17β-estradiol pellet (subcutaneous, 0.72 mg) were injected in the mammary fat pad with MCF-7-VECTOR, MCF-7-ZEB1, or MCF-7-ZEB2 cells (5 × 106 cells per injection). a Tumors were measured twice weekly with digital caliper. Data points represent average tumor volume (mm3) ± S.E.M. (***p < 0.001). Tumors were removed at day 40 and mice were monitored for another 30 days at which point lungs were removed to observe metastases. Lungs were paraffin embedded, sectioned, and stained with Hematoxylin and Eosin (H&E). b Total number and c area of metastatic lesions were quantified and showed increased metastases in ZEB1- and ZEB2-overexpressing groups compared to vector control. d Representative images of H&E stained lungs from MCF-7-VECTOR, MCF-7-ZEB1, and MCF-7-ZEB2 xenografts. e Transwell migration assays were performed with MCF-7-ZEB2 and VECTOR-expressing cells. Migrated cells were quantified using ImageJ. Data show mean ± S.E.M., n = 3. (*p < 0.05 **; p < 0.01)
Additionally, we performed assays measuring alterations in cell migration in ZEB1- or ZEB2-overexpressing cells in vitro. Contrary to what was observed in vivo, MCF-7 cell migration was increased by both ZEB1 and ZEB2 (Fig. 3e). ZR-75–1 cells overexpressing ZEB1 or ZEB2 were also significantly more migratory than vector cells (Fig. 3e). As these luminal A cell lines have little to no detectable endogenous ZEB2 expression and present with low motility, these data show that forced ZEB2 expression in these cell systems drove a motile and metastatic phenotype. These data are consistent with previous studies which have characterized ZEB2 as a driver of cell motility and metastasis in numerous cancers [19, 34].
Transcriptomic analysis reveals alterations in estrogen signaling pathways in ZEB2-expressing cells
To further explore the effects of ZEB2 on the phenotype of luminal breast cancer cells, we performed RNA seq analysis of MCF-7 cells expressing vector or ZEB2. We performed upstream regulator analysis using Ingenuity Pathway Analysis (IPA) using a z-score cutoff of 1.5. Several compounds regulating estrogen signaling were predicted to be inactivated in ZEB2-expressing cells. β-estradiol and estrogen were predicted to be inhibited in ZEB2, but this difference was only significant in ZEB2-overexpressing cells (Table 1). We then analyzed the data using GSEA for altered hallmark pathways. Notably, downregulation of estrogen response pathways was observed in ZEB2-overexpressing cells (Fig. 4a, Supplementary Figure S2), demonstrating divergence in function between these two homologous family members. Furthermore, examination of breast cancer patient data based on DNA microarray analysis using bc-GenExMiner v4.5 [26] revealed significantly higher expression of ZEB2 in ER negative (ER-) tumors (Fig. 4b). We next interrogated the correlation in gene expression of these factors with a subset of estrogen signaling genes ESR1, FOXA1, GATA3, PGR, TFF1, and BCL2 based on our RNA sequencing data, using the IPA program. This analysis revealed a significant negative trend and inverse association ZEB2 expression with ESR1, FOXA1, GATA3, and BCL2 (Table 2). PGR and TFF1 associations were not significant. These findings were similar to correlation plots that demonstrated a negative correlation between ZEB2 and the estrogen signaling genes based on DNA microarrays in a cohort of breast cancer patients with ER + tumors, as obtained using the publicly available bc-GenExMiner v3.0 database [27] (Supplementary Figure S3).

ZEB2 alters the estrogen response in ER + breast cancer. a GSEA hallmark pathway analysis from generated RNA sequencing data of ZEB2 transcriptional profiles compared to vector controls shows decreased early and late estrogen response in ZEB2-expressing MCF7 cells when compared to vector control (FDR < 0.05). b BC-GenExMiner data from breast cancer patient tumors reveals a decrease in ZEB2 expression in ER + tumors when compared to ER− tumors (p < 0.0001)
Estrogen response is abrogated by ZEB2 in ER + breast cancer
The ERα signaling pathway plays a critical role in cell proliferation, motility, and survival in response to estrogen stimulation [35, 36]. We sought to confirm these predicted alterations in ERα signaling in vitro. Baseline gene expression of ERα complex members was assessed by qPCR. ESR1, FOXA1, and GATA3 expression was repressed by ZEB2 compared to vector control in both MCF-7 and ZR-75-1 cells (Fig. 5a). The members of this complex have been shown to be essential to ERα genomic function in breast cancer cells [37]. Furthermore, FOXA1 is necessary for transcription of ERα [37, 38]. We then sought to interrogate the role of ZEB2, specifically, in estrogen response as characterized by transcriptional alterations in ERα signaling target genes. MCF-7 and ZR-75-1 vector- and ZEB2-expressing cells were treated with estrogen for 24 h and analyzed for expression of the estrogen responsive genes BCL2, PGR, and TFF1 using qPCR. ZEB2-expressing cells had a diminished estrogen response when compared to estrogen treated vector control cells, though the response was not eliminated entirely (Fig. 5b, c). Additionally, MCF-7 cells were analyzed for estrogen response element promoter activity using luciferase assay. We found that ZEB2 cells had diminished activity of the ERE promoter upon estrogen treatment (Supplementary Figure S4). Notably, baseline ERE activity should not change dramatically because it is theoretically very lowly activated in the DMSO condition so it cannot be decreased in any meaningful way. In this line of thought, it should only be activated after estrogen treatment. We observed ERE to be activated in both vector and ZEB2-overexpressing cells, but to a decreased extent in the ZEB2 group. Taken together, these results demonstrate a novel role for ZEB2 in the regulation of estrogen response in luminal A breast cancer cells.

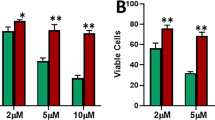
ZEB2-expressing cells have a decreased transcriptional and proliferative response to estrogen treatment. a qPCR analysis of expression of ER complex gene expression in ZEB2 compared to vector control cells. b, c Expression of estrogen responsive genes in vector and ZEB2-expressing cells in response to 24 h treatment with 10 nM E2. (*p < 0.05, ** p < 0.01, *** p < 0.001). d Proliferation of vector and ZEB2-expressing MCF-7 and ZR-75-1 cells was quantified using Ki-67 staining (red) after 24 h treatment with 10 nM E2 or DMSO control. DAPI was used as a nuclear counterstain (blue). e Representative images were captured using fluorescent microscopy and images are shown at 200X magnification. Scale bars represent 0.25 mm
We then investigated the effects of ERα signaling on MCF7 and ZR75 proliferation in vector and ZEB2-expressing cells. E2 treatment increased cell proliferation in vector control cells (Fig. 5c, d); conversely, ZEB2-expressing cells showed no alterations in proliferation upon E2 stimulation. These data reflect the pathway activation patterns predicted in our RNA seq data, as well as the patterns of gene expression seen in breast cancer patient tumors in vivo. It is of note that in cells expressing ZEB2, baseline growth did not change compared to vector control indicating that the cells are not only insensitive to estrogen growth signals, but perhaps more importantly, that they are capable of functioning without ER signaling. This estrogen independent proliferation led us to investigate the response of ZEB2 specifically to estrogen targeted therapies, and whether this decrease in estrogen response induced by ZEB2 conferred resistance to those therapies.
To address this, we investigated the effects of ERα inhibition in ZEB2-expressing cells compared to cells expressing the vector control in our ER + cell lines in a colony assay. We found that ZEB2-expressing cells had a significantly higher proportion of clonogenic survival in the presence of ICI 182,780 when compared to the vector control (Fig. 6a), indicating that ZEB2 confers a distinct survival advantage in ER + cells when estrogen signaling is abrogated. Next, we found that expression of ZEB2 protected cells from ICI 182,780 induced suppression of proliferation in both MCF-7 and ZR-75-1 cell lines (Fig. 6b, c). Additionally, migration of cells expressing ZEB2 was not affected by ICI 182,780 or estrogen treatment when measured by transwell migration assay (Supplementary Figure S5), demonstrating that ZEB2 mediated changes in migration occur independent of estrogen signaling.

ZEB2 decreases endocrine sensitivity in ER + breast cancer cells. a Percent clonogenic survival of vector and ZEB2-expressing cells when treated with ICI 182,780 or DMSO control for 10 days. (***p < .001). b Proliferation of vector and ZEB2-expressing MCF-7 and ZR-75-1 cells were quantified using Ki-67 staining after 24 h treatment with 100 nM ICI 182,780, or DMSO control. c Representative images were captured using fluorescent microscopy and images are shown at 200X magnification. Scale bars represent 0.25 mm. ZR-75-1 is abbreviated to ZR-75 in the graphs within the figure
Discussion
Endocrine resistance and metastasis remain significant obstacles to breast cancer patient survival. These two processes are often linked, but shared mechanisms between them are less clearly elucidated. In the present study, we found that the ZEB2 transcription factor act independently of their canonical role as EMT inducers to mediate migration and metastasis in luminal A breast cancer. Furthermore, ZEB2 was shown to play a significant role as a repressor of estrogen signaling and hormone therapy response.
The role of ZEB2 as a driver of cell motility, migration, and metastatic spread has been documented in numerous malignancies, including glioma, oral, ovarian, gastric, lung, and pancreatic cancers [19, 28, 39,40,41,42,43]. Until recent years, the implication of ZEB2 in EMT associated with tumor cell invasion has been rooted in their ability to repress CDH1 and miR200 expression [31, 44]. This defining aspect of ZEB2 function was characterized in previous studies showing this repression in MDCK, hepatocellular and colon epidermoid carcinoma cells with forced exogenous expression of ZEB2 [21, 45, 46].
Many factors play into the signaling and biology involved in breast cancer metastasis. In contrast to existing literature, which maintains that ZEB2 expression promotes a mesenchymal phenotype, we see that in this study ZEB2-expressing cells present with a distinctly non-fibroblast like shape. Furthermore, the cells did not lose their cell contacts, a characteristic that is observed in mesenchymal cell types. While EMT has been shown to play a driving role in breast cancer metastasis in many studies, canonical EMT-inducing factors, are fully capable of driving metastasis through EMT-independent mechanisms [11, 12, 47]. Metastatic progression occurs through a myriad of differing process and studies have shown that cells are not required to shed all epithelial characteristics to achieve a metastatic phenotype [48, 49]. Furthermore, EMT is a “plastic” process, and complete EMT is not necessary for metastasis [50, 51]. This is exemplified by our finding that while ZEB2 expression successfully initiated cell migration and metastasis in MCF-7 and ZR-75–1 cells, our in vitro data suggest this is not through canonical EMT pathways. Our preliminary genomic analyses in Figs. 1 and 2 demonstrated no trends in canonical EMT genes and do not suggest that ZEB2 transcriptionally regulates canonical EMT pathways, more extensive genomic arrays would elucidate non-canonical EMT pathways that may have contributed to acquisition of this phenotype. Furthermore, protein analyses of EMT markers would determine more concrete associations between ZEB2-mediated regulation of EMT pathways. In this manuscript, we explored non-EMT driven mechanisms that ZEB2 utilized to gain migratory and metastatic cell behavior.
While metastasis contributes significantly to mortality in ER + breast cancer patients, its detrimental effects are often compounded by the concurrent acquisition of resistance to endocrine targeted therapies, the mainstay of ER + breast cancer treatment. A myriad of mechanisms exist by which ER + breast cancer cells progress to an endocrine-resistant phenotype [52]. Studies have shown that resistance to these therapies can arise from increased estrogen signaling initiated after treatment with the inhibitors [53]. However, the downregulation of ER transcriptional co-repressors, has also been implicated in endocrine resistance. FOXA1 expression has been demonstrated to be necessary for both ESR1 transcription and ER-α binding and function as a transcriptional regulator [38]. Similarly, GATA3 has also been shown to participate in this functional protein complex with FOXA1 [37, 54]. In this study, we see a significant decrease in expression of these important ER complex members upon expression of ZEB2. However, it is important to consider that the changes in luciferase activity could possibly be due to reduced ER transcription or reduced transcription of important ER complex members. Furthermore, we show that ZEB2 specifically, decreases estrogen signaling in MCF-7 and ZR-75-1 cells, resulting in estrogen independent proliferation. The increased cell migration due to ZEB2 overexpression in the non-invasive luminal cell line MCF-7 was abrogated in the presence of estrogen, and ICI 182,780 treatment did not suppress ZEB2-mediated migration. These data indicated ZEB2 resulted in estrogen independent migration in addition to proliferation. Loss of FOXA1 and GATA3 ER complex members in hormone positive breast cancers have been associated with metastasis and worse patient outcomes and prognoses [55, 56] and are important for ER functionality; ZEB2 significantly suppressed both FOXA1 and GATA3 expression. While our data suggests migration of ZEB2-expressing cells may be independent of ER signaling, we cannot rule out that these cells adapted alternative signaling of estrogen-related pathways.
Conclusion
This work presents a novel role for ZEB2 in breast cancer biology in the luminal A subtype. Future studies will examine the activity of the ZEB family of transcription factors in breast cancers with acquired resistance to endocrine therapy.
References
Kohler BA, Sherman RL, Howlander N, Jemal A, Ryerson AB, Henry KA, Boscoe FP, Cronin KA, Lake A, Noone AM et al (2015) Annual report to the nation on the status of cancer, 1975–2011, featuring incidence of breast cancer subtypes by race/ethnicity, poverty, and state. J Natl Cancer Inst 107(6):djv048.
Siegel RL, Miller KD, Jemal A (2018) Cancer statistics, 2018. CA Cancer J Clin 68:7–30
Guo F, Kuo Y-f, Shih YCT, Giordano SH, Berenson AB (2018) Trends in breast cancer mortality by stage at diagnosis among young women in the United States. Cancer 124(17):3500–3509
Hashmi AA, Aijaz S, Khan SM, Mahboob R, Irfan M, Zafar NI, Nisar M, Siddiqui M, Edhi MM, Faridi N et al (2018) Prognostic parameters of luminal A and luminal B intrinsic breast cancer subtypes of Pakistani patients. World J Surg Oncol 16(1).
Perou CM, Sorlie T, Eisen MB, Van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA et al (2000) Molecular portraits of human breast tumours. Nature 406:747–752
Meisel JL, Venur VA, Gnant M & Carey L (2018) Evolution of targeted therapy in breast cancer: where precision medicine began. Am Soc Clin Oncol Educ Book 78–86.
Musgrove EA, Sutherland RL (2009) Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer 9:631
Giulliano M, Schifp R, Osborne CK, Trivedi MV (2011) Biological mechanisms and clinical implications of endocrine resistance in breast cancer. Breast 20(Suppl 3):S42–S49
Hiscox S, Gee J, Nicholson RI (2007) Endocrine resistance and breast cancer invasion. In: Mansel RE, Fodstad O, Jiang WG (Eds.) Metastasis of Breast Cancer. Dordrecht: Springer Netherlands.
Yuan J, Liu M, Yang L, Tu G, Zhu Q, Chen M, Cheng H, Lou H, Fu W, Li Z, Yang G (2015) Acquisition of epithelial-mesenchymal transition phenotype in the tamoxifen-resistant breast cancer cell: a new role for G protein-coupled estrogen receptor in mediating tamoxifen resistance through cancer-associated fibroblast-derived fibronectin and beta1-integrin signaling pathway in tumor cells. Breast Cancer Res 17(1):69
Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. J Clin Invest 119:1420–1428
Savagner PP (2010) The epithelial-mesenchymal transition (EMT) phenomenon. Ann Oncol 21(Suppl 7):vii89-vii92.
Lamouille S, Xu J, Derynck R (2014) Molecular mechanisms of epithelial–mesenchymal transition. Nat Rev Mol Cell Biol 15(3):178–196
Thiery JP, Acloqque H, Huang RY, Nieto MA (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139(5):871–890
Welch DR, Hurst DR (2019) Defining the hallmarks of metastasis. Cancer Res 79(12).
Aigner K, Dampier B, Descovich L, Mikula M, Sultan A, Schreiber M, Mikulits W, Brabletz T, Strand D, Obrist P et al (2007) The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 26(49):6979–6988
Korpal M, Lee ES, Hu G, Kang Y (2008) The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by directly targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem 283:14910–14914
Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, Goodall GJ (2008) A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res 68:7846–7854
Kurahara H, Takao S, Maemura K, Mataki Y, Kuwahata T, Maeda K, Ding Q, Sakoda M, Iino S, Ishigami S et al (2012) Epithelial–mesenchymal transition and mesenchymal–epithelial transition via regulation of ZEB-1 and ZEB-2 expression in pancreatic cancer. J Surg Oncol 105:655–661
Voutsadakis IA (2016) Epithelial-mesenchymal transition (EMT) and regulation of EMT factors by steroid nuclear receptors in breast cancer: A review and in silico investigation. J Clin Med 5(1):11
Vandewalle C, Comijn J, De Craene B, Vermassen P, Bruyneel E, Andersen H, Tulchinsky E, Van Roy F, Berx G (2005) SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell–cell junctions. Nucleic Acids Res 33:6566–6578
Rhodes LV, Antoon JW, Muir SE, Elliott S, Beckman BS, Burow ME (2010) Effects of human mesenchymal stem cells on ER-positive human breast carcinoma cells mediated through ER-SDF-1/CXCR4 crosstalk. Mol Cancer 9:295
Rhodes LV, Tate CR, Segar HC, Burks HE, Phamduy TB, Hoang V, Elliott S, Gilliam D, Pounder FN, Anbalagan M et al (2014) Suppression of triple-negative breast cancer metastasis by pan-DAC inhibitor panobinostat via inhibition of ZEB family of EMT master regulators. Breast Cancer Res Treat 145:593–604
Martin EC, Rhodes LV, Elliott S, Krebs AE, Nephew KP, Flemington EK, Collins-Burow BM, Burow ME (2014) microRNA regulation of mammalian target of rapamycin expression and activity controls estrogen receptor function and RAD001 sensitivity. Mol Cancer 13:229
Miller DFB, Yan PS, Buechlein A, Rodriguez BA, Yilmaz AS, Goel S, Lin H, Collins-Burow B, Rhodes LV, Braun C, Pradeep S et al (2013) A new method for stranded whole transcriptome RNA-seq. Methods (San Diego, Calif.) 63:126–134.
Jezequel P, Campone M, Gouraud W, Guerin-Charbonnel C, Leux C, Ricolleau G, Campion L (2012) bc-GenExMiner: an easy-to-use online platform for gene prognostic analyses in breast cancer. Breast Cancer Res Treat 131:765–775
Jézéquel P, Gouraud W, Ben Azzouz F, Guérin-Charbonnel C, Juin P, Lasla H, Campone M (2021) bc-GenExMiner 4.5: new mining module computes breast cancer differential gene expression analyses. Database (Oxford) baab007
Sánchez-Tillό E, Siles L, De Barrios O, Cuatrecasas M, Vaquero EC, Castells A, Postigo A (2011) Expanding roles of ZEB factors in tumorigenesis and tumor progression. Am J Cancer Res 1:897–912
Hill L, Browne G, Tulchinsky E (2013) ZEB/miR-200 feedback loop: at the crossroads of signal transduction in cancer. Int J Cancer 132:745–754
Friedl P, Gilmour D (2009) Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol 10:445–457
Friedl P, Locker J, Sahai E, Segall JE (2012) Classifying collective cancer cell invasion. Nat Cell Biol 14:777–783
Yilmaz M, Christofori G (2010) Mechanisms of motility in metastasizing cells. Mol Cancer Res 8:629–642
Yilmaz M, Christofori G, Lehembre F (2007) Distinct mechanisms of tumor invasion and metastasis. Trends Mol Med 13:535–541
Qi S, Song Y, Peng Y, Wang H, Long H, Yu X et al (2012) ZEB2 mediates multiple pathways regulating cell proliferation, migration, invasion and apoptosis in glioma. PLoS ONE. https://doi.org/10.1371/journal.pone.0038842
Marino M, Galluzzo P, Ascenzi P (2006) Estrogen signaling multiple pathways to impact gene transcription. Curr Genomics 7:497–508
Saha Roy S, Vadlamudi RK (2012) Role of estrogen receptor signaling in breast cancer metastasis. Int J Breast Cancer 2012:654698.
Theodorou V, Stark R, Menon S, Carroll JS (2013) GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res 23:12–22
Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS (2011) FOXA1 is a critical determinant of estrogen receptor function and endocrine response. Nat Genet 43:27–33
Mejlvang J, Kriajevska M, Vandewalle C, Chernova T, Saayan AE, Berx G, Mellon JK, Tulchinsky E (2007) Direct repression of cyclin D1 by SIP1 attenuates cell cycle progression in cells undergoing an epithelial mesenchymal transition. Mol Biol Cell 18:4615–4624
Dai YH, Tang YP, Zhu HY, Lv L, Chu Y, Zhou YQ, Hou JR (2012) ZEB2 promotes the metastasis of gastric cancer and modulates epithelial mesenchymal transition of gastric cancer cells. Dig Dis Sci 57:1253–1260
You J, Li Y, Fang N, Liu B, Zu L, Chang R, Li X, Zhou Q (2014) MiR-132 Suppresses the migration and invasion of lung cancer cells via targeting the EMT regulator ZEB2. PLOS One 9:e91827
Kong YH, Syed Zanaruddin SN, Lau SH, Ramanathan A, Kallarakkal TG, Vincent-Chong VK, Wan Mustafa WM, Abraham MT, Abdul Rahman ZA, Zain RB et al (2015) Co-expression of TWIST1 and ZEB2 in oral squamous cell carcinoma is associated with poor survival. PLOS One 10:e0134045
Wang Q, Jiang H, Deng X, Fang W, Guo S (2015) Expressions of ZEB2 and C-myc in epithelial ovarian cancer and their clinical significance Nan Fang Yi Ke Da Xue Xue Bao 35:1765–9.
Brabletz S, Brabletz T (2010) The ZEB/miR-200 feedback loop—a motor of cellular plasticity in development and cancer? EMBO Rep 11:670–677
Comijn J, Berx G, Vermassen P, Verschueren K, Van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, Van Roy F (2001) The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell 7:1267–1278
Miyoshi A, Kitajima Y, Sumi K, Sato K, Hagiwara A, Koga Y, Miyazaki K (2004) Snail and SIP1 increase cancer invasion by upregulating MMP family in hepatocellular carcinoma cells. Br J Cancer 90:1265–1273
Tsai JH, Yang J (2013) Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev 27:2192–2206
Krakhmal NV, Zavyalova MV, Denisov EV, Vtorushin SV, Perelmuter VM (2015) Cancer invasion: patterns and mechanisms. Acta Naturae 7:17–28
Campbell K, Casanova J (2016) A common framework for EMT and collective cell migration. Development 143:4291–4300
Garg M (2017) Epithelial, mesenchymal and hybrid epithelial/mesenchymal phenotypes and their clinical relevance in cancer metastasis. Expert Rev Mol Med 19:e3.
Jolly MK, Boareto M, Huang B, Jia D, Lu M, Ben-Jacob E, Onuchic JN, Levine H (2015) Implications of the hybrid epithelial/mesenchymal phenotype in metastasis. Front Oncol 5:155
Tokunaga E, Hisamatsu Y, Tanaka K, Yamashita N, Saeki H, Oki E, Kitao H, Maehara Y (2014) Molecular mechanisms regulating the hormone sensitivity of breast cancer. Cancer Sci 105:1377–1383
Osborne CK, Schiff R (2011) Mechanisms of endocrine resistance in breast cancer. Annu Rev Med 62:233–247
Albergaria A, Paredes J, Sousa B, Milanezi F, Carneiro V, Bastos J, Costa S, Viera D, Lopes N, Lam EW et al (2009) Expression of FOXA1 and GATA-3 in breast cancer: the prognostic significance in hormone receptor-negative tumours. Breast Cancer Res 11(3):R40
Hosoda M, Yamamoto M, Nakano K, Hatanaka KC, Takakuwa E, Hatanaka Y, Matsuno Y, Yamashita H (2014) Differential expression of progesterone receptor, FOXA1, GATA3, and p53 between pre- and postmenopausal women with estrogen receptor-positive breast cancer. Breast Cancer Res Treat 144:249–261
Hisamatsu Y, Tokunaga E, Yamashita N, Akiyoshi S, Okada S, Nakashima Y, Taketani K, Aishima S, Oda Y, Morita M, Maehara Y (2015) Impact of GATA-3 and FOXA1 expression in patients with hormone receptor-positive/HER2-negative breast cancer. Breast Cancer 22:520–528
Acknowledgements
Thank you to Dr. Van T. Hoang, PhD and the animal vivarium staff at Tulane University School of Medicine for their assistance with the in vivo experiments. High throughput sequencing was performed at the Center for Genomics and Bioinformatics at Indiana University, Bloomington.
Funding
This project was funded by the National Institutes of Health R01-CA174785-01A1 (BMC-B) and the National Institutes of Health R01-CA125806-02 (MEB).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to disclose.
Ethical approval
All procedures involving these animals were conducted in compliance with State and Federal laws, standards of the U.S. Department of Health and Human Services, and guidelines established by the Tulane University Animal Care and Use Committee. The Tulane University Animal Care and Use Committee approved the use of animals in this specific study. The facilities and laboratory animal program of Tulane University are accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Burks, H.E., Matossian, M.D., Rhodes, L.V. et al. ZEB2 regulates endocrine therapy sensitivity and metastasis in luminal a breast cancer cells through a non-canonical mechanism. Breast Cancer Res Treat 189, 25–37 (2021). https://doi.org/10.1007/s10549-021-06256-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-021-06256-x