
Abstract
Purpose
Seviteronel (INO-464) is an oral, selective cytochrome P450c17a (CYP17) 17,20-lyase (lyase) and androgen receptor inhibitor with in vitro and in vivo anti-tumor activity. This open-label phase 1 clinical study evaluated safety, tolerability, pharmacokinetics (PK), and activity of once-daily (QD) seviteronel in women with locally advanced or metastatic TNBC or ER+ breast cancer.
Methods
Seviteronel was administered in de-escalating 750, 600, and 450 mg QD 6-subject cohorts. The 750 mg QD start dose was a phase 2 dose determined for men with castration-resistant prostate cancer in (Shore et al. J Clin Oncol 34, 2016). Enrollment at lower doses was initiated in the presence of dose-limiting toxicities (DLTs). The primary objective of this study was to determine seviteronel safety, tolerability, and MTD. The secondary objectives included description of its PK in women and its initial activity, including clinical benefit rate at 4 (CBR16) and 6 months (CBR24).
Results
Nineteen women were enrolled. A majority of adverse events (AEs) were Grade (Gr) 1/2, independent of relationship; the most common were tremor (42%), nausea (42%), vomiting (37%), and fatigue (37%). Four Gr 3/4 AEs (anemia, delirium, mental status change, and confusional state) deemed possibly related to seviteronel occurred in four subjects. DLTs were observed at 750 mg (Gr 3 confusional state with paranoia) and 600 mg (Gr 3 mental status change and Gr 3 delirium) QD, with none at 450 mg QD. The recommended phase 2 dose (RP2D) was 450 mg QD, and at the RP2D, 4 of 7 subjects reached at least CBR16 (2 TNBC subjects and 2 ER+ subjects achieved CBR16 and CBR24, respectively); no objective tumor responses were reported.
Conclusions
Once-daily seviteronel was generally well tolerated in women with and 450 mg QD was chosen as the RP2D.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The underlying molecular heterogeneity of breast cancer is well recognized in the literature. Each breast cancer subtype is characterized by varied risk factors, clinicopathologic and molecular features, responses to therapy, patterns of recurrence, and clinical outcomes [1]. Molecular profiling has led to the classification of breast cancer into reproducible subgroups [2,3,4]. Furthermore, these advances have enabled investigators to identify novel targets for the development of potential therapeutic interventions. The androgen receptor (AR) is one such target. AR is expressed in 70–90% of all invasive breast cancers [5,6,7]. Emerging data suggest that the AR-signaling pathway may play a critical role in breast cancer pathogenesis.
Several groups have described a population of triple-negative breast cancer (TNBC) which demonstrates a gene expression signature resembling that of endocrine responsive tumors. This subtype is characterized by expression of AR and exhibits androgen-dependent, estrogen-independent growth in preclinical models [8,9,10]. The current standard of care in the treatment of TNBC is limited to traditional cytotoxic chemotherapy. The development of well-tolerated, effective, and targeted regimens that delay the need for cytotoxic chemotherapy and its side effects is an unmet need. Three early-phase prospective clinical studies investigating antiandrogen therapy have demonstrated clinical benefit of AR-targeted agents in women with metastatic AR+ TNBC [11,12,13].
Similar to TNBC, the role of AR in the management of estrogen receptor-positive (ER+) breast cancer is an area of active research. AR is expressed in up to 90% of ER+ tumors and preclinical data suggest that AR expression is associated with resistance to both tamoxifen and aromatase inhibitors in ER+ cell lines [14,15,16]. While initially beneficial, resistance to endocrine therapy eventually develops in the majority of patients with ER+ breast cancer. As a result, the question of how best to manage patients in this endocrine-resistant setting has become a significant clinical concern. Tamoxifen-resistant breast tumors have been shown in preclinical models to have elevated AR expression along with reduced ERα mRNA levels, and treatment with anti-androgens in this setting resulted in reversal of tamoxifen resistance [14,15,16]. Aromatase inhibitors are widely used in women with postmenopausal hormone receptor-positive (HR+) breast cancer in both the adjuvant and metastatic settings. Investigators have reported that resistance to aromatase inhibition may involve AR activation, as sensitivity to anastrozole in preclinical models was restored using the antiandrogen, enzalutamide, and the CYP17 inhibitor, abiraterone acetate [17]. Furthermore, AR contributed to ERα transcriptional activity in MCF7 cells co-expressing AR and aromatase, which could then be inhibited with use of bicalutamide [17]. These data suggest a role for AR in resistance to estrogen targeted therapies that may be overcome by inhibition of AR. We hypothesize that an agent that combines AR inhibition with decreased sex steroid production (e.g., androgens and estrogens) will demonstrate clinical benefit in both HR− and HR+ breast cancer, including the subset of women with endocrine-resistant tumors.
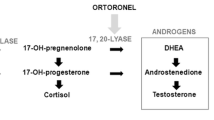
Seviteronel (INO-464) is an orally bioavailable, selective cytochrome P450c17a (CYP17) 17,20 lyase (lyase) and AR inhibitor. It is approximately 10-fold selective for CYP17 lyase versus CYP17 17-α hydroxylase (hydroxylase) inhibition [18]. Seviteronel inhibits the AR through competitive antagonism of both wild-type and mutated forms (e.g., F876L and T877A) of the receptor [19]. Through this unique dual mechanism of action, seviteronel inhibits androgen production, thus reducing downstream estrogen aromatization from androgens, while also inhibiting AR binding and activation.
Seviteronel has been shown to inhibit the growth in multiple breast cancer models, both in vitro and in vivo [20]. Seviteronel inhibited estrogen-stimulated proliferation and cell growth in a soft agar assay of MCF7 (ER+/low AR expression), H16N2 (AR−/PR−/low ER expression) cellular proliferation, the growth of tamoxifen-resistant MCF7 cells (TAMR), and DHT-stimulated growth of MDA-MB-453 cells (ER−/AR+), all with higher potency/efficacy than enzalutamide. Furthermore, seviteronel inhibited tumor growth and increased survival compared to enzalutamide in a tamoxifen-resistant MCF7 mouse xenograft model.
This report provides safety, tolerability, and pharmacokinetic findings from a phase 1 study of seviteronel in women with advanced TNBC or ER+ breast cancer and also provides preliminary insight into the endocrine response and clinical benefit of dual inhibition of CYP17 lyase and the AR.
Subjects and methods
Major eligibility criteria
Women with documented histological or cytological evidence of unresectable locally advanced or metastatic breast cancer that was either ER−, PR− and HER2−, or ER+ and HER2− were enrolled. There was no requirement for subjects to be AR+ for phase 1 study entry. Archival tumor samples were collected when available for future AR status determination to allow for potential inclusion in the planned phase 2 expansion. Female subjects with ER+ breast cancer must have been postmenopausal (or currently undergoing ovarian suppression using LHRH agonists) and had disease progression following at least one line of prior endocrine therapy, which may have included progression within 6 months of adjuvant endocrine therapy. There was no restriction on the number of prior therapies for women with TNBC. Prior use of investigational agents that inhibited CYP17 or the AR was allowed. The presence of CNS metastases was not exclusionary, provided the subject was asymptomatic. Eligibility required an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, WBC ≥ 3000/µl, ANC ≥ 1500/µl, platelet count ≥ 100,000/µl, HGB ≥ 10 g/dl and not transfusion dependent, AST and ALT levels ≤ 3X the upper limit of normal (ULN), bilirubin levels of ≤ 2.0 mg/dl, serum creatinine of ≤ 2.0 mg/dl and K + > 3.5 mEq/l. Subjects who required pharmacological or replacement doses of systemic corticosteroids or were administered systemic corticosteroids within 7 days of study drug administration were ineligible.
Study design and treatment
The primary objective of this phase 1 study was to determine the safety, tolerability, and maximum tolerated dose (MTD) of oral seviteronel administered once daily (QD) in women with triple-negative unresectable locally advanced or metastatic breast cancer, or postmenopausal women with ER+/HER2− unresectable locally advanced or metastatic breast cancer. The secondary objectives of the phase 1 study were to describe the pharmacokinetics (PK) of QD seviteronel and estimate its efficacy using clinical benefit rate at 16 weeks (CBR16 for TNBC) and clinical benefit rate at 24 weeks (CBR24 for ER+ breast cancer). Clinical benefit was defined as either stable disease, partial response, or complete response based upon RECIST 1.1. The study was approved by the institutional review board at each site. Informed consent was obtained from all individual participants included in the study.
Seviteronel was administered orally as 150-mg tablets in de-escalating dose cohorts of 750, 600, and 450 mg QD. The 750 mg QD start dose was a phase 2 dose determined for men with castration-resistant prostate cancer [1]. Study drug was administered in 28-day continuous dosing cycles. Study drug was discontinued if they were no longer clinically benefitting, an adverse event that precluded further participation in the study, or withdrawal of consent.
Six subjects were enrolled in sequential dose-level cohorts starting at 750 mg QD. Dose-limiting toxicity (DLT) was defined as any Grade 3 or greater adverse event possibly/probably/definitely related to seviteronel that occurred from the first dose of study drug through the end of the first 28-day continuous dosing cycle (Cycle 1). Two or more DLTs in a cohort resulted in opening the next lower dose-level cohort to six subjects. MTD was defined as the highest dose-level in which the incidence of DLTs was less than 33%. Toxicity was graded using the NCI Common Terminology Criteria for Adverse Events (NCI CTCAE), version 4.03.
Evaluations
Baseline evaluations were conducted within 28-days of study drug initiation and included medical history, physical exam, vital signs, ECOG performance status, ECG, CBC and serum chemistries, urinalysis and imaging assessments.
Repeat evaluations were conducted bi-weekly for Cycle 1 and then monthly thereafter through the end of treatment visit. Efficacy assessments by imaging were repeated every 2 months for the first year on study then every 3 months thereafter. The evaluation included all appropriate radiographic or scintigraphic procedures to document areas of metastatic disease, including bone scans, computed tomography scans and/or magnetic resonance imaging dependent upon what modality was utilized at baseline to assess metastatic disease.
Endocrine analysis
Blood samples were collected for serum estradiol and testosterone concentration determination at baseline and the end of Cycle 1. Endocrine samples were analyzed using a central lab (inVentiv Health Clinical Lab, Inc., Princeton, NJ). The lower limits of quantitation for serum estradiol and testosterone assessed using gas chromatography–tandem mass spectrometry were 2.3 and 86.7 pmol/L, respectively.
Pharmacokinetic analysis
Blood samples for pharmacokinetic (PK) analysis were collected prior to first dose of seviteronel and 0.5, 1, 2, 4, 6, 8, and 24 h after seviteronel dose. Additional samples were collected at Cycle 1 Day 14, Cycle 2 Day 1 and then every even-numbered cycle for spot PK analysis. The lower limit of quantification of seviteronel concentration in plasma when analyzed using liquid chromatography–tandem mass spectrometry was 20 ng/mL (0.05 uM) (Tandem Laboratories, Durham, NC). Dense PK parameters were analyzed by noncompartmental methods using WinNonlin (Certara, Princeton, NJ).
Results
Subject characteristics
A total of 19 women with ER+ (n = 14) or TNBC (n = 5) were enrolled from August 2015 to March 2016 with data presented as of 26 October 2017. Baseline characteristics, including disease status and prior lines of therapy are presented in Table 1. Most subjects (83–100% depending upon dose-level cohort) had received two or more lines of prior therapy for advanced disease. Only 1 subject (5%) had locally advanced disease and none had bone only disease; a majority of women (84%) had visceral disease.
Dose de-escalation
Out of the 6 subjects enrolled at 750 mg QD, one DLT reported (Gr 3 confusional state with paranoia, considered possibly related to study drug). Six subjects were then enrolled at 600 mg QD; two DLTs reported (Gr 3 mental status change and Gr 3 delirium, both considered possibly related to study drug). Seven subjects were enrolled at 450 mg QD; one subject was replaced at Day 22 of Cycle 1 due to dyspnea associated with disease progression. No DLTs were observed at 450 mg QD. The recommended phase 2 dose (R2PD) for seviteronel in women was determined to be 450 mg QD.
Tolerability
An overview of treatment-emergent AEs is presented in Tables 2 and 3. Treatment with seviteronel was generally well tolerated, with the majority of AEs Gr 1 or 2. The most common AEs were tremor (42%), nausea (42%), vomiting (37%), and fatigue (37%), independent of relationship. Four subjects reported AEs Gr 3 or greater. As per the investigator possibly related to seviteronel [anemia (450 mg QD), delirium (600 mg QD), mental status change (600 mg QD), and confusional state (750 mg QD)]. Four 4 deaths were reported and all were considered unrelated to study drug by the investigator [disease progression (n = 2), hepatic failure (n = 1), and ischemic stroke (n = 1)]. Fifteen serious adverse events (SAE) Gr 3 or greater were reported in 12 subjects with 3 events considered at least possibly related to seviteronel [delirium (600 mg QD), mental status change (600 mg QD) and confusional state (750 mg QD)].
Overall median treatment duration was 43 days (13, 257) and with the longest median duration observed in the 450 mg QD cohort [136 days (22, 257)]. All subjects are discontinued from study (Table 4). Treatment duration and reason for discontinuation are presented in Fig. 1. Progressive disease was the most prevalent reason for treatment discontinuation across all dose cohorts. Twenty-one percent (4/19) of subjects underwent a dose reduction due to an AE, which typically resulted in an improvement in the AE.

Duration of seviteronel therapy
Pharmacodynamic effects
Serum estradiol and testosterone concentrations were determined at baseline and Cycle 2 Day 1. Across dose-level cohorts, there were 6 subjects evaluable for analysis. Median decline in estradiol across dose-level cohorts was − 52% (− 29, − 89), and median testosterone decline was − 59% (− 45, − 76). Individual steroid reductions are presented in Supplemental Fig. 1.
Pharmacokinetics
A noncompartmental PK summary is presented in Table 5. Plasma concentrations after a single dose of seviteronel from subjects in the 450, 600, and 750 mg cohorts are presented in Supplemental Fig. 2. A comparison of seviteronel exposure to body surface area (BSA) and body mass index (BMI) in women at 450, 600, and 750 mg QD is presented in Supplemental Fig. 3.
Efficacy
Five of 19 subjects (26.3%) and 2 of 19 (11%) across dose groups reached at least CBR16 and CBR24, respectively (Fig. 1). In the 450 mg QD cohort, which was the RP2D, 4 of 7 subjects reached at least CBR16 (2 TNBC subjects and 2 ER+ subjects achieved CBR16 and CBR24, respectively). No objective tumor responses were reported.
Discussion
The results of this phase 1 clinical study demonstrate that seviteronel is generally well tolerated in subjects with advanced breast cancer. Based upon the safety and tolerability in the current phase 1 study, the recommended phase 2 dose of seviteronel in women was determined to be 450 mg QD. Pharmacodynamic effect was observed with decline in estrogen and testosterone consistent with CYP17 lyase inhibition, and preliminary evidence of clinical benefit was noted in a heavily pretreated population warranting further evaluation in breast cancer.
The most common adverse events with seviteronel were tremors, nausea, vomiting, fatigue, blurred vision, and dizziness. Some of these adverse effects, such as nausea, fatigue, and decreased cognition have been reported with other anti-androgens [11, 12, 21]. However, unlike bicalutamide, seviteronel was not associated with hepatic transaminase elevation [11]. One of the most common toxicities of abiraterone is hypokalemia, secondary to hypermineralocorticoidism. This was not seen with seviteronel, highlighting the selectivity of CYP17 lyase inhibition [21].
Human genetic mutations that lead to isolated CYP17 lyase deficiency or combined CYP17 hydroxylase/lyase deficiency result in potent sex steroids decreases, whereas only the latter results in significant progesterone increases and significant cortisol decreases [22, 23]. While seviteronel is a potent and selective CYP17 lyase inhibitor, its lyase activity is not completely isolated from hydroxylase; it still harbors some activity against CYP17 hydroxylase. Significant CYP17 hydroxylase inhibition results in cortisol suppression and an increase in ACTH, which can drive upstream steroid accumulation, including corticosterone and the associated mineralocorticoid excess syndrome (MES) [24]. Significant CYP17 hydroxylase inhibition does not appear to be occurring in men or women treated with seviteronel as the common signs and symptoms of MES, which include hypertension, hypokalemia, and fluid overload, were not observed in the current study or in men with CRPC treated with seviteronel [25] (Gupta et al. submitted). However, the most common AEs observed with seviteronel, including those that appear to have a CNS origin, are also found in patients experiencing adrenal glucocorticoid insufficiency [26,27,28], suggesting minor CYP17 hydroxylase inhibition. Accordingly, to ameliorate associated AEs, the addition of the glucocorticoid mimetic dexamethasone to seviteronel is currently being investigated in ongoing breast and prostate cancer studies.
Across doses examined in women, there was a dose-proportional relationship for peak and trough plasma drug concentrations, and overall exposure (AUC), after a single dose of seviteronel. Half-life ranged from 6.4 to 8.4 h, supporting once daily dosing. These results are similar to phase 1 seviteronel PK results in men with CRPC with regard to plasma half-life and dose-proportional drug exposure relationship (Gupta et al, submitted).
In the current study, the median decline in estradiol was 52% with seviteronel after 1 month of treatment across dose groups. By comparison, in postmenopausal women with ER + BC treated with letrozole, a nonsteroidal aromatase inhibitor, there is an approximately 36% decline in estradiol by 3 months of treatment [29]. In a similar group of women treated with abiraterone + prednisone, estradiol concentrations declined to an extent similar to women receiving exemestane alone [30]. This is in contrast to enzalutamide where there is 13% increase in estradiol when given in combination with exemestane and a 40% increase when given in combination with anastrozole [31].
The median decline in testosterone across dose groups was 59% in the current study. Potent (94%) declines in testosterone were observed after 1 month of treatment with abiraterone/prednisone, but this was accompanied by significant increases in progesterone concentrations (2,666%), which was considered a major reason for a lack of overall clinical benefit of abiraterone/prednisone in this population [21, 30].
Abiraterone is a steroidal CYP17 inhibitor with potent hydroxylase activity, and given its lack of lyase selectivity it is not surprising that progesterone concentrations were elevated, as is the case with men treated with abiraterone/prednisone [24]. Significant progesterone elevations do not occur in men (Gupta et al, submitted) or women treated with seviteronel [data on file], which is in line with the CYP17 lyase activity of seviteronel. The same is true for castrate male rhesus monkeys: progesterone concentrations remained unchanged with seviteronel treatment, but were significantly increased with abiraterone [32].
The maximum plasma concentration of seviteronel in women at 450 mg QD (recommended phase 2 dose) was similar to that achieved with the recommended phase 2 dose in men of 600 mg QD (Gupta et al. submitted). When exposure results from the three dose levels were combined, there was a moderate relationship between exposure and body surface area and body mass index. Given the moderate relationship and for patient convenience, a single fixed dose is being chosen for further phase 2 development in women.
In this study, which enrolled a heavily pretreated population with most (84%) having visceral disease, the 16- and 24-week CBR was 26.3 and 11%, respectively. The primary objective of the current study was to determine the safety and tolerability of seviteronel and not clinical activity; therefore, AR status determination or positivity was not required. That being said, these results are comparable with other early-phase breast cancer studies investigating anti-androgens as monotherapy where subjects were AR+. In a phase 2 study (TBCRC011), investigating bicalutamide in subjects with AR+ (ER-/PR-) metastatic breast cancer (N = 26), the 24-week CBR was 19%, median progression-free survival (PFS) was 12 weeks, and no objective responses were seen [11]. Similarly, in the MDV3100-11 study, investigating enzalutamide in subjects with AR + TNBC (N = 118; intent to treat population with AR > 0%), the 16-week and 24-week CBR was 25% and 20%, respectively and median PFS was 12.6 weeks (12). Regarding abiraterone, a 24-month CBR of 22% was reported in subjects with metastatic ER+/AR + breast cancer (N = 32; evaluable population with AR ≥ 1%) [33], and 24-month CBR of 20% among subjects with metastatic AR + TNBC (N = 30; evaluable population with AR ≥ 10%) [12].
Besides monotherapy, clinical studies have also investigated the combination of anti-androgens with endocrine therapy in ER+ advanced breast cancer. In a phase 1/1b study, investigating enzalutamide in combination with endocrine therapy among subjects with advanced ER+ breast cancer (N = 70), the CBR 16 was noted to be 9%, but one subject experienced stable disease for more than 3 years. Similarly, in a randomized phase 2 study investigating abiraterone in subjects with metastatic ER+ breast cancer (N = 297), there was no significant difference in median PFS in subjects receiving abiraterone plus exemestane (4.5 months) versus abiraterone alone (3.7 months) or exemestane alone (3.7 months) [21]. However, subjects who had dual AR+/ER+ disease, as detected by fresh biopsies or circulating tumor cells, had a significantly higher PFS with abiraterone plus exemestane, as compared to exemestane alone [30] highlighting the need of predictive biomarker for subject selection in studies investigating AR directed therapy in breast cancer.
Furthermore, the landscape of ER+ breast cancer has changed with the approval of mTOR and CDK 4/6 inhibitors [34,35,36,37]. The combination of seviteronel and everolimus demonstrated synergy in breast cancer cellular proliferation models including AR + TNBC [38]. Whether the combination of seviteronel with everolimus or investigational targeted therapies will result in better outcomes is unclear and warrants further studies.
In conclusion, oral seviteronel is generally well tolerated at 450 mg daily dosing and is the recommended phase 2 dose for women with breast cancer. Seviteronel’s dual mechanism of action with reduced sex-steroid production and AR antagonism may provide a unique treatment option, even in the endocrine-failure population. Given the preliminary evidence of clinical benefit in a heavily pretreated population with high disease burden, further evaluation is warranted and continues in the ongoing phase 2 clinical study.
References
Shore ND, Gupta S, Fleming MT, Berry WR, Zhang J, Kurman MR, Eisner JR, Moore WR (2016) Once-nightly (QD) dual CYP17-Lyase (L) inhibitor / androgen receptor (AR) antagonist VT-464 in patients with CRPC. J Clin Oncol 34 (2): [2016 Genitourinary Cancers Symposium])
Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D (2000) Molecular portraits of human breast tumours. Nature 406(6797):747–752. https://doi.org/10.1038/35021093
Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Eystein Lonning P, Borresen-Dale AL (2001) Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 98 (19):10869–10874. https://doi.org/10.1073/pnas.19136709898/19/10869
Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, Demeter J, Perou CM, Lonning PE, Brown PO, Borresen-Dale AL, Botstein D (2003) Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA 100 (14):8418–8423. https://doi.org/10.1073/pnas.09326921000932692100
Soreide JA, Lea OA, Varhaug JE, Skarstein A, Kvinnsland S (1992) Androgen receptors in operable breast cancer: relation to other steroid hormone receptors, correlations to prognostic factors and predictive value for effect of adjuvant tamoxifen treatment. Eur J Surg Oncol 18(2):112–118
Kimura N, Mizokami A, Oonuma T, Sasano H, Nagura H (1993) Immunocytochemical localization of androgen receptor with polyclonal antibody in paraffin-embedded human tissues. J Histochem Cytochem 41(5):671–678
Lea OA, Kvinnsland S, Thorsen T (1989) Improved measurement of androgen receptors in human breast cancer. Cancer Res 49(24 Pt 1):7162–7167
Farmer P, Bonnefoi H, Becette V, Tubiana-Hulin M, Fumoleau P, Larsimont D, Macgrogan G, Bergh J, Cameron D, Goldstein D, Duss S, Nicoulaz AL, Brisken C, Fiche M, Delorenzi M, Iggo R (2005) Identification of molecular apocrine breast tumours by microarray analysis. Oncogene 24(29):4660–4671. https://doi.org/10.1038/sj.onc.1208561
Doane AS, Danso M, Lal P, Donaton M, Zhang L, Hudis C, Gerald WL (2006) An estrogen receptor-negative breast cancer subset characterized by a hormonally regulated transcriptional program and response to androgen. Oncogene 25(28):3994–4008. doi:1209415 [pii] https://doi.org/10.1038/sj.onc.1209415
Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA (2011) Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 121(7):2750–2767. https://doi.org/10.1172/JCI45014
Gucalp A, Tolaney S, Isakoff SJ, Ingle JN, Liu MC, Carey LA, Blackwell K, Rugo H, Nabell L, Forero A, Stearns V, Doane AS, Danso M, Moynahan ME, Momen LF, Gonzalez JM, Akhtar A, Giri DD, Patil S, Feigin KN, Hudis CA, Traina TA, Translational Breast Cancer Research C (2013) Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin Cancer Res 19(19):5505–5512. https://doi.org/10.1158/1078-0432.CCR-12-3327
Bonnefoi H, Grellety T, Tredan O, Saghatchian M, Dalenc F, Mailliez A, L’Haridon T, Cottu P, Abadie-Lacourtoisie S, You B, Mousseau M, Dauba J, Del Piano F, Desmoulins I, Coussy F, Madranges N, Grenier J, Bidard FC, Proudhon C, MacGrogan G, Orsini C, Pulido M, Goncalves A (2016) A phase II trial of abiraterone acetate plus prednisone in patients with triple-negative androgen receptor positive locally advanced or metastatic breast cancer (UCBG 12 – 1). Ann Oncol 27(5):812–818. https://doi.org/10.1093/annonc/mdw067
Traina TA, Miller K, Yardley DA, O’Shaughnessy J, Cortes J, Awada A, Kelly CM, Trudeau ME, Schmid P, Gianni L, Garcia-Estevez L, Nanda R, Ademuyiwa FO, Chan S, Steinberg JL, Blaney ME, Tudor IC, Uppal H, Peterson AC, Hudis CA (2015) Results from a phase 2 study of enzalutamide (ENZA), an androgen receptor (AR) inhibitor, in advanced AR + triple-negative breast cancer (TNBC). ASCO Meet Abstr 33 (15_suppl):1003
De Amicis F, Thirugnansampanthan J, Cui Y, Selever J, Beyer A, Parra I, Weigel NL, Herynk MH, Tsimelzon A, Lewis MT, Chamness GC, Hilsenbeck SG, Ando S, Fuqua SA (2010) Androgen receptor overexpression induces tamoxifen resistance in human breast cancer cells. Breast Cancer Res Treat 121(1):1–11. https://doi.org/10.1007/s10549-009-0436-8
Ciupek A, Rechoum Y, Gu G, Gelsomino L, Beyer AR, Brusco L, Covington KR, Tsimelzon A, Fuqua SA (2015) Androgen receptor promotes tamoxifen agonist activity by activation of EGFR in ERalpha-positive breast cancer. Breast Cancer Res Treat 154(2):225–237. https://doi.org/10.1007/s10549-015-3609-7
Cochrane DR, Bernales S, Jacobsen BM, Cittelly DM, Howe EN, D’Amato NC, Spoelstra NS, Edgerton SM, Jean A, Guerrero J, Gomez F, Medicherla S, Alfaro IE, McCullagh E, Jedlicka P, Torkko KC, Thor AD, Elias AD, Protter AA, Richer JK (2014) Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res 16(1):R7. https://doi.org/10.1186/bcr3599
Rechoum Y, Iacopetta D, Barone I, Rovito D, Ando S, Weigel N, Fuqua SAW (2013) Collaboration of AR and ERα in conferring resistance to an aromatase inhibitor. ASCO Meet Abstr 31:579
Rafferty SW, Eisner JR, Moore WR, Schotzinger RJ, Hoekstra WJ (2014) Highly-selective 4-(1,2,3-triazole)-based P450c17a 17,20-lyase inhibitors. Bioorg Med Chem Lett 24(11):2444–2447. https://doi.org/10.1016/j.bmcl.2014.04.024
Norris JD, Ellison SJ, Baker JG, Stagg DB, Wardell SE, Park S, Alley HM, Baldi RM, Yllanes A, Andreano KJ, Stice JP, Lawrence SA, Eisner JR, Price DK, Moore WR, Figg WD, McDonnell DP (2017) Androgen receptor antagonism drives cytochrome P450 17A1 inhibitor efficacy in prostate cancer. J Clin Invest 127(6):2326–2338. https://doi.org/10.1172/JCI87328
Ellison SJNJ., Wardell S, Eisner JR, Hoekstra WJ, Stagg DB, Alley HM, Moore WR, McDonnell DP (2016) Effects of the dual selective CYP17 lyase inhibitor and androgen receptor (AR) antagonist, VT-464, on AR+ and ER+ tumor models in vitro and in vivo. [abstract]. Cancer Res 76 P3–14
O’Shaughnessy J, Campone M, Brain E, Neven P, Hayes D, Bondarenko I, Griffin TW, Martin J, De Porre P, Kheoh T, Yu MK, Peng W, Johnston S (2016) Abiraterone acetate, exemestane or the combination in postmenopausal patients with estrogen receptor-positive metastatic breast cancer. Ann Oncol 27(1):106–113. https://doi.org/10.1093/annonc/mdv487
Martin RM, Lin CJ, Costa EM, de Oliveira ML, Carrilho A, Villar H, Longui CA, Mendonca BB (2003) P450c17 deficiency in Brazilian patients: biochemical diagnosis through progesterone levels confirmed by CYP17 genotyping. J Clin Endocrinol Metab 88(12):5739–5746. https://doi.org/10.1210/jc.2003-030988
Kok RC, Timmerman MA, Wolffenbuttel KP, Drop SL, de Jong FH (2010) Isolated 17,20-lyase deficiency due to the cytochrome b5 mutation W27X. J Clin Endocrinol Metab 95(3):994–999. https://doi.org/10.1210/jc.2008-1745
Attard G, Reid AH, Auchus RJ, Hughes BA, Cassidy AM, Thompson E, Oommen NB, Folkerd E, Dowsett M, Arlt W, de Bono JS (2012) Clinical and biochemical consequences of CYP17A1 inhibition with abiraterone given with and without exogenous glucocorticoids in castrate men with advanced prostate cancer. J Clin Endocrinol Metab 97(2):507–516. https://doi.org/10.1210/jc.2011-2189
de Bono J, Pezaro CJ, Gillessen S, Shore ND, Nordquist LT, Efstathiou E, Araujo JC, Berry WR, Liu G, Vogelzang NJ, Omlin AG, Schotzinger RJ, Eisner JR, Moore WR (2015) The oral CYP17-Lyase (L) inhibitor VT-464 in patients with CRPC. J Clin Oncol 33 ((suppl 7)):abstr 187
Charmandari E, Nicolaides NC, Chrousos GP (2014) Adrenal insufficiency. Lancet 383(9935):2152–2167. https://doi.org/10.1016/S0140-6736(13)61684-0
Longo DL FA, Kasper DL, Hauser SL, Jameson J, Loscalzo J (eds) (2012) Harrison’s principles of internal Medicine. 18 edn. McGraw-Hill, New York
Michels A, Michels N (2014) Addison disease: early detection and treatment principles. Am Fam Physician 89(7):563–568
Zidan J, Chetver L, Hussein O, Zucker M (2010) Effect of letrozole on plasma lipids, triglycerides, and estradiol in postmenopausal women with metastatic breast cancer. Oncologist 15(11):1159–1163. https://doi.org/10.1634/theoncologist.2009-0222
Li W, O’Shaughnessy J, Hayes D, Campone M, Bondarenko I, Zbarskaya I, Brain E, Stenina M, Ivanova O, Graas MP, Neven P, Ricci D, Griffin T, Kheoh T, Yu M, Gormley M, Martin J, Schaffer M, Zelinsky K, De Porre P, Johnston SR (2016) Biomarker associations with efficacy of abiraterone acetate and exemestane in postmenopausal patients with estrogen receptor-positive metastatic breast cancer. Clin Cancer Res 22(24):6002–6009. https://doi.org/10.1158/1078-0432.CCR-15-2452
Schwartzberg LS, Yardley DA, Elias AD, Patel M, LoRusso P, Burris HA, Gucalp A, Peterson AC, Blaney ME, Steinberg JL, Gibbons JA, Traina TA (2017) A phase I/Ib study of enzalutamide alone and in combination with endocrine therapies in women with advanced breast cancer. Clin Cancer Res 23(15):4046–4054. https://doi.org/10.1158/1078-0432.CCR-16-2339
Eisner J, Abbott DH, Bird IM, Rafferty SW, Moore WR, Schotzinger RJ (2012) Assessment of steroid hormones upstream of P450c17 (CYP17) in chemically castrate male rhesus monkeys following treatment with the CYP17 inhibitors VT-464 and abiraterone acetate (AA). In: The Endocrine Society’s 94th Annual Meeting and Expo:abstr SAT-266
Ng CHMMI., Rea D et al. Phase I/II study of abiraterone acetate (AA) in estrogen receptor (ER) or androgen receptor (AR) positive metastatic breast cancer (MBC). European Society for Medical Oncology Congress, Vienna, September 28–October 2 2012
Baselga J, Campone M, Piccart M, Burris 3rd HA, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, Beck JT, Ito Y, Yardley D, Deleu I, Perez A, Bachelot T, Vittori L, Xu Z, Mukhopadhyay P, Lebwohl D, Hortobagyi GN (2012) Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med 366(6):520–529. https://doi.org/10.1056/NEJMoa1109653
Finn RS, Aleshin A, Slamon DJ (2016) Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers. Breast Cancer Res 18(1):17. https://doi.org/10.1186/s13058-015-0661-5
Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS, Paluch-Shimon S, Campone M, Blackwell KL, Andre F, Winer EP, Janni W, Verma S, Conte P, Arteaga CL, Cameron DA, Petrakova K, Hart LL, Villanueva C, Chan A, Jakobsen E, Nusch A, Burdaeva O, Grischke EM, Alba E, Wist E, Marschner N, Favret AM, Yardley D, Bachelot T, Tseng LM, Blau S, Xuan F, Souami F, Miller M, Germa C, Hirawat S, O’Shaughnessy J (2016) Ribociclib as first-line therapy for HR-positive, advanced breast cancer. N Engl J Med 375(18):1738–1748. https://doi.org/10.1056/NEJMoa1609709
Sledge GW Jr, Toi M, Neven P, Sohn J, Inoue K, Pivot X, Burdaeva O, Okera M, Masuda N, Kaufman PA, Koh H, Grischke EM, Frenzel M, Lin Y, Barriga S, Smith IC, Bourayou N, Llombart-Cussac A (2017) MONARCH 2: abemaciclib in combination with fulvestrant in women with HR+/HER2- advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol:JCO2017737585. https://doi.org/10.1200/JCO.2017.73.7585
Gordon MA, D’Amato NC, Gu H, Babbs B, Wulfkuhle J, Petricoin EF, Gallagher I, Dong T, Torkko K, Liu B, Elias A, Richer JK (2017) Synergy between androgen receptor antagonism and inhibition of mTOR and HER2 in breast cancer. Mol Cancer Ther 16(7):1389–1400. https://doi.org/10.1158/1535-7163.MCT-17-0111
Funding
This study was funded by Innocrin Pharmaceuticals. E.S.B-B and J.R.E are compensated employees of Innocrin Pharmaceuticals. T.A.T. receives compensation as a Steering Committee member for Innocrin Pharmaceuticals.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All other authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Additional information
Aditya Bardia and Ayca Gucalp contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bardia, A., Gucalp, A., DaCosta, N. et al. Phase 1 study of seviteronel, a selective CYP17 lyase and androgen receptor inhibitor, in women with estrogen receptor-positive or triple-negative breast cancer. Breast Cancer Res Treat 171, 111–120 (2018). https://doi.org/10.1007/s10549-018-4813-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-018-4813-z