
Abstract
Purpose
Male breast cancer (MBC) is a rare cancer entity, with mutations in BRCA1 and BRCA2 genes accounting for ~ 10% of patients. Multiple-gene sequencing has already entered clinical practice for female breast cancer, whereas the performance of panel testing in MBC has not been studied extensively. Therefore, the aim of this study was to evaluate the clinical utility of panel testing for MBC, by the largest gene panel used so far, through investigation of patients deriving from a population with known founder effects.
Methods
Genomic DNA from one hundred and two Greek MBC patients, unselected for age and family history, was used to prepare libraries which capture the entire coding regions of 94 cancer genes.
Results
Loss-of-function (LoF) mutations were found in 12.7% of the cases, distributed in six genes: BRCA2, ATM, BRCA1, CHEK2, PMS2, and FANCL. BRCA2 mutations were the most frequent, followed by ATM mutations, accounting for 6.9 and 2%, respectively, while mutations in other genes were detected in single cases. Age at diagnosis or family history was not predictive of mutation status. Beyond mutations in established breast cancer predisposing genes, LoF mutations in PMS2 and FANCL among MBC patients are reported here for the first time.
Conclusions
Our findings, using the largest gene panel for MBC patients so far, indicate that BRCA testing should be the primary concern for MBC patients. Until sufficient evidence arises from larger studies, multiple-gene panels may be of limited benefit for MBC and their families, at least for MBC patients of specific descent.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Male breast cancer (MBC) is a rare cancer entity, representing just 1% of all breast cancer cases [1, 2] and with the tendency to be diagnosed at an advanced stage, in part due to lack of awareness. Dedicated research studies on male breast cancer are limited, mainly due to the rarity of the disease, and therefore, MBC is currently being treated based on the knowledge extrapolated from female breast cancer (FMB). Quite recently, a large set of MBC cases was retrospectively centrally analyzed, showing that breast cancers arising in men are usually ER-, PR-, and AR-positive, Luminal B-like/HER2-negative [3].
MBC risk increases with hormonal abnormalities and advanced age, as well as with the number of first-degree relatives diagnosed with breast cancer [4]. More importantly, cancer-predisposing mutations, specifically in BRCA1 and BRCA2 genes, have been long-standing the key genetic risk factor for MBC diagnosis. Loss-of-function (LoF) mutations in BRCA1 and BRCA2 genes confer cumulative lifetime MBC risks of 1–5 and 5–0%, respectively, with BRCA2 mutations occurring more frequently [5, 6].
The prevalence of mutations in MBC in the post-BRCA era has been mainly assessed by individual efforts, with LoF mutations in PALB2 and CHEK2 mainly been reported. Specifically, through population-specific studies, the CHEK2 c.1100delC mutation was linked to tenfold increased risk for MBC [7, 8], while PALB2 mutations are reported to increase MBC risk by eight times [9]. In addition to that, males carrying LoF mutations in the syndromic genes NF1 and PTEN have been reported to be at increased risk for MBC diagnosis, but these studies are small and have ascertainment bias [10, 11].
The evolution of sequencing technologies, along with the implementation of panel testing in clinical practice, has enabled the identification of multiple breast cancer-predisposing mutations in additional genes, the majority of which involved female breast cancer (FBC) cases. Depending on the stringency of the selection criteria, mutation in genes other than BRCA1 and BRCA2 can account for 4–11% of FBC cases [12,13,14,15].
On the contrary, data deriving from comprehensive testing among MBC cases are limited, with the recent report by Pritzlaff et al., being the largest published, so far [16]. Through utilization of various gene panels, pathogenic or likely pathogenic variants were identified in sixteen genes in 18.1% of MBC patients, of various ethnicities, tested. The great majority of variants (12.3%) lied within BRCA1 and BRCA2, while CHEK2 was the second more frequent gene, detected in 4.1% of the cases.
To better understand the contribution and association of mutations in cancer genes to MBC, we interrogated a cohort of 102 Greek MBC patients, unselected for family history or age at diagnosis, by the largest tested so far, comprehensive gene panel that includes 94 cancer genes. The aim of this study was to evaluate the clinical utility of implementation of panel testing, at least for some genes, among MBC cases through investigation of patients deriving from a specific population with known founder effects.
Patients and methods
Patient cohort
One hundred and two male individuals diagnosed with breast cancer and treated mainly at Papageorgiou Hospital of Thessaloniki and the University Hospital of Heraklion, both in Greece, were invited to participate and to donate their biological material for future research purposes. Patients were unselected for family history and age at diagnosis. The study was approved by the Bioethics committee of NCSR “Demokritos,” as well as both hospitals’ ethic committees, in agreement with the 1975 Helsinki statement, revised in 1983. Genetic counseling was mandatory prior to genetic testing, and written informed consent was obtained from all individuals before performing genetic analysis.
Analysis of 94 cancer genes through next-generation sequencing
Genomic DNA, extracted from whole blood using the salt-extraction procedure [17], was used to prepare indexed libraries to target the sequence of 94 cancer predisposing genes using the Illumina Trusight Cancer Panel and was sequenced on a MiSeq analyzer (Illumina, San Diego, USA). FASTQ, BAM, and VCF files were produced through Basespace (Illumina, San Diego, USA). The minimum base and amplicon coverage was 50× and 100× respectively. All called variants of interest were confirmed by Sanger sequencing.
Variant annotation and classification
Called variants were annotated by VariantStudio version 3 (Illumina, San Diego, USA) against the human reference genome GRCh38. Variants were filtered and classified based on the recommendations published by American College of Medical Genetics and Genomics (ACMG) [18]. More specifically, all variants considered of unknown significance (VUS), with minor allele frequency (MAF) lower than 1%, have been assessed for their pathogenicity with the use of five in silico software (Align-GVGD, SIFT, PolyPhen, Mutation Taster and PhastCons). Therefore, the evolutionary amino acid conservation, biochemical and transactivation consequence of the amino acid change, as well as the effect on canonical splicing have been interrogated, while testing by functional assays has been also considered. Based on the data collected from all the aforementioned methods, each VUS has been categorized with a scale 1–5, with 1 and 5 can be considered polymorphic and pathogenic, respectively.
Multiplex ligation-dependent probe amplification (MLPA)
All samples were subsequently tested by MLPA for the detection of large genomic rearrangements. Specifically, SALSA MLPA KIT P002 and SALSA MLPA KIT P090 (MRC-Holland, Netherlands) were used for the BRCA1 and BRCA2 genes, respectively, according to the manufacturer’s instructions.
Results
Patient characteristics
This study cohort included 102 MBC cases (mean age at diagnosis: 62.8 ± 12 years). Among them, a small proportion (16/102; 15.6%) was diagnosed at a young age (< 50 years). Histology reports were available for eighty-two individuals; information on histology type, hormone receptors, and HER2 status was extrapolated. The majority of tumors (73/82; 89%) were estrogen receptor-positive; three cases were triple negative (3/82; 3.65%), while 14.6% (12/83) showed HER2 amplification. The most common histology type was ductal (80/82; 97.5%, of which two in situ diagnoses), with other types being rare (papillary and lobular, seen once and twice, respectively).
In total, 14.7% (15/102) of the patients were diagnosed with a second primary cancer, of which gastrointestinal malignancies (colorectal and duodenal cancer), represented one-third of them. Other cancer types involved are prostate, thyroid, pancreatic, bladder, laryngeal, and non-Hodgkin’s lymphoma. Interestingly, two of these patients were diagnosed with a metachronous breast cancer.
Mutation prevalence
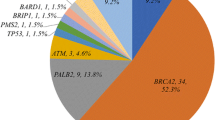
Definitely pathogenic mutations were identified in 12.7% (13/102) of the patients tested, distributed in six cancer susceptibility genes. BRCA2 was the most commonly mutated gene, accounting for more than half of the mutations identified (7/102; 6.9%), followed by ATM (2/102; 1.96%). LoF mutations in all other genes (BRCA1, CHEK2, PMS2, and FANCL) were single events. The mutation distribution is illustrated in Fig. 1.

Proportions of male breast cancer patients with germline loss-of-function mutations in BRCA2, ATM, BRCA1, CHEK2, PMS2, and FANCL genes
Two individuals (2/102; 1.96%) were found to carry a MUTYH monoallelic variant (p.Arg245His) and an APC allele p.Ile1307Lys, respectively, which are both associated with slight increase in lifetime colorectal cancer risk. Pathogenic variants in PALB2, NF1, and PTEN genes, which have been previously associated with MBC, have not been detected during this study. All definitely pathogenic mutations identified during this study, along with age at diagnosis, available clinicopathological data, and family history, are summarized in Table 1.
Tumor pathology and characteristics of mutation carriers
Among the thirteen MBC carriers, the histology subtypes were ductal (10/12 available; 83.3%), lobular and papillary (1/12 available; 8.3%, each). The clinical subtypes included ten (90.9%) luminal and one (9.1%) HER2-positive breast cancers.
Additional characteristics of mutation carriers
The median age of breast cancer diagnosis in mutation carriers was 61.3 ± 11.3 years, which was not statistical significantly different to that of non-carriers (63 ± 12.1 years; p = 0.64). Second primary cancer diagnosis was reported in two cases, pancreatic cancer and multiple myeloma, both in BRCA2 carriers. Strong family history was reported in three cases, again all of which carried BRCA2 mutations, while four cases reported at least one family relative to breast cancer diagnosis. Selected pedigrees are illustrated in Fig. 2.

Selected pedigrees of male breast cancer families, carrying LoF mutations. Probands are represented by the arrow, while breast cancer patients are colored in black. BrCa breast cancer, Ca cancer, CRC colorectal cancer, PrCa prostate cancer, WT/MT allele carriers of mutations, WT/WT wild type for the familial mutation
Change of clinical care based on panel test result
In total, an additional 2% of the patients tested will be offered amended surveillance protocols based on their test result. More specifically, PMS2 and CHEK2 carriers are potential candidates for gastrointestinal and colorectal cancer screening, respectively. This proportion can be doubled if specific guidelines are proposed for male ATM mutation carriers, specifically for prostate and pancreas surveillance for which at the moment there is insufficient evidence.
Variants of unknown clinical significance—variant classification
Assessment and classification have been performed for all detected variants located in gene coding regions, having MAF below 1%. Nine variants were identified in eight cancer susceptibility genes, have been considered suspicious, and/or have a potential to be associated with pathogenicity. Of these, BRCA1 p.Gly1727Ala has been classified as likely pathogenic, due to its rarity and the aggravating predictions of all tools used. The remaining missense changes were considered as variants of unknown significance, due to the conflicting evidence that is currently available, and were seen as follows: ATM (2), BRIP1 (1), CHEK2 (1), MLH1 (1), MSH2 (1), NBN (1), and PALB2 (1). All the above information is summarized in Table 2.
Discussion
In this study of unselected males with breast cancer, of Greek descent, 12.7% carried germline LoF alleles in six genes, previously associated with cancer predisposition. Traditionally, early age at cancer diagnosis, multiple primary cancers, and breast cancer family history are red flags for genetic testing referral. Interestingly, this does not seem to be the case for MBC. In line with previously published studies [16, 19], age at MBC diagnosis cannot be used as indicator of an underlying genetic defect. The study was designed to collect unselected MBC cases and, therefore, patient follow-up for secondary diagnosis, as well as detailed family history were not collected prior to analysis. Therefore, a limitation of our study is that statistical associations cannot be accurately performed and appropriately addressed for these cofactors.
BRCA2 mutations dominate the mutation spectrum, with a prevalence of 6.9%. Previous studies have reported BRCA2 mutations in 4–20% of MBC patients [13, 20, 21], with the upper end most likely involving ascertainment bias due to selection based on family history and population-specific alleles. The prevalence reported as 11 and 12%, in a recent large series of MBC [16] and in an Italian multicenter study [22], respectively, can be considered representative for mixed populations due to a large number of patients included and is higher than ours. This can be attributed to the genetic peculiarity of Greeks, characterized by BRCA1 strong founder effects, as shown through the extensive studies performed on female breast and ovarian cancer patients [23, 24]. Similarly, the BRCA2 mutation rate in Finnish MBC patients, another population with strong founder effects, was reported as 7.8% [25], which is comparable to ours.
The most frequent mutations in genes other than BRCA1 and BRCA2 were seen in ATM and CHEK2, which have been previously associated with both MBC and FBC. In our study, ATM mutations were the second most prevalent, accounting for 2% of the cases. Heterozygous ATM mutations have been associated with two- to fourfold increased risk for FBC [26, 27], while they also seem to increase pancreatic [28] and, possibly, ovarian cancer risk [29]. While there is no evidence for elevated risk for other cancers in ATM heterozygotes, this is the third published report of ATM mutation in MBC patients. Through multigene panel testing, ATM mutations were detected in six MBC patients (1.5%), of which one was an ataxia telangiectasia patient and two carried a pathogenic BRCA2 mutation [16]. Therefore, it seems that ATM mutations can be rare predisposing events for MBC, indicating that increased breast cancer surveillance can be proposed for male carriers, as for female carriers, based on National Comprehensive Cancer Network (NCCN) guidelines (https://www.nccn.org/).
Whether CHEK2 mutations confer increased risk for breast cancer is in the spotlight for many years. Clear associations and risks mostly derive from data on c.1100delC mutation, which is associated with a fourfold increased breast cancer risk in women and can be found in significant proportion among high-risk breast cancer patients in populations with founder effects. More specifically, among Finnish FBC and MBC patients, the prevalence is approximately 5 [30] and 5.9%, respectively [31]. In our series, a single CHEK2 LoF, but not truncating, allele (p.Gly167Arg) was identified. The low prevalence of CHEK2 mutations can be due to the rarity of c.1100delC allele in Mediterranean populations and especially, in individuals of Greek descent [32]. Quite recently, protein-truncating CHEK2 variants were associated with a 3.8-fold increased MBC risk and identified in ~ 2% of the MBC patients studied, indicating a clear association among CHEK2 truncating variants and MBC [16]. On the contrary, association with other LoF CHEK2 alleles is possible but questionable, mainly due to small number of carriers, and requires further investigation.
Interestingly, LoF mutations in two genes, which encode for proteins that are involved in DNA repair, and more specifically FANCL (Fanconi anemia pathway) and PMS2 (mismatch repair (MMR) pathway), that are not established predisposing breast cancer genes, have been identified. To the best of our knowledge, this is the first report of inactivating mutations among MBC patients. Specifically, the change in the initiator codon of FANCL has been detected in a MBC patient diagnosed at a relatively young age, who also had family history of breast cancer. A borderline association with increased FBC has been reported among Czech high-risk breast cancer patients carrying a four-base pair duplication in FANCL (c.1096_1099dupATTA) [33], located in the last exon of FANCL. This association was further doubted in a subsequent study on German breast cancer patients, where it was reported as a possible low-risk allele [34], mostly due to the likely hypomorphic nature of the specific allele. On the contrary, the mutation reported herein is pathogenic based on ACMG guidelines [35] and is predicted to alter normal protein FANCL production, therefore can be associated with both FBC and MBC susceptibilities.
Moreover, breast cancer predisposition conferred by mutations in MMR genes, including PMS2, has been questioned for long time. Quite recently, through a modified segregation analysis in a significant number of PMS2 carriers, the standardized incidence ratio of 3.8 for FBC risk showed to be significant. Due to the low number of carriers, this cannot be a definite association, but can be taken into account, especially in families that show breast cancer clustering [36]. Consistently, through a retrospective evaluation of patients who undergone panel testing, 11.9% carried MMR mutations alone and had breast cancer diagnosis only, with MSH6 and PMS2 mutations being statistically significant more frequent than MLH1 and MSH2 mutations, suggesting a possible breast cancer predisposition [37].
Germline LoF mutations among MLH1, MSH2, MSH6, and PMS2 genes are now known to confer variable lifetime cancer risks, with PMS2 mutations proposed to be associated with an attenuated Lynch syndrome phenotype, characterized by lower penetrance, later age at diagnosis, and lower risks for extracolonic Lynch syndrome-associated cancers [37,38,39]. Therefore, the cancer phenotypic spectrum associated with PMS2 mutations may be different, involving predisposition to breast cancer, as well. In the absence of a clear association of breast cancer to PMS2 mutations, the surveillance of this PMS2 carrier and his family relatives who will be tested positive for this mutation possibly need to be altered. Following the NCCN guidelines, gastrointestinal and gynecological surveillance is proposed for carriers, while chemoprevention by aspirin intake can be also proposed (https://www.nccn.org/). As a whole, clinical actionability will be proposed for ~ 2% of the MBC patients included in this study, based on the available evidence and current guidelines.
Interestingly, pathogenic variants in the genes PALB2, NF1, and PTEN, those have been previously reported among MBC cases [10, 11, 40], have not been identified through our study. This can be attributed to the relatively small size of our cohort, but can be indicative of the rarity or non-association of these genes with MBC pathogenesis.
In conclusion, this is an extensive evaluation of a comprehensive multicancer gene panel in a cohort of Greek MBC cases, unselected for family history or age at diagnosis. Our findings indicate that genetic testing for BRCA2 and BRCA1 can be sufficient for these patients, if taking into account prompt for change of care, which will be offered to only 2% of patients in our cohort beyond BRCA carriers. Although mutations in new genes that can be possibly associated with breast cancer predisposition emerged from this study, genetic events in genes, other than BRCA2 and BRCA1, involve rare cases. Until the production of sufficient evidence and clinical guidelines, from larger studies, it seems that gene panel testing may be of limited benefit for MBC patients of specific descent.
References
Korde LA, Zujewski JA, Kamin L, Giordano S, Domchek S, Anderson WF, Bartlett JM, Gelmon K, Nahleh Z, Bergh J, Cutuli B, Pruneri G, McCaskill-Stevens W, Gralow J, Hortobagyi G, Cardoso F (2010) Multidisciplinary meeting on male breast cancer: summary and research recommendations. J Clin Oncol 28(12):2114–2122. https://doi.org/10.1200/JCO.2009.25.5729
Weiss JR, Moysich KB, Swede H (2005) Epidemiology of male breast cancer. Cancer Epidemiol Biomark Prev 14(1):20–26
Cardoso F, Bartlett JMS, Slaets L, van Deurzen CHM, van Leeuwen-Stok E, Porter P, Linderholm B, Hedenfalk I, Schroder C, Martens J, Bayani J, van Asperen C, Murray M, Hudis C, Middleton L, Vermeij J, Punie K, Fraser J, Nowaczyk M, Rubio IT, Aebi S, Kelly C, Ruddy KJ, Winer E, Nilsson C, Dal Lago L, Korde L, Benstead K, Bogler O, Goulioti T, Peric A, Litiere S, Aalders KC, Poncet C, Tryfonidis K, Giordano SH (2017) Characterization of male breast cancer: results of the EORTC 10085/TBCRC/BIG/NABCG International Male Breast Cancer Program. Ann Oncol. https://doi.org/10.1093/annonc/mdx651
Brinton LA, Richesson DA, Gierach GL, Lacey JV Jr, Park Y, Hollenbeck AR, Schatzkin A (2008) Prospective evaluation of risk factors for male breast cancer. J Natl Cancer Inst 100(20):1477–1481. https://doi.org/10.1093/jnci/djn329
Tai YC, Domchek S, Parmigiani G, Chen S (2007) Breast cancer risk among male BRCA1 and BRCA2 mutation carriers. J Natl Cancer Inst 99(23):1811–1814. https://doi.org/10.1093/jnci/djm203
Breast Cancer Linkage C (1999) Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst 91(15):1310–1316
Meijers-Heijboer H, van den Ouweland A, Klijn J, Wasielewski M, de Snoo A, Oldenburg R, Hollestelle A, Houben M, Crepin E, van Veghel-Plandsoen M, Elstrodt F, van Duijn C, Bartels C, Meijers C, Schutte M, McGuffog L, Thompson D, Easton D, Sodha N, Seal S, Barfoot R, Mangion J, Chang-Claude J, Eccles D, Eeles R, Evans DG, Houlston R, Murday V, Narod S, Peretz T, Peto J, Phelan C, Zhang HX, Szabo C, Devilee P, Goldgar D, Futreal PA, Nathanson KL, Weber B, Rahman N, Stratton MR, Consortium CH-BC (2002) Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet 31(1):55–59. https://doi.org/10.1038/ng879
Wasielewski M, den Bakker MA, van den Ouweland A, Meijer-van Gelder ME, Portengen H, Klijn JG, Meijers-Heijboer H, Foekens JA, Schutte M (2009) CHEK2 1100delC and male breast cancer in the Netherlands. Breast Cancer Res Treat 116(2):397–400. https://doi.org/10.1007/s10549-008-0162-7
Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkas K, Roberts J, Lee A, Subramanian D, De Leeneer K, Fostira F, Tomiak E, Neuhausen SL, Teo ZL, Khan S, Aittomaki K, Moilanen JS, Turnbull C, Seal S, Mannermaa A, Kallioniemi A, Lindeman GJ, Buys SS, Andrulis IL, Radice P, Tondini C, Manoukian S, Toland AE, Miron P, Weitzel JN, Domchek SM, Poppe B, Claes KB, Yannoukakos D, Concannon P, Bernstein JL, James PA, Easton DF, Goldgar DE, Hopper JL, Rahman N, Peterlongo P, Nevanlinna H, King MC, Couch FJ, Southey MC, Winqvist R, Foulkes WD, Tischkowitz M (2014) Breast-cancer risk in families with mutations in PALB2. N Engl J Med 371(6):497–506. https://doi.org/10.1056/NEJMoa1400382
Fackenthal JD, Marsh DJ, Richardson AL, Cummings SA, Eng C, Robinson BG, Olopade OI (2001) Male breast cancer in Cowden syndrome patients with germline PTEN mutations. J Med Genet 38(3):159–164
Lakshmaiah KC, Kumar AN, Purohit S, Viveka BK, Rajan KR, Zameer MAL, Namitha P, Saini ML, Azim HA Jr, Saini KS (2014) Neurofibromatosis type I with breast cancer: not only for women! Hered Cancer Clin Pract 12(1):5. https://doi.org/10.1186/1897-4287-12-5
Kurian AW, Hare EE, Mills MA, Kingham KE, McPherson L, Whittemore AS, McGuire V, Ladabaum U, Kobayashi Y, Lincoln SE, Cargill M, Ford JM (2014) Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J Clin Oncol 32(19):2001–2009. https://doi.org/10.1200/JCO.2013.53.6607
Tung N, Battelli C, Allen B, Kaldate R, Bhatnagar S, Bowles K, Timms K, Garber JE, Herold C, Ellisen L, Krejdovsky J, DeLeonardis K, Sedgwick K, Soltis K, Roa B, Wenstrup RJ, Hartman AR (2015) Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer 121(1):25–33. https://doi.org/10.1002/cncr.29010
Kapoor NS, Curcio LD, Blakemore CA, Bremner AK, McFarland RE, West JG, Banks KC (2015) Multigene panel testing detects equal rates of pathogenic BRCA1/2 mutations and has a higher diagnostic yield compared to limited BRCA1/2 analysis alone in patients at risk for hereditary breast cancer. Ann Surg Oncol 22(10):3282–3288. https://doi.org/10.1245/s10434-015-4754-2
Desmond A, Kurian AW, Gabree M, Mills MA, Anderson MJ, Kobayashi Y, Horick N, Yang S, Shannon KM, Tung N, Ford JM, Lincoln SE, Ellisen LW (2015) Clinical actionability of multigene panel testing for hereditary breast and ovarian cancer risk assessment. JAMA Oncol 1(7):943–951. https://doi.org/10.1001/jamaoncol.2015.2690
Pritzlaff M, Summerour P, McFarland R, Li S, Reineke P, Dolinsky JS, Goldgar DE, Shimelis H, Couch FJ, Chao EC, LaDuca H (2017) Male breast cancer in a multi-gene panel testing cohort: insights and unexpected results. Breast Cancer Res Treat 161(3):575–586. https://doi.org/10.1007/s10549-016-4085-4
Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucl Acids Res 16(3):1215
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424. https://doi.org/10.1038/gim.2015.30
Silvestri V, Barrowdale D, Mulligan AM, Neuhausen SL, Fox S, Karlan BY, Mitchell G, James P, Thull DL, Zorn KK, Carter NJ, Nathanson KL, Domchek SM, Rebbeck TR, Ramus SJ, Nussbaum RL, Olopade OI, Rantala J, Yoon SY, Caligo MA, Spugnesi L, Bojesen A, Pedersen IS, Thomassen M, Jensen UB, Toland AE, Senter L, Andrulis IL, Glendon G, Hulick PJ, Imyanitov EN, Greene MH, Mai PL, Singer CF, Rappaport-Fuerhauser C, Kramer G, Vijai J, Offit K, Robson M, Lincoln A, Jacobs L, Machackova E, Foretova L, Navratilova M, Vasickova P, Couch FJ, Hallberg E, Ruddy KJ, Sharma P, Kim SW, kConFab I, Teixeira MR, Pinto P, Montagna M, Matricardi L, Arason A, Johannsson OT, Barkardottir RB, Jakubowska A, Lubinski J, Izquierdo A, Pujana MA, Balmana J, Diez O, Ivady G, Papp J, Olah E, Kwong A, Hereditary B, Ovarian Cancer Research Group N, Nevanlinna H, Aittomaki K, Perez Segura P, Caldes T, Van Maerken T, Poppe B, Claes KB, Isaacs C, Elan C, Lasset C, Stoppa-Lyonnet D, Barjhoux L, Belotti M, Meindl A, Gehrig A, Sutter C, Engel C, Niederacher D, Steinemann D, Hahnen E, Kast K, Arnold N, Varon-Mateeva R, Wand D, Godwin AK, Evans DG, Frost D, Perkins J, Adlard J, Izatt L, Platte R, Eeles R, Ellis S, Embrace Hamann U, Garber J, Fostira F, Fountzilas G, Pasini B, Giannini G, Rizzolo P, Russo A, Cortesi L, Papi L, Varesco L, Palli D, Zanna I, Savarese A, Radice P, Manoukian S, Peissel B, Barile M, Bonanni B, Viel A, Pensotti V, Tommasi S, Peterlongo P, Weitzel JN, Osorio A, Benitez J, McGuffog L, Healey S, Gerdes AM, Ejlertsen B, Hansen TV, Steele L, Ding YC, Tung N, Janavicius R, Goldgar DE, Buys SS, Daly MB, Bane A, Terry MB, John EM, Southey M, Easton DF, Chenevix-Trench G, Antoniou AC, Ottini L (2016) Male breast cancer in BRCA1 and BRCA2 mutation carriers: pathology data from the Consortium of Investigators of Modifiers of BRCA1/2. Breast Cancer Res 18(1):15. https://doi.org/10.1186/s13058-016-0671-y
Susswein LR, Marshall ML, Nusbaum R, Vogel Postula KJ, Weissman SM, Yackowski L, Vaccari EM, Bissonnette J, Booker JK, Cremona ML, Gibellini F, Murphy PD, Pineda-Alvarez DE, Pollevick GD, Xu Z, Richard G, Bale S, Klein RT, Hruska KS, Chung WK (2016) Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet Med 18(8):823–832. https://doi.org/10.1038/gim.2015.166
Frank TS, Deffenbaugh AM, Reid JE, Hulick M, Ward BE, Lingenfelter B, Gumpper KL, Scholl T, Tavtigian SV, Pruss DR, Critchfield GC (2002) Clinical characteristics of individuals with germline mutations in BRCA1 and BRCA2: analysis of 10,000 individuals. J Clin Oncol 20(6):1480–1490. https://doi.org/10.1200/JCO.2002.20.6.1480
Ottini L, Silvestri V, Rizzolo P, Falchetti M, Zanna I, Saieva C, Masala G, Bianchi S, Manoukian S, Barile M, Peterlongo P, Varesco L, Tommasi S, Russo A, Giannini G, Cortesi L, Viel A, Montagna M, Radice P, Palli D (2012) Clinical and pathologic characteristics of BRCA-positive and BRCA-negative male breast cancer patients: results from a collaborative multicenter study in Italy. Breast Cancer Res Treat 134(1):411–418. https://doi.org/10.1007/s10549-012-2062-0
Konstantopoulou I, Tsitlaidou M, Fostira F, Pertesi M, Stavropoulou AV, Triantafyllidou O, Tsotra E, Tsiftsoglou AP, Tsionou C, Droufakou S, Dimitrakakis C, Fountzilas G, Yannoukakos D (2014) High prevalence of BRCA1 founder mutations in Greek breast/ovarian families. Clin Genet 85(1):36–42. https://doi.org/10.1111/cge.12274
Fostira F, Tsitlaidou M, Papadimitriou C, Pertesi M, Timotheadou E, Stavropoulou AV, Glentis S, Bournakis E, Bobos M, Pectasides D, Papakostas P, Pentheroudakis G, Gogas H, Skarlos P, Samantas E, Bafaloukos D, Kosmidis PA, Koutras A, Yannoukakos D, Konstantopoulou I, Fountzilas G (2012) Prevalence of BRCA1 mutations among 403 women with triple-negative breast cancer: implications for genetic screening selection criteria: a Hellenic Cooperative Oncology Group Study. Breast Cancer Res Treat 134(1):353–362. https://doi.org/10.1007/s10549-012-2021-9
Syrjakoski K, Kuukasjarvi T, Waltering K, Haraldsson K, Auvinen A, Borg A, Kainu T, Kallioniemi OP, Koivisto PA (2004) BRCA2 mutations in 154 finnish male breast cancer patients. Neoplasia 6(5):541–545. https://doi.org/10.1593/neo.04193
Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, North B, Jayatilake H, Barfoot R, Spanova K, McGuffog L, Evans DG, Eccles D, Breast Cancer Susceptibility C, Easton DF, Stratton MR, Rahman N (2006) ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet 38(8):873–875. https://doi.org/10.1038/ng1837
Couch FJ, Shimelis H, Hu C, Hart SN, Polley EC, Na J, Hallberg E, Moore R, Thomas A, Lilyquist J, Feng B, McFarland R, Pesaran T, Huether R, LaDuca H, Chao EC, Goldgar DE, Dolinsky JS (2017) Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol 3(9):1190–1196. https://doi.org/10.1001/jamaoncol.2017.0424
Roberts NJ, Jiao Y, Yu J, Kopelovich L, Petersen GM, Bondy ML, Gallinger S, Schwartz AG, Syngal S, Cote ML, Axilbund J, Schulick R, Ali SZ, Eshleman JR, Velculescu VE, Goggins M, Vogelstein B, Papadopoulos N, Hruban RH, Kinzler KW, Klein AP (2012) ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov 2(1):41–46. https://doi.org/10.1158/2159-8290.CD-11-0194
Lilyquist J, LaDuca H, Polley E, Davis BT, Shimelis H, Hu C, Hart SN, Dolinsky JS, Couch FJ, Goldgar DE (2017) Frequency of mutations in a large series of clinically ascertained ovarian cancer cases tested on multi-gene panels compared to reference controls. Gynecol Oncol 147(2):375–380. https://doi.org/10.1016/j.ygyno.2017.08.030
Vahteristo P, Bartkova J, Eerola H, Syrjakoski K, Ojala S, Kilpivaara O, Tamminen A, Kononen J, Aittomaki K, Heikkila P, Holli K, Blomqvist C, Bartek J, Kallioniemi OP, Nevanlinna H (2002) A CHEK2 genetic variant contributing to a substantial fraction of familial breast cancer. Am J Hum Genet 71(2):432–438. https://doi.org/10.1086/341943
Hallamies S, Pelttari LM, Poikonen-Saksela P, Jekunen A, Jukkola-Vuorinen A, Auvinen P, Blomqvist C, Aittomaki K, Mattson J, Nevanlinna H (2017) CHEK2 c.1100delC mutation is associated with an increased risk for male breast cancer in Finnish patient population. BMC Cancer 17(1):620. https://doi.org/10.1186/s12885-017-3631-8
Apostolou P, Fostira F, Papamentzelopoulou M, Michelli M, Panopoulos C, Fountzilas G, Konstantopoulou I, Voutsinas GE, Yannoukakos D (2015) CHEK2 c.1100delC allele is rarely identified in Greek breast cancer cases. Cancer Genet 208(4):129–134. https://doi.org/10.1016/j.cancergen.2015.02.006
Zemankova P, Lhota F, Kleiblova P, Soukupova J, Vocka M, Janatova M, Kleibl Z (2016) RE: frameshift variant FANCL*c.1096_1099dupATTA is not associated with high breast cancer risk. Clin Genet 90(4):387–389. https://doi.org/10.1111/cge.12842
Pfeifer K, Schurmann P, Bogdanova N, Neuhauser K, Kostovska IM, Plaseska-Karanfilska D, Park-Simon TW, Schindler D, Dork T (2016) Frameshift variant FANCL*c.1096_1099dupATTA is not associated with high breast cancer risk. Clin Genet 90(4):385–386. https://doi.org/10.1111/cge.12837
Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, Lyon E, Ward BE, Molecular Subcommittee of the ALQAC (2008) ACMG recommendations for standards for interpretation and reporting of sequence variations: revisions 2007. Genet Med 10(4):294–300. https://doi.org/10.1097/gim.0b013e31816b5cae
ten Broeke SW, Brohet RM, Tops CM, van der Klift HM, Velthuizen ME, Bernstein I, Capella Munar G, Gomez Garcia E, Hoogerbrugge N, Letteboer TG, Menko FH, Lindblom A, Mensenkamp AR, Moller P, van Os TA, Rahner N, Redeker BJ, Sijmons RH, Spruijt L, Suerink M, Vos YJ, Wagner A, Hes FJ, Vasen HF, Nielsen M, Wijnen JT (2015) Lynch syndrome caused by germline PMS2 mutations: delineating the cancer risk. J Clin Oncol 33(4):319–325. https://doi.org/10.1200/JCO.2014.57.8088
Espenschied CR, LaDuca H, Li S, McFarland R, Gau CL, Hampel H (2017) Multigene panel testing provides a new perspective on lynch syndrome. J Clin Oncol 35(22):2568–2575. https://doi.org/10.1200/JCO.2016.71.9260
Bonadona V, Bonaiti B, Olschwang S, Grandjouan S, Huiart L, Longy M, Guimbaud R, Buecher B, Bignon YJ, Caron O, Colas C, Nogues C, Lejeune-Dumoulin S, Olivier-Faivre L, Polycarpe-Osaer F, Nguyen TD, Desseigne F, Saurin JC, Berthet P, Leroux D, Duffour J, Manouvrier S, Frebourg T, Sobol H, Lasset C, Bonaiti-Pellie C, French Cancer Genetics N (2011) Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 305(22):2304–2310. https://doi.org/10.1001/jama.2011.743
Senter L, Clendenning M, Sotamaa K, Hampel H, Green J, Potter JD, Lindblom A, Lagerstedt K, Thibodeau SN, Lindor NM, Young J, Winship I, Dowty JG, White DM, Hopper JL, Baglietto L, Jenkins MA, de la Chapelle A (2008) The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology 135(2):419–428. https://doi.org/10.1053/j.gastro.2008.04.026
Ding YC, Steele L, Kuan CJ, Greilac S, Neuhausen SL (2011) Mutations in BRCA2 and PALB2 in male breast cancer cases from the United States. Breast Cancer Res Treat 126(3):771–778. https://doi.org/10.1007/s10549-010-1195-2
Acknowledgements
We are indebted to the patients who participated in our research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no disclosures/conflict of interest to declare.
Rights and permissions
About this article

Cite this article
Fostira, F., Saloustros, E., Apostolou, P. et al. Germline deleterious mutations in genes other than BRCA2 are infrequent in male breast cancer. Breast Cancer Res Treat 169, 105–113 (2018). https://doi.org/10.1007/s10549-018-4661-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-018-4661-x