
Abstract
Purpose
Vantictumab is a monoclonal antibody that binds to frizzled (FZD) receptors and inhibits canonical WNT signaling. This phase Ib dose escalation study enrolled patients with locally recurrent or metastatic HER2-negative breast cancer who were treated with weekly paclitaxel in combination with escalating doses of vantictumab.
Methods
Patients were enrolled in dose escalation cohorts treated with weekly paclitaxel 90 mg/m2 on days 1, 8 and 15 in combination with vantictumab 3.5–14 mg/kg days 1 and 15 or 3–8 mg/kg day 1 of every 28-day cycle. Primary endpoints were safety, dose-limiting toxicities (DLTs). Secondary endpoints included pharmacokinetics, efficacy and an exploratory biomarker analysis.
Results
Forty-eight female patients with a mean age of 54 were enrolled. The majority (66.6%) received prior chemotherapy for recurrent or metastatic disease; 45.8% were hormone receptor (HR)-positive, HER2-negative and 54.2% triple-negative. The most frequent adverse events related to any study treatment were nausea (54.2%), alopecia (52.1%), fatigue (47.9%), and peripheral neuropathy (43.8%). No DLTs occurred; however, 6 patients experienced fractures outside of the DLT window. The overall response rate was 31.3% and the clinical benefit rate was 68.8%. A 6-gene WNT pathway signature showed significant association with progression-free survival (PFS) and overall survival (OS) for the biomarker high versus biomarker low groups (PFS: p = 0.029 and OS: p = 0.00045, respectively).
Conclusions
The combination of vantictumab and weekly paclitaxel was generally well tolerated with promising efficacy; however, the incidence of fractures limits future clinical development of this particular WNT inhibitor in metastatic breast cancer.
Clinical Trial Registration:
ClinicalTrials.gov registration: NCT01973309
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Metastatic breast cancer remains a leading cause of cancer-related death in women worldwide [1]. Although many agents can prolong progression-free survival (PFS), and even overall survival (OS) in some cases, acquired resistance to these therapies is nearly universal and there remains a critical need for novel treatment strategies to address chemotherapy resistance [2].
The WNT/ß-catenin signaling pathway is involved in the control of multiple cellular processes including cellular proliferation, determination of cell fate, self-renewal and maintenance of cancer stem cells [3]. WNT signaling is dysregulated in many human cancers through overexpression of WNT ligands, epigenetic silencing of negative regulators, and mutations in APC, Axin 1 or ß-catenin [4]. Targeting cancer stem cells with agents that inhibit WNT/ß-catenin signaling is a promising strategy to overcome chemotherapy resistance.
Vantictumab (OMP-18R5) is a fully human immunoglobulin G2 (IgG2) monoclonal antibody that binds to Frizzled (FZD) receptors 1, 2, 5, 7, and 8 and inhibits canonical WNT signaling [5]. In human cancer cell line and patient-derived xenograft models of breast cancer, vantictumab treatment resulted in tumor growth inhibition that was potentiated by the addition of paclitaxel [5, 6]. Vantictumab treatment reduced the frequency of tumor-initiating cells and led to down-regulation of gene expression programs associated with epithelial-mesenchymal transition [5, 6]. Sequential dosing of vantictumab followed by paclitaxel resulted in more robust mitotic cell death, tumor growth inhibition and decreased cancer stem cell frequency [6]. These findings support the hypothesis that treatment with vantictumab can sensitize cancer stem cells to paclitaxel.
Vantictumab was investigated in a first-in-human, phase I dose escalation study where the drug was generally well tolerated at escalating doses administered weekly, every 2 weeks or every 3 weeks [7]. Bone fractures were observed in a subset of patients and were considered target-mediated toxicity due to the role of WNT pathway signaling in bone homeostasis [8].
This open-label, phase Ib dose escalation study enrolled patients with locally recurrent or metastatic HER2-negative breast cancer who were treated with weekly paclitaxel in combination with escalating doses of vantictumab to evaluate safety, tolerability, immunogenicity, and preliminary efficacy.
Methods
Patients
Eligible patients had locally recurrent or metastatic HER2-negative breast cancer treated with ≤ 2 prior lines of chemotherapy for locally recurrent or metastatic disease. Patients had evaluable or measurable disease by RECIST v1.1 (Response Evaluation Criteria in Solid Tumors) [9] and an Eastern Cooperative Oncology Group (ECOG) performance status of 0–1. Patients were ≥ 18 years old with adequate hematopoietic, hepatic and kidney function. Key exclusion criteria included: dose delays during prior taxane treatment; pregnant or breastfeeding; anti-cancer therapy within 3 weeks or 5 half-lives; bone radiation < 2 weeks; untreated brain metastasis or leptomeningeal disease; HIV; bleeding diathesis or therapeutic warfarin; osteoporosis; bone metastases with pathologic fracture, planned orthopedic intervention or unable to receive bisphosphonate or denosumab; chronic glucocorticoid steroids; ß-C-terminal telopeptide (ß-CTX) > 1000 pg/mL and metabolic bone disease.
Additional bone-specific exclusions were added for cohorts 4–7: prior radiation to the spine or pelvis; gastric bypass; fragility fracture; moderate morphometric fracture identified on imaging; and risk of major osteoporotic fracture > 20% or hip fracture > 3% by FRAX. Normal calcium, 25-OH-vitamin D and TSH were required.
Written informed consent was obtained prior to study-related procedures in accordance with federal and institutional guidelines. The institutional review boards of participating institutions approved the protocol prior to patient enrollment.
Study design
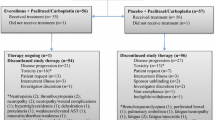
This was a phase Ib, dose escalation study of weekly paclitaxel in combination with escalating doses of vantictumab. In cohorts 1–3, vantictumab was administered at increasing doses (3.5 mg/kg, 7 mg/kg, and 14 mg/kg) intravenously (IV) on days 1 and 15 of each 28-day cycle (every 2 week dosing; Q2W). In cohorts 4–6, vantictumab was administered at increasing doses (3 mg/kg, 5 mg/kg, and 8 mg/kg) IV on day 1 only (every 4 week dosing; Q4W). Cohort 7 evaluated sequential dosing of vantictumab 8 mg/kg IV Q4W administered 2 days prior to the first dose of paclitaxel (Fig. 1).

Vantictumab and paclitaxel administration schedule. In cohorts 1–3, vantictumab was administered every 2 weeks on day 1 and day 15 of each cycle. Paclitaxel was administered on day 1, 8 and 15 of each cycle. In cohorts 4–6, vantictumab was administered every 4 weeks on day 1 only of each cycle. Paclitaxel was administered as in cohorts 1–3. In cohort 7, vantictumab was administered every 4 weeks on day 1 of each cycle and paclitaxel was administered on day 3, 10 and 17 of each cycle. Q2W, every 2 weeks; Q4W, every 4 weeks; D1, day 1
For Cohorts 1–6, paclitaxel (90 mg/m2) was administered IV on Days 1, 8, and 15 of each 28-day cycle and Days 3, 10, and 17 in cohort 7 (Fig. 1). The starting dose of vantictumab was based on safety of 5 mg/kg and 10 mg/kg IV every 3 weeks in a phase I study [7].
Dose escalation of vantictumab was conducted using a modified 3 + 3 design with 3–6 patients in cohorts 1–3 and ≥ 6 patients in cohorts 4–6. The maximum tolerated dose (MTD) was defined as the highest dose level at which < 2 of 6 evaluable patients experienced dose-limiting toxicity (DLT) or any grade fragility fracture. The DLT window was 28 and 31 days in cohorts 1–6 and 7, respectively. Patients could receive one dose reduction of paclitaxel (from 90 mg/m2 to 70 mg/m2) and still be DLT evaluable. Patients who did not receive all three planned doses of paclitaxel during the first cycle for any reason other than a DLT were not considered DLT evaluable and were replaced.
Following cohorts 1–3, emerging data from other trials of vantictumab Q2W indicated increased risk of fragility fractures. As a result, all patients discontinued vantictumab and for cohorts 4–7 vantictumab was administered Q4W. Additional eligibility criteria were added and a bone safety window of 56 days following the first dose of vantictumab was added. The bone safety window resulted in a longer observation period to surveil for fragility fractures.
Repeat imaging was performed every 8 weeks and patients continued on treatment until progression, start of other anti-cancer therapy, patient decision, investigator decision based on patient’s best interest or protocol non-compliance.
Safety monitoring
At screening, patients underwent physical examination, ECOG, ECG, bone scan, lateral thoracolumbar spine x-ray, DEXA and calculation of FRAX score. Labs included: complete blood count; fasting chemistry, bone turnover markers and lipids; 25-hydroxy vitamin D; coagulation tests, urinalysis; and serum pregnancy test as applicable.
Safety assessments during study treatment included physical examination, ECG and labs. AEs were assessed using the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. DEXA was repeated every 2 cycles. Fasting bone turnover markers (ß-CTX, bone-specific alkaline phosphatase, osteocalcin and procollagen type 1 amino-terminal propeptide [P1NP]) were determined at a central laboratory on day 1 of each cycle.
Additional bone-specific safety procedures
At screening and treatment termination, a FRAX score was calculated (https://www.shef.ac.uk/FRAX/tool.jsp) and patients with a 10-year fracture risk of > 20% for any bone or > 3% for the hip received zoledronic acid 5 mg IV prior to cycle 1 day 1 repeated every 12 months while on vantictumab. Patients with a history of fragility fracture of the hip or symptomatic vertebral fracture also received zoledronic acid as above. All patients received at least vitamin D 1000 IU/day and calcium 1000–1200 mg/day.
Following initiation of study treatment, any patient experiencing a ≥ twofold increase in ß-CTX from baseline or a T-score decline to < -2.5 on DEXA received zoledronic acid 5 mg IV repeated every 12 months while on vantictumab. For patients with a creatinine clearance < 35 mL/min, denosumab 60 mg SQ every 6 months could be administered. Patients receiving an IV bisphosphonate or denosumab prior to the study were continued on this therapy.
For patients enrolled in cohorts 4–7, all postmenopausal women received zoledronic acid at baseline and ß-CTX and DEXA were sampled between day 22–28 of each cycle allowing review prior to the next cycle. A decrease in P1NP ≥ 50% from baseline triggered zoledronic acid which could be repeated.
DLT definitions
DLTs were AEs related to vantictumab: grade ≥ 3 non-hematologic, non-hepatic AEs with the exception of grade 3 rash, nausea, vomiting, diarrhea, electrolyte disturbances or infusion reactions responding promptly to supportive care; grade ≥ 4 thrombocytopenia or neutropenia ≥ 7 days; grade ≥ 3 total bilirubin or hepatic transaminases; and new fragility fracture during the bone safety window.
Vantictumab and paclitaxel treatment modifications and discontinuation
Vantictumab was discontinued in the following situations: 1) patients with bone metastasis on chronic zoledronic acid or denosumab with a ≥ twofold increase in ß-CTX from baseline or a T-score decline to < -2.5 on DEXA, 2) patients experiencing a vantictumab-related fragility fracture or 3) patients with persistent changes in bone turnover markers or bone density not responsive to zoledronic acid or denosumab. Changes in bone turnover markers were confirmed with repeat measurement prior to vantictumab discontinuation. In patients receiving chronic therapy with zoledronic acid or denosumab, an increase in ß-CTX of up to 300 pg/mL did not lead to discontinuation as this value is associated with profound suppression of osteoclast activity.
Patients who experienced a DLT or clinically significant grade 2 toxicity or grade 3/4 toxicity considered related to vantictumab after the DLT window could hold vantictumab for up to 28 days following discussion with the study team.
Dose modifications of paclitaxel were allowed in accordance with the package insert. Standard premedications were used to prevent hypersensitivity reactions. Supportive care medications including hematopoietic growth factors and transfusion could be used per standard clinical practice, but not prophylactically. Paclitaxel could be discontinued permanently and vantictumab continued; however, if vantictumab was discontinued permanently the patient came off study.
Pharmacokinetic and immunogenicity assessments
Vantictumab drug levels were measured on cycle 1 day 1 prior to and 30 min post the end of the vantictumab infusion. Additionally, pharmacokinetic (PK) samples were obtained on cycle 1 day 8 prior to paclitaxel, cycle 1 day 15 prior to vantictumab, cycle 3 day 1 after the end of the vantictumab infusion, cycle 3 day 8 prior to paclitaxel, cycle 3 day 15 prior to vantictumab, and before vantictuamb on day 1 of alternating subsequent cycles. Blood samples for assessment of anti-drug antibodies (ADA’s) were obtained on day 1 of cycles 1, 3 and every other subsequent cycle and treatment termination.
Biomarker assessments
Formalin-fixed, paraffin-embedded (FFPE) tumor tissue was collected as archival tissue or a fresh tumor biopsy for all patients at the time of trial entry. RNA was isolated and quantitative polymerase chain reaction (qPCR) was performed for 6 Wnt pathway genes identified from preclinical models as potentially predictive biomarkers (DKK1, FBXW2, CCND2, RHOU, CTBP2, and WIF1). Optimal assay sensitivity was achieved using cDNA synthesis by gene specific priming followed by 18 cycles of pre-amplification. The delta Cq value of each gene was calculated by subtracting the mean of the Cq values of four reference genes (PUM1, SDHA, TOP1 and GUSB) from the Cq value of each of the 6 genes. The Lasso model with 500 × tenfold cross-validation was used to select the optimal model and measure the performance with PFS or OS as the response variable. Each of the 6 genes was includes as a binary variable by setting the cut-off at either 25%, or 50% or 75%, based on best performance by the Lasso analysis method. The model with the best performance (C index = 0.797) for OS was: CCND2 at 50%, CTBP2 at 50%, DKK1 at 75%, FBXW2 at 50%, RHOU at 25%, WIF1 at 50%. Using these cutoffs, the gene signature score was calculated with each gene as a binary variable and the cut-off of 55% was identified.
Statistical analysis
Descriptive statistics were used for baseline characteristics, adverse event frequency, tumor response and PK parameters. The safety population included all patients who received at least one partial or complete dose of vantictumab and who had at least one post-dosing safety evaluation. The immunogenicity population included all subjects who had a baseline and at least one follow-up vantictumab sample obtained. The Kaplan–Meier method was utilized for time-to-event variables and all analysis were completed using Statistical Analysis Software (SAS) version 9.2 or higher.
Results
Patients
A total of 48 patients were enrolled in the study and all received vantictumab and paclitaxel. Patients were enrolled at 5 sites in the United States between October 2013 and April 2017. Patient demographics and baseline characteristics are described in Table 1. The mean age of patients was 54 years old (range 32–78), all were female and 66.6% were previously treated with at least one prior line of chemotherapy for recurrent or metastatic disease; 45.8% had HR-positive, HER2-negative breast cancer and 54.2% TNBC.
Patients received on average 4.0 (range 1–16) and 11.0 (range 1–58) doses of vantictumab and paclitaxel, respectively. The average duration of treatment was 2.5 (range 0.03–13.6) and 3.2 (range 0.03–18.0) months for vantictumab and paclitaxel, respectively. Reasons for study treatment discontinuation included disease progression by RECIST (n = 29, 60.4%), withdrawal of consent/patient decision (n = 6, 12.5%), investigator decision based on patient’s best interest (n = 4, 8.3%), study terminated by the sponsor, (n = 4, 8.3%), adverse events (n = 3, 6.3%), clinical disease progression (n = 1, 2.1%) and death (n = 1, 2.1%).
Dose escalation and dose-limiting toxicities
No DLTs or bone fractures were observed in cohorts 1–3 (Table 2). However, based on a comprehensive analysis of emerging evidence of increased incidence of fragility fractures related to vantictumab administered at this schedule in other ongoing phase Ib studies, Q2W dosing of vantictumab was not considered safe. Vantictumab treatment was discontinued in patients enrolled in cohorts 1–3 and the protocol was amended as described above to include additional eligibility criteria, bone safety monitoring and administration of zoledronic acid or denosumab in high risk patients.
In cohorts 4–6, 8–10 patients were enrolled per cohort and treated with 3.0 mg/kg, 5.0 mg/kg and finally 8.0 mg/kg Q4W (Table 2). These patients experienced no DLTs and no bone fractures during the DLT period. The maximum administered dose following the bone mitigation plan was 8.0 mg/kg Q4W and the MTD of vantictumab according to protocol-defined criteria was not reached.
Based on preclinical data demonstrating greater synergy between vantictumab and paclitaxel with sequential dosing, cohort 7 was opened to include vantictumab dosing prior to paclitaxel. 10 patients were enrolled in cohort 7; however, a patient developed a grade 3 fragility fracture of the hip with non-union requiring hip replacement surgery that occurred after the DLT assessment and bone safety windows. This event was considered related to vantictumab by the investigator and following the event, the study was terminated.
Safety
All patients included in the safety analysis (n = 48) experienced at least 1 treatment-emergent adverse event (AE); 29 (60.4%) and 44 (91.7%) patients experienced an AE considered by the study investigators to be related to vantictumab or paclitaxel, respectively. The most frequent all-grade AEs related to any study treatment were nausea (54.2%), alopecia (52.1%), fatigue (47.9%), peripheral neuropathy (43.8%), and neutropenia (31.3%) (Table 3). The most frequent grade 3/4 AEs related to any study treatment were neutropenia (20.1%) and peripheral neuropathy (6.3%). The most frequent AEs related to vantictumab were fatigue (25%), nausea (22.9%), and constipation (16.7%) (Table 3).
Twelve patients (25%) experienced at least one serious adverse event (SAE), irrespective of causality, while on study. One patient experienced an SAE of dyspnea with a fatal outcome that was attributed by the investigator to disease progression. Vantictumab treatment-related SAEs occurred in 3 patients and included: grade 2 non-displaced fracture of the superior pubic ramus and non-displaced fracture of the right and left sacral ala in one patient at day 70; non-displaced bilateral sacral ala fractures in one patient at day 36; and fragility fracture of the hip and closed fracture of the left hip with non-union requiring hip replacement surgery in one patient at day 100 and day 127, respectively. Two patients experienced SAEs related to paclitaxel that included grade 2 enteritis and grade 3 nausea and vomiting in one patient each.
All patients underwent a baseline DEXA scan and 87.5% (n = 42) underwent at least one repeat DEXA. One patient had a decrease in bone density with a T-score of < − 1 to > − 2.5 during the study and two patients had a decrease to ≤ 2.5 on study. One of these patients experienced non-displaced bilateral sacral ala fractures at day 36. This patient received zoledronic acid prior to experiencing a fracture for an increase in ß-CTX ≥ twofold over baseline. Overall, 43 patients received bone protective therapy with zoledronic acid or denosumab prior to day 1 or during study treatment; 22 patients prior to day 1 for bone metastasis, 13 patients prior to day 1 for being postmenopausal at the time of study entry, 5 patients during study treatment for an increase in ß-CTX ≥ twofold over baseline, and 3 patients during study treatment for P1NP decrease ≤ 50% from baseline. Overall, positive correlations were found between ß-CTX and P1NP (correlation coefficient = 0.128), BSAP (correlation coefficient = 0.052) and osteocalcin (correlation coefficient = 0.245).
Overall, 6 patients experienced fractures; 3 fragility fractures related to vantictumab in 1 patient in cohorts 2, 3 and 7 and 3 pathologic or traumatic fractures unrelated to vantictumab in 1 patient in cohorts 4, 6 and 7 (Table 2). The majority of these patients received prior zoledronic acid. The incidence of fractures in this study was considered high enough to outweigh potential benefits from the addition of vantictumab to paclitaxel and the study was closed for this reason. All patients discontinued vantictumab at the time of study closure; however, some patients remained on paclitaxel outside of the study.
Pharmacokinetic and immunogenicity analysis
Vantictumab serum concentrations were within the expected drug exposure levels. Two of 48 patients (4.2%) developed ADAs at some point during the study; however, this did not impact exposure.
Efficacy
The best response was partial response in 31.3% (n = 15), stable disease 37.5% (n = 18) and progressive disease 22.9% (n = 11) (Fig. 2). Four patients (8.3%) were not evaluable for the efficacy endpoint. The overall response rate (ORR) was 31.3% and the clinical benefit rate (CR + PR + SD) was 68.8%. The ORR was similar for HR + HER2- 31.8% (n = 7/22) and TNBC 30.8% (n = 8/26). The median duration of response was 3.4 months (95% CI 1.8, 4.5). The median PFS was 3.8 months (95% CI 3.1, 5.3) and median OS was 12.7 months (95% CI 9.3, 16.4).

Waterfall plot. Maximal % change in tumor measurements for patients enrolled in all cohorts by investigator assessment. n = 44; 4 patients were not evaluable
Exploratory biomarker analysis
The most recent available metastatic or primary tumor archival FFPE tumor sample was collected from patients (n = 40). A 6-gene Wnt pathway biomarker signature showed significant association with PFS and OS for the biomarker high versus biomarker low groups (PFS: p = 0.029 and OS: p = 0.00045, respectively) (Fig. 3).

6-Gene Wnt signature. Analysis for progression-free survival (PFS) and overall survival (OS) outcome on FFPE tissue collected at baseline from 40 patients treated with paclitaxel and vantictumab in various cohorts. A) PFS and B) OS for patients characterized as biomarker high (≥ 55%) and low (< 55%). Baseline archival tumor tissue was obtained, RNA isolated and quantitative PCR performed to determine expression of CCND2, CTBP2, DKK1, FBXW2, RHOU and WIF1. Patients with high biomarker levels (green line, n = 18); low biomarker levels (red line, n = 22)
Discussion
In this open-label, multisite, phase Ib dose escalation study, patients with locally advanced or metastatic HER2-negative breast cancer were treated with weekly paclitaxel and increasing doses of vantictumab administered Q2W or Q4W with concurrent paclitaxel or Q4W with sequential paclitaxel. The observed toxicity profile of paclitaxel and vantictumab was consistent with the known safety profile of paclitaxel; however, multiple patients experienced fractures related to vantictumab which ultimately led to study closure. The maximum administered dose of vantictumab in this study was 14.0 mg/kg Q2W and 8.0 mg/kg Q4W; however, these doses were not deemed tolerable due to this higher than accepted rate of bone fractures occurring after the DLT observation period.
The most common treatment-emergent toxicities included nausea, alopecia, fatigue, peripheral neuropathy and neutropenia; all consistent with the known safety profile of paclitaxel. Mechanism-based toxicity with fragility fractures were reported in 3 patients and pathologic or traumatic fractures in another 3 patients despite incorporation of an aggressive bone safety plan comprised of carefully selected eligibility criteria, monitoring of bone turnover markers, frequent DEXA scans, and the mandatory use of bisphosphonates or RANKL inhibitors for patients identified as at risk at baseline or on study based on protocol-specified monitoring.
Many pharmacologic agents are in clinical development to target and inhibit WNT signaling for the treatment of cancer. Vantictumab is a fully humanized monoclonal antibody that binds to FZD receptors 1, 2, 5, 7, and 8 and inhibits canonical WNT signaling [5]. Ipafricept is a recombinant fusion protein made up of a human FZD 8 receptor extracellular ligand domain and human IgG1 and acts as a decoy receptor to inhibit WNT signaling [6]. Fragility fractures were also observed in patients treated with ipafricept as a single agent and in combination with paclitaxel and carboplatin and in patients treated with vantictumab in combination with nab-paclitaxel and gemcitabine, pointing to a class effect of these canonical WNT signaling inhibitors [10, 11].
The role of WNT signaling in bone remodeling is an area of active research by many groups; however, it remains incompletely characterized [12]. WNT ligands promote osteoblast differentiation from mesenchymal stem cells and inhibit osteoblast apoptosis [13]. Global deletion of the WNT co-receptor Lrp5 in mouse models results in decreased osteoblasts and a resulting decrease in bone mass whereas overexpression of Lrp5 leads to an increase in bone mass [14]. Similar results have been observed for single allele Lrp6 deletion; however, deletion of both alleles is fatal in mice [15]. Further work is needed to determine the impact of targeted inhibition of particular WNTs, FZDs, Lrps or downstream mediators on bone remodeling with a goal of assisting the development of WNT pathway inhibitors without treatment-related bone loss. Interestingly, in this study prophylactic administration of zoledronic acid or denosumab was not sufficient to prevent bone loss in patients administered vantictumab.
Promising clinical activity was observed in this trial with the combination of vantictumab and paclitaxel in patients with locally advanced or metastatic breast cancer previously treated with up to 2 prior lines of chemotherapy in the locally advanced or metastatic setting. The overall response rate was 31.8% for patients with HR + HER2-negative breast cancer and 30.8% for patients with TNBC in this trial. This is comparable to paclitaxel in the first line setting where the ORR was 33.2% in MERiDiAN and 21.2% in E2100 [16, 17]. The majority of patients in this trial had previously received 2–4 prior lines of chemotherapy. We performed an exploratory analysis using a 6-gene WNT pathway signature and found that expression of these genes correlated with PFS and OS for patients treated with vantictumab and paclitaxel. While this remains hypothesis-generating based on the small sample size and single arm nature of our trial, it raises the possibility of being able to identify biomarkers predictive of response to WNT inhibitors that have a more favorable safety profile in patients with breast cancer in the future.
References
Cardoso F, Senkus E, Costa A et al (2018) 4th ESO-ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 4)dagger. Ann Oncol 29:1634–1657
Scimeca M, Urbano N, Bonfiglio R et al (2019) Novel insights into breast cancer progression and metastasis: A multidisciplinary opportunity to transition from biology to clinical oncology. Biochim Biophys Acta Rev Cancer 1872:138–148
Clevers H, Loh KM, Nusse R (2014) Stem cell signalling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 346:1248012
Reya T, Clevers H (2005) Wnt signalling in stem cells and cancer. Nature 434:843–850
Gurney A, Axelrod F, Bond CJ et al (2012) Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci U S A 109:11717–11722
Fischer MM, Cancilla B, Yeung VP et al (2017) WNT antagonists exhibit unique combinatorial antitumor activity with taxanes by potentiating mitotic cell death. Sci Adv 3:e1700090
Smith DC, Rosen L.S., Chugh R, Goldman J.W., Xu L, Kapoun A, Brachman R.K., Dupont J, Stagg R.J., Tolcher A.W., Papadopoulos K.P. First-in-human evaluation of the human monoclonal antibody vantictumab (OMP-18R5; anti-Frizzled) targeting the WNT pathway in a phase I study for patients with advanced solid tumors. In American Society of Clinical Oncology (ASCO) Annual Meeting. J Clin Oncol 2013; 2540–2540.
Goldring SR, Goldring MB (2007) Eating bone or adding it: the Wnt pathway decides. Nat Med 13:133–134
Therasse P, Arbuck SG, Eisenhauer EA et al (2000) New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst 92:205–216
Jimeno A, Gordon M, Chugh R et al (2017) A First-in-Human Phase I Study of the Anticancer Stem Cell Agent Ipafricept (OMP-54F28), a Decoy Receptor for Wnt Ligands, in Patients with Advanced Solid Tumors. Clin Cancer Res 23:7490–7497
Moore KN, Gunderson CC, Sabbatini P et al (2019) A phase 1b dose escalation study of ipafricept (OMP54F28) in combination with paclitaxel and carboplatin in patients with recurrent platinum-sensitive ovarian cancer. Gynecol Oncol 154:294–301
Grigorie D, Lerner UH (2018) The Crucial Role of the Wnt System in Bone Remodelling. Acta Endocrinol (Buchar) 14:90–101
Baron R, Kneissel M (2013) WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med 19:179–192
Kato M, Patel MS, Levasseur R et al (2002) Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J Cell Biol 157:303–314
Holmen SL, Giambernardi TA, Zylstra CR et al (2004) Decreased BMD and limb deformities in mice carrying mutations in both Lrp5 and Lrp6. J Bone Miner Res 19:2033–2040
Miles D, Cameron D, Bondarenko I et al (2017) Bevacizumab plus paclitaxel versus placebo plus paclitaxel as first-line therapy for HER2-negative metastatic breast cancer (MERiDiAN): A double-blind placebo-controlled randomised phase III trial with prospective biomarker evaluation. Eur J Cancer 70:146–155
Miller K, Wang M, Gralow J et al (2007) Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med 357:2666–2676
Acknowledgements
The authors would like to thank the patients and their families for their participation in this study, as well as the study staff for managing the study.
Funding
This study was funded by OncoMed Pharmaceuticals. This work was also supported by the National Institutes of Health (NIH) and the National Cancer Institute (NCI) through 5P30CA046934-25 (University of Colorado Cancer Center Support Grant) and 1K23CA172691-01A1 (J.R. Diamond).
Author information
Authors and Affiliations
Contributions
JRD contributed to trial design, conduct of the trial, patient enrollment, data analysis and manuscript writing. CB, DR, AM, CO, JO, AF and MM contributed to trial design, patient enrollment and data acquisition. CZ, RH, AMK, LX, SU, BS and RKB contributed to trial design and execution, data analysis and biomarker studies. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
CZ, RH, AMK, LX, SU, BS and RKB are present or former employees of OncoMed Pharmaceuticals. AF received consulting fees from OncoMed.
Research involving human participants and/or animals
This article does not contain any studies with animals performed by any of the authors. All procedures performed in the studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. This study was conducted according to ICH guidelines and all sites had IRB/EC approval prior to enrolling patients.
Informed consent
Informed consent was obtained from all participants included in the study. All participants gave their informed consent in writing prior to inclusion in the study.
Consent for publication
Patients enrolled on the clinical provided consent for publication of results. This manuscript does not contain data from any individual person.
Availability of data and materials
The datasets generated and/or analyzed during the current study are not publicly available due to restrictions in the informed consent document signed by the patients enrolled in this trial.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Diamond, J.R., Becerra, C., Richards, D. et al. Phase Ib clinical trial of the anti-frizzled antibody vantictumab (OMP-18R5) plus paclitaxel in patients with locally advanced or metastatic HER2-negative breast cancer. Breast Cancer Res Treat 184, 53–62 (2020). https://doi.org/10.1007/s10549-020-05817-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-020-05817-w