
Abstract
Purpose
Non-invasive blood-based molecular markers have been investigated for cancer diagnosis and prognosis. Circulating free or cell-free DNA (cfDNA) variables have been shown to be putative markers in breast cancer prognosis.
Methods
Here, we investigated the potential prognostic ability of cfDNA concentration and cfDNA integrity (cfDI) in a study cohort of 268 patients by quantitative PCR. We compared cfDNA concentration and cfDI at baseline and after one cycle of therapy in metastatic breast cancer (MBC) patients.
Results
A significantly increased cfDI (P = 1.21E-7 for ALU and P = 1.87E-3 for LINE1) and decreased cfDNA concentration (P = 1.17E-3 for ALU and P = 1.60E-2 for LINE1) in both repetitive DNA elements after one cycle of therapy was observed. A multiple Cox regression model indicated that cfDI and cfDNA concentration can serve as independent prognostic markers in patients at baseline with HR (95% CI) of 0.70 (0.48–1.01) for ALU cfDI, 0.63 (0.44–0.92) for LINE1 cfDI, 2.44 (1.68–3.53) for ALU cfDNA concentration, and 2.12 (1.47–3.06) for LINE1 cfDNA concentration and after one cycle of therapy with HR (95% CI) of 0.59 (0.42–0.84) for ALU cfDI, 0.51 (0.36–0.74) for LINE1 cfDI, 1.59 (1.31–1.92) for ALU cfDNA concentration, and 1.30 (1.17–1.45) for LINE1 cfDNA concentration, respectively. By comparing integrated prediction error of different models, cfDNA variables were shown to improve the prognostic power of the CTC status.
Conclusions
We hereby show that cfDNA variables, especially in combination with other markers, can serve as attractive prognostic markers for MBC patients at baseline and during the systematic therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Breast cancer is the most common female cancer, with more than 230,000 new cases diagnosed in the United States alone in 2016 [1]. Metastatic breast cancer (MBC) can spread to distant organs of the body, with bone, liver, and lung being the most common sites [2]. Distant metastases cause about 90% of deaths due to breast cancer [3]. The average survival time for MBC patients is less than 3 years, although new treatments have been shown to improve the outcome of patients [4].
In recent years, the development of therapy for metastatic breast cancer such as chemotherapy, radiotherapy, endocrine therapy, and targeted therapy gives patients as well as scientists hope [5]. Patients with MBC can be treated and controlled for some time before the cancer recurs. Thus, systematic therapy is needed to treat the recurred breast cancer. Meanwhile, radiographic inspection is needed to monitor the response of the systematic therapy. However, radiographic inspection is difficult to have a real-time radiologic imaging these days in order to monitor the progress of the disease. Therefore, prognostic and predictive biomarkers for MBC are prominent these days, as well as biomarkers for therapy response in personalized anticancer.
Recently, the investigation of circulating molecular markers in peripheral blood (“liquid biopsies”) has developed fast because they are easily accessible, reproducible and can achieve real-time monitoring in cancer [6]. Biomarkers such as circulating tumor cells (CTCs), microRNAs, and circulating DNA have been explored in many types of cancer as potential diagnostic and prognostic markers for personalized medicine [7,8,9,10,11,12,13,14,15]. Among them, cell-free or circulating free DNA concentration and cell-free DNA integrity (cfDI) are emerging biomarkers. Elevated cfDNA concentrations have been shown in many types of cancers compared to healthy controls [16, 17]. cfDI is calculated as the ratio of longer DNA fragment concentration to shorter ones of a specific genetic locus and indicates the extent of cfDNA fragmentation. cfDNA concentration and cfDI, which represent the quantity and quality of cfDNA, have been investigated as diagnostic or prognostic markers in many cancers for a wide range of research applications [18,19,20,21,22,23].
In previous study, we have shown the prognostic capacity of cfDNA variables in respect of MBC for itself as well as in combination with the CTC status [12]. Meanwhile, other studies also confirmed the significant difference of cfDNA concentration between MBC and locally confined breast cancer and benign controls and healthy controls. cfDI difference was also observed between MBC and benign patients [24]. However, no studies have ever compared the difference of cfDNA variables at the time point of enrollment into the study (MBCBL) and after the first cycle of systematic therapy (MBC1C) in patients. In this study, we investigated whether cfDNA variables can be a useful prognostic marker accompanying to therapy in MBC study. Here, we show that cfDNA variables could improve the prognostic power in MBC patients when used in combination with the determination of the CTC status.
Methods and materials
Study subjects
This study was approved by the Ethical Committee of the University of Heidelberg (Heidelberg, Germany). The study was conducted at the National Center for Tumor Diseases (NCT), Heidelberg, Germany and the Department of Obstetrics and Gynecology, Women’s Clinic of Heidelberg University, Heidelberg, Germany. All subjects were metastatic breast cancer patients who were continuously recruited throughout May 2010 and December 2014. All subjects were females and Caucasians. Written informed consent was obtained from all participants.
Blood samples were collected for CTC enumeration and cfDNA extraction from patients when enrolled about to start the first cycle of systematic therapy at MBCBL and MBC1C patients. Only patients were included where blood samples were available at both time points of MBCBL and MBC1C. In total, 268 patients were included in this study. Here, essential elements related with tumor marker studies were described as listed before [25].
Sample processing and cfDNA extraction
For cfDNA extraction, peripheral blood was collected from all patients in 9-ml EDTA tubes (S-Monovette R, Sarstedt, Nümbrecht, Germany). Blood was centrifuged at 1300 g for 20 min at 10 °C within 2 h after blood withdrawal. The supernatant was transferred and centrifuged again at 15500 g for 10 min at 10 °C. This step was done to make sure that the plasma was free of cells or cell debris. The blood plasma supernatant was snap frozen in liquid nitrogen and stored at − 80 °C until further use. cfDNA was extracted from 800 µl blood plasma using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) with minor modifications as described before [12]. Extracted cfDNA was eluted in 30 µl of AE elution buffer. The eluate was re-applied onto the column, and the final eluate was collected and stored at − 20 °C. Blood samples from MBCBL and MBC1C patients were extracted together to avoid any batch effects. Method for enumeration of CTCs is described in Supplemental Data.
Measurement of cfDNA concentration and cfDI
Concentration and integrity of circulating DNA in blood plasma were evaluated by measuring the short and long fragments of two repetitive DNA elements, ALU (ALU-111 bp, ALU-260 bp) and LINE1 (LINE1-97 bp, LINE1-266 bp) by quantitative PCR using ABsolute qPCR SYBR Green Mix (Thermo Scientific, Carlsbad, USA) and the LightCycler480 system (Roche Diagnostics, Mannheim, Germany). The methods were described elsewhere before [12]. The cfDNA eluate was diluted into 1:20 to achieve optimal PCR efficiency. Concentrations of DNA fragments were calculated using the absolute quantification method according to the Light Cylcer 480 software instructions. cfDI was calculated as the ratio of long divided by short fragment concentrations for each of the elements: ALU-260/111, LINE1-266/97 as described before [12]. As short amplicons were nested within the long fragments, cfDI values should always be in the range of 0–1. The short fragment concentrations were regarded as overall cfDNA concentrations.
Methods of enumeration of CTCs
Enrichment and enumeration of CTCs using the CellSearch technology (CellSearchTM Epithelial Cell Kit/CellSpotter™ Analyzer; Janssen Diagnostics LLC, Raritan, NJ, USA) were processed as described before [26, 27]. Briefly, 7.5-mL samples of peripheral whole blood were collected in CellSave tubes (Janssen Diagnostics LLC, Raritan, NJ, USA) containing ethylenediaminetetraacetic acid (EDTA) and a cellular preservative. Samples were maintained at room temperature and processed within 96 h. Epithelial cells were immunomagnetically enriched using ferrofluid nanoparticles coated with antibodies against epithelial cell adhesion molecule (EpCAM). Subsequently, EpCAM-positive cells were labeled with the nuclear dye 4′,6-diamidino-2-phenylindole (DAPI) and immunostained with monoclonal antibodies specific for keratins and for the leukocyte common antigen CD45. Cells with intact nuclei that were CD45 negative and keratin positive were defined as CTCs and enumerated by trained operators.
Statistical analysis
All statistical analyses were carried out using the IBM SPSS Statistics 24.0 (SPSS, Chicago, IL) package and R package “survival”, version 2.40-1, “survcomp”, version 1.25.0 and “pec”, version 2.5.3, respectively. cfDNA concentrations were not normally distributed and thus were log2-transformed for further data analysis. Differences of cfDNA concentrations and cfDI between the two groups were evaluated by the paired sample Wilcoxon rank sum tests. Correlation between ALU and LINE1 results were determined by Spearman correlation. Kaplan–Meier curves were constructed for progress-free survival (PFS) and overall survival (OS), after stratifying the data based on their cfDI or cfDNA concentration. Here, PFS was defined as time from patients’ enrollment or after the first cycle of systematic therapy to disease progression. OS was defined as time from patient’s enrollment or after the first cycle of systematic therapy until death. Median values were used as the cut-off point for ALU and LINE1 cfDNA concentration. cfDI cut-off points were selected using the turning points on the curves of the study set according to the survival hazard ratio curve. CTC status was determined by CTC enumeration as CTC positive for ≥ 5 CTC or CTC negative < 5 CTC per 7.5 ml of peripheral blood as defined before [28]. PFS and OS times were estimated at medians with 95% confidence intervals (CIs).
To assess correlation to PFS or OS, Cox proportional hazard models were built for cfDI, cfDNA concentration, CTC status, and other clinical parameters and the corresponding hazard ratios (HR) with 95% CIs were calculated. Cox models with the corresponding variables were evaluated by calculating their prediction accuracy as assessed by integrated prediction error (IPE) scores computed after 10, 20, 30, and 40 months. The IPE of different models was compared. P values less than 0.05 are regarded as statistically significant.
Results
Altered cfDNA concentration, cfDNA integrity from baseline to one cycle of therapy
To evaluate the consistency of ALU and LINE1 results, Spearman’s rank correlation method was applied for both cfDI and cfDNA concentration. The results between the independently measured ALU and LINE1 elements were consistent with high correlation coefficient values both for log2cfDNA concentration (r = 0.92) and cfDI (r = 0.66).
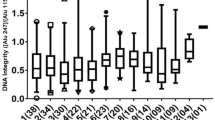
Using the paired sample Wilcoxon rank sum tests, we analyzed the results between MBCBL and MBC1C patients. Generally, the cfDNA concentration was higher in MBCBL than in MBC1C. The concentrations (mean ± SD) of ALU cfDNA concentration between MBCBL and MBC1C patients were 0.49 ± 1.25 ng/µl and 0.28 ± 0.50 ng/µl. Same trend was obtained from LINE1 cfDNA concentration (0.67 ± 2.08 for MBCBL patients and 0.42 ± 0.91 for MBC1C patients), as shown in Table 1 and Fig. 1a, b. The differences were significant for both (ALU: P = 1.17E-03; LINE1: P = 1.60E-02). Patients at baseline had generally a significantly lower cfDI (median ALU cfDI = 0.53, median LINE1 cfDI = 0.45) compared to MBC1C patients (median ALU cfDI = 0.63, median LINE1 cfDI = 0.49) (P = 1.21E-07 for ALU and P = 1.87E-03 for LINE1) (Table 1, Fig. 1c, d). In detail, we also did analysis according to breast cancer molecular subtype. The results are shown in Supplemental Table 1. However, because of the limited patient number in different groups, we still focus on the aim whether cfDNA variables can be a prognostic marker in the whole MBC patients.

Box and whisker plots of markers in MBC patients with baseline and after one cycle therapy estimated (a) log2ALUcfDNA concentration, (b) log2LINE1cfDNA concentration, (c) ALU cfDI and (d) LINE1 cfDI
Prognostic value of cfDNA integrity and cfDNA concentration in MBCBL and MBC1C patients
In MBCBL patients, those with low cfDNA concentration had significant longer PFS time by Kaplan–Meier analysis (log-rank test P = 2.5 × 10E-4 for ALU, P = 6.7 × 10E-4 for LINE1). Patients with high cfDI showed a significant longer PFS time (log-rank test P = 2.4 × 10E-2 for ALU, P = 1.5 × 10E-2 for LINE1) (Table 2). Further, MBCBL patients with high cfDNA concentration showed significant shorter OS time compared to those with low cfDNA concentration (log-rank test P = 4.3 × 10-7 for ALU, P = 6.9 × 10-7 for LINE1) (Table 2). Patients with higher cfDI had a significant longer OS time than patients with lower cfDI (log-rank test P = 0.04 for ALU, P = 0.03 for LINE1).
Similarly, in MBC1C patients, those with a lower cfDNA concentration presented longer PFS time than those with a higher cfDNA concentration (log-rank test P = 1.70 × 10E-2 for ALU, P = 4.94 × 10E-3 for LINE1). The group with higher cfDI had a significant longer PFS time than those with a higher cfDI (log-rank test P = 8.69 × 10E-3 for ALU, P = 3.56 × 10E-2 for LINE1) (Table 2). As for OS in MBC1C patients, the same trend was observed of OS in MBC1C patients (P < 0.001) (Table 2).
cfDNA variables with clinical variables and prognosis
Next, we investigated if associations between the cfDI or cfDNA concentration and the clinical and pathological characteristics can be confirmed (Table 3). We found in both MBCBL and MBC1C, cfDI was significantly decreased in patients with visceral metastasis (especially liver metastasis) compared to patients with non-visceral metastasis (Supplemental Table 2 and Supplemental Fig. 1). Univariate and multivariate Cox Regression with variables in MBCBL and MBC1C patients were investigated. Univariate Cox regression analysis showed that variables like cfDNA concentration, cfDI, ER status, PR status, therapy lines given, numbers of metastatic sites, endocrine therapy, and antibody therapy were associated with unfavorable outcomes of MBCBL and MBC1C patients (Table 4).
Multivariate Cox regression based on proportional hazards assumption was employed here. The model was constructed with the clinicopathological factors that had shown a P value < 0.05 in univariate Cox regression. Here, we observed that cfDNA concentration showed an independent prognostic value on both OS and PFS in MBCBL patients (Table 5). Nevertheless, the P value indicated that cfDI had no significant effect on OS and PFS. Furthermore, ER/PR status, first or more line of therapy, and number of metastatic sites showed independent prognostic power. In MBC1C patients, similar results were observed (Table 6). The P values here indicated that the difference between cfDI on overall survival in MBL1C patients was bordering on statistical significance (P = 0.06 for ALU and P = 1.65 E-2 for LINE1). Results showed that cfDI and cfDNA concentration can be independent prognostic markers of survival.
Furthermore, we investigated the prognostic power of all four cfDNA markers combined by Cox regression. All cfDNA markers combined showed an HR (95% CI) of 2.91 (1.85–4.58) for OS and an HR (95% CI) of 1.70 (1.21–2.39) for PFS in MBCBL patients and an HR (95% CI) of 2.53 (1.77–3.62) for OS and HR (95% CI) of 1.81 (1.25–2.63) for PFS in MBC1C patients (Table 7). Kaplan–Meier Curves also showed that the cfDNA marker combination was significantly correlated to the OS (P = 1.94E-6) and PFS (P = 5E-4) in MBCBL patients and OS (P = 3.60E-8) and PFS (P = 6.14E-4) with log-rank test in MBC1C patients (Fig. 2).

Prognostic value of all cfDNA markers combined by Kaplan–Meier curve of overall survival and progression-free survival in MBCBL and MBC1C patients. (a) OS of all cfDNA markers combined in MBCBL patients. 0: < cut-off, 1: > cut-off; (b) PFS of all cfDNA markers combined in MBCBL patients. 0: < cut-off, 1: > cut-off; (c) OS of all cfDNA markers combined in MBC1C patients. 0: < cut-off, 1: > cut-off; (d) PFS of all cfDNA markers combined in MBC1C patients. 0: < cut-off, 1: > cut-off
We also investigated the prognostic value of cell-free DNA variables kinetics in MBC patients in supplemental data (Supplemental Figs. 2, 3). No significant differences were observed between all groups for cfDI kinetics (P > 0.1 of all). Meanwhile, we also investigated the kinetics of cell-free DNA variables from MBCBL patients to MBC1C according to specific therapy (Supplemental Table 3). Results showed a significant decrease of cfDNA concentration and a significant increase in cfDI in patients treated with chemotherapy from baseline to the first cycle of therapy (P < 0.001 for all).
Comparison of the prognostic value of cell-free DNA variables and CTC status in MBC1C patients
First, we analyzed the correlation of cfDNA variables to CTC status. We observed that cfDNA concentration was significantly correlated with CTC status for ALU and LINE1 both in MBCBL and MBC1C patients (P < 0.001 for all). cfDI was not correlated with CTC status (P = 0.651 for ALU and P = 0.325 for LINE1 in MBCBL, P = 0.341 for ALU and P = 0.317 for LINE1 in MBC1C). There was a significant decrease of mean CTC values of 27.69 in 7.5 ml blood from MBCBL patients in MBC1C patients (mean 18.78 in 7.5 ml blood) (P = 3.58E-10). Results also showed that patients with CTC-negative status had longer PFS and OS time compared to CTC-positive patients in MBC1C patients (P < 0.0001).
Integrated prediction error (IPE) scores were determined to compare the prognostic ability of different models in MBCBL and MBC1C patients. In MBC1c patients, the Cox model with all four cfDNA variables had the lowest IPE scores at 10 months (0.085 for PFS and 0.196 for OS) and better performance than the model with CTC status alone (0.088 for PFS and 0.202 for OS) (Table 8, Fig. 3). The same trend was observed for the observation periods of 20 months, 30 months, and 40 months for PFS and OS. Combining cfDNA variables and CTC status of patients showed the best prediction accuracy (0.081 for PFS and 0.195 for OS). This was also observed for observation periods of 20 months, 30 months, and 40 months (Table 8, Fig. 3).

Comparison of integrated prediction error curves up to 40 months of models with all four cfDNA variables or circulating tumor cell (CTC) status, or cfDNA variables with CTC status, for (a) progression-free survival (PFS), (b) overall survival (OS) in MBCBL patients, (c) PFS and (d) OS in MBC1C patients
Also in MBCBL patients, the Cox model with all four cfDNA variables had lower IPE scores than the Cox model with CTC status and the lowest IPE score was observed by combining cfDNA variables and CTC status (Table 8, Fig. 3). Remarkably, the prognostic accuracy was generally even higher (IPE scores lower) in MBCBL measurements than in MBC1C. IPE scores for 10 months were 0.066 for PSF and 0.176 for OS in MBCBL patients while IPE scores were 0.085 for PSF and 0.196 for OS in MBC1C patients for all cfDNA variables at same time. The same holds true for other observation periods (20, 30, 40 months) and for the prognostic power of the CTC status (Table 8).
Discussion
In this study, we analyzed the cfDNA integrity (cfDI) and cfDNA concentration (cfDNA conc) of ALU and LINE1 genomic elements in metastatic breast cancer patients before and after the first cycle of systematic therapy. To the best of our knowledge, this is the first study comparing the prognostic power of cfDNA variables in MBC patients before and after the first cycle of systematic therapy. Generally, a decreased level of cfDNA concentration and an increased value of cfDNA integrity after the first cycle of systematic therapy were observed. We also confirmed that cfDNA variables (cfDNA concentration and cfDNA integrity) can be independent prognostic marker in MBC patients and can significantly improve the prognostic power of CTC status determination.
Here, we observed decreased cfDNA concentrations and increased cfDI after the first cycle of systematic therapy of MBC patients. In former study, Madhavan et al. have found that lower cfDI and higher cfDNA concentration in MBC patients compared to primary breast cancer patients and healthy individuals and that these cfDNA variables were associated with prognosis of MBC patients [12]. No comparisons of the difference of cfDI and cfDNA concentration before and after one cycle of therapy were reported so far. Treatments can relief the burden of circulating tumor DNA circulation. Leon et al. found a decrease of serum DNA concentration when the treatment was beneficial [29]. Deligezer et al. also found that some patients (21/41) showed elevated cfDNA value and others (20/41) had declined cfDNA value when completing the adjuvant chemotherapy [30].
The size distribution of cfDNA fragments within plasma or serum has been poorly studied. There are many controversial results about cfDI [31]. Many studies observed a reduced cfDI in malignant cancer patients [12, 32, 33], while others reported an increased cfDI compared to healthy controls [21, 22, 34]. At first, it has been hypothesized that in healthy controls, DNA fragments were released mainly by apoptotic cells which range at about 180–200 base pairs. While in cancer patients, DNA fragments released by malignant cells undergoing different pathophysiological processes including necrosis, autophagy, or mitotic catastrophe vary a lot in length size [35]. Recently, studies confirmed the short fragments of DNA observed in cancer patients compared to healthy individuals [36, 37]. In our study, cfDI is especially reduced in patients with visceral metastasis, especially liver metastasis, which has also been observed by Jiang et al. using paired-end sequencing and identification of tumor originated DNA by copy number aberrations [38].
In this study, we confirmed the significance and independence of the prognostic value of the cfDNA variables cfDI and cfDNA concentration in MBCBL and MBC1C patients. Although cfDNA concentration has been confirmed to be an independent biomarker in MBC patients, the varied amount of cfDNA concentration and the lacking specificity such as increased cfDNA concentration can also be observed in other cancers and benign diseases or under physiological conditions limited its clinic usage as a single marker [19, 39, 40]. Therefore, the combination of cfDNA variables is critical. The combination of four cfDNA variables as a marker showed an HR of 2.91 for OS and an HR of 1.70 for PFS in MBCBL patients and an HR of 2.53 for OS and an HR of 1.81 for PFS in MBC1C patients. Madhavan et al. have shown that combination of cfDI and cfDNA concentration had prognostic power in MBC patients and could differentiate MBC patients from healthy controls (AUC = 0.93 for CTCpos-MBC; AUC = 0.81 for CTCneg-MBC) as a diagnostic marker [12]. In a prospective clinical study of primary BC patients, we also showed that cfDI was an independent predictor of impending breast cancer recurrence [15]. Umetani et al. also claimed that serum cfDI can be a prognostic biomarker for predicting breast cancer progression. However, the study only observed that cfDI was correlated to the size of breast cancer and lymph node metastasis [41]. Recently, the cell-free tumor DNA mutations have been investigated in MBC. Chandarlapaty et al. found that ESR1 mutations were associated with worse outcomes in patients with metastatic breast cancer who were previously treated with an aromatase inhibitor [42]. Nevertheless, we hold the advantages like easily accessible, inexpensive, and reliable markers.
Here, we also compared the relation of cfDNA with the known prognostic biomarker of circulating tumor cells (CTCs). CellSearch technique has been approved by FDA for quantifying CTCs in patients with metastatic breast cancer as a prognostic biomarker [43]. Integrated Prediction Error (IPE) score is an overall measure for the prediction of the model at all times [44]. The lower IPE score is more accurate and stable is the respective model. The IPE scores for the cfDNA variables combination were lower than the IPE score for the CTC status for both OS and PFS, which indicates cfDNA variables to be a more accurate model. The lowest IPE scores were achieved when combining cfDNA variables with the CTC status. This confirms that cfDNA variables especially in combination with other markers such as CTC status can serve as attractive prognostic markers in MBC patients at baseline and during systematic therapy.
The strengths of this study are the large study population, standardized sample procedures, and comprehensive clinical data analysis. Limitations of the study should also be noted. To be able to compare the results MBCBL and MBC1C time points, we only enrolled patients who survived the first cycle of systemic therapy, which affects MBC1C patients’ survival time. Factors like time between sample collection and processing, plasma purification, the number of freeze–thaw cycles, and the employed cfDNA extraction methods can all affect cfDNA quality and quantity [45]. Here, we applied same standardized sample processing procedures to all samples. Furthermore, larger and multicenter sample cohorts are needed to be investigated to confirm the results.
In summary, our results show a decreased cfDNA concentration, increased cfDNA integrity, and a decreased CTC number from the enrollment of the study to the first cycle of systematic therapy in MBC patients. The cfDNA variables’ combination can be an independent prognostic marker in MBC patients at baseline and after the first cycle of systematic therapy and especially in combination with other markers such as CTC status.
Abbreviations
- AUC:
-
Area under the curve
- BL:
-
Baseline
- cfDI:
-
Circulating free or cell-free DNA integrity
- cfDNA:
-
Circulating free or cell-free DNA
- CI:
-
Confidence interval
- CTC:
-
Circulating tumor cell
- HR:
-
Hazard ratio
- IPE:
-
Integrated prediction errors
- MBC:
-
Metastatic breast cancer
- NCT:
-
National Center for Tumor Diseases, Heidelberg, Germany
- PFS:
-
Progression-free survival
- OS:
-
Overall survival
References
Siegel RL et al (2016) Cancer statistics, 2016. CA Cancer J Clin 66:7–30
Weigelt B et al (2005) Breast cancer metastasis: markers and models. Nat Rev Cancer 5:591–602
Mehlen P et al (2006) Metastasis: a question of life or death. Nat Rev Cancer 6:449–458
Swain SM et al (2015) Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N Engl J Med 372:724–734
Walker AJ et al (2016) FDA approval of palbociclib in combination with fulvestrant for the treatment of hormone receptor-positive, HER2-negative metastatic breast cancer. Clin Cancer Res 22:4968
Schwarzenbach H et al (2011) Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer 11:426–437
Cuk K et al (2013) Circulating microRNAs in plasma as early detection markers for breast cancer. Int J Cancer 132:1602–1612
Madhavan D et al (2013) Cancer diagnosis and prognosis decoded by blood-based circulating microRNA signatures. Front Genet 4:116
Cuk K et al (2013) Plasma microRNA panel for minimally invasive detection of breast cancer. PLoS ONE 8:e76729
Madhavan D et al (2016) Circulating miRNAs with prognostic value in metastatic breast cancer and for early detection of metastasis. Carcinogenesis 37:461–470
Madhavan D et al (2012) Circulating miRNAs as surrogate markers for circulating tumor cells and prognostic markers in metastatic breast cancer. Clin Cancer Res 18:5972–5982
Madhavan D et al (2014) Plasma DNA integrity as a biomarker for primary and metastatic breast cancer and potential marker for early diagnosis. Breast Cancer Res Treat 146:163–174
Wallwiener M et al (2014) Serial enumeration of circulating tumor cells predicts treatment response and prognosis in metastatic breast cancer: a prospective study in 393 patients. BMC Cancer 14:512
Dawson SJ et al (2013) Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med 368:1199–1209
Cheng J et al (2017) Cell-free circulating DNA integrity is an independent predictor of impending breast cancer recurrence. Oncotarget 8:54537
Schwarzenbach H et al (2011) Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer 11:426–437
Diaz LA Jr et al (2014) Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol 32:579–586
Hao TB et al (2014) Circulating cell-free DNA in serum as a biomarker for diagnosis and prognostic prediction of colorectal cancer. Br J Cancer 111:1482–1489
Szpechcinski A et al (2015) Cell-free DNA levels in plasma of patients with non-small-cell lung cancer and inflammatory lung disease. Br J Cancer 113:476–483
Huang ZH et al (2006) Quantitative analysis of plasma circulating DNA at diagnosis and during follow-up of breast cancer patients. Cancer Lett 243:64–70
Jiang WW et al (2006) Increased plasma DNA integrity index in head and neck cancer patients. Int J Cancer 119:2673–2676
Wang BG et al (2003) Increased plasma DNA integrity in cancer patients. Cancer Res 63:3966–3968
El-Shazly SF et al (2010) Evaluation of serum DNA integrity as a screening and prognostic tool in patients with hepatitis C virus-related hepatocellular carcinoma. Int J Biol Markers 25:79–86
Stötzer OJ et al (2014) Diagnostic relevance of plasma DNA and DNA integrity for breast cancer. Tumor Biol 35:1183–1191
McShane LM et al (2005) REporting recommendations for tumour MARKer prognostic studies (REMARK). Br J Cancer 93:387–391
Riethdorf S et al (2007) Detection of circulating tumor cells in peripheral blood of patients with metastatic breast cancer: a validation study of the cell search system. Clin Cancer Res 13:920–928
Wallwiener M et al (2015) The impact of HER2 phenotype of circulating tumor cells in metastatic breast cancer: a retrospective study in 107 patients. BMC Cancer 15:403
Cristofanilli M et al (2005) Circulating tumor cells: a novel prognostic factor for newly diagnosed metastatic breast cancer. J Clin Oncol 23:1420–1430
Leon SA et al (1977) Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res 37:646–650
Deligezer U et al (2008) Effect of adjuvant chemotherapy on integrity of free serum DNA in patients with breast cancer. Ann N Y Acad Sci 1137:175–179
Cheng J et al. (2017) Cell-free circulating DNA Integrity based on peripheral blood as a biomarker for diagnosis of cancer: a systematic review. Cancer Epidemiol Biomark
Mead R et al (2011) Circulating tumour markers can define patients with normal colons, benign polyps, and cancers. Br J Cancer 105:239–245
Huang A et al (2016) Plasma circulating cell-free DNA integrity as a promising biomarker for diagnosis and surveillance in patients with hepatocellular carcinoma. J Cancer 7:1798–1803
Umetani N et al (2006) Prediction of breast tumor progression by integrity of free circulating DNA in serum. J Clin Oncol 24:4270–4276
Jahr S et al (2001) DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res 61:1659–1665
Heitzer E et al (2015) Circulating tumor DNA as a liquid biopsy for cancer. Clin Chem 61:112–123
Underhill HR et al (2016) Fragment Length of Circulating Tumor DNA. PLoS Genet 12:e1006162
Jiang P et al (2015) Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc Natl Acad Sci USA 112:E1317–E1325
Atamaniuk J et al (2004) Increased concentrations of cell-free plasma DNA after exhaustive exercise. Clin Chem 50:1668–1670
van der Vaart M et al (2007) The origin of circulating free DNA. Clin Chem 53:2215
Umetani N et al (2006) Prediction of breast tumor progression by integrity of free circulating DNA in serum. J Clin Oncol 24:4270–4276
Chandarlapaty S et al (2016) Prevalence of ESR1 Mutations in Cell-Free DNA and Outcomes in Metastatic Breast Cancer: a secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol 2:1310–1315
Budd GT et al (2006) Circulating tumor cells versus imaging—predicting overall survival in metastatic breast cancer. Clin Cancer Res 12:6403
Kronek L-P et al (2008) Logical analysis of survival data: prognostic survival models by detecting high-degree interactions in right-censored data. Bioinformatics 24:i248–i253
El Messaoudi S et al (2013) Circulating cell free DNA: preanalytical considerations. Clin Chim Acta 424:222–230
Acknowledgements
We thank the study participants and all our colleagues who helped us with patient recruitment, blood collection, and processing. The study has been supported by cellgene.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cheng, J., Holland-Letz, T., Wallwiener, M. et al. Circulating free DNA integrity and concentration as independent prognostic markers in metastatic breast cancer. Breast Cancer Res Treat 169, 69–82 (2018). https://doi.org/10.1007/s10549-018-4666-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-018-4666-5