
Abstract
Purpose
Aromatase inhibitor-associated musculoskeletal symptoms (AIMSS) frequently occur in women being treated for breast cancer. Prior studies suggest high prevalence of vitamin D deficiency in breast cancer patients with musculoskeletal (MS) pain. We conducted a randomized, placebo-controlled trial to determine if 30,000 IU vitamin D3 per week (VitD3) would prevent worsening of AIMSS in women starting adjuvant letrozole for breast cancer.
Methods
Women with stage I–III breast cancer starting adjuvant letrozole and 25(OH)D level ≤40 ng/ml were eligible. All subjects received standard daily supplement of 1200 mg calcium and 600 IU vitamin D3 and were randomized to 30,000 IU oral VitD3/week or placebo. Pain, disability, fatigue, quality of life, 25(OH)D levels, and hand grip strength were assessed at baseline, 12, and 24 weeks. The primary endpoint was incidence of an AIMSS event.
Results
Median age of the 160 subjects (80/arm) was 61. Median 25OHD (ng/ml) was 25 at baseline, 32 at 12 weeks, and 31 at 24 weeks in the placebo arm and 22, 53, and 57 in the VitD3 arm. There were no serious adverse events. At week 24, 51% of women assigned to placebo had a protocol defined AIMSS event (worsening of joint pain using a categorical pain intensity scale (CPIS), disability from joint pain using HAQ-II, or discontinuation of letrozole due to MS symptoms) vs. 37% of women assigned to VitD3 (p = 0.069). When the brief pain inventory (BPI) was used instead of CPIS, the difference was statistically significant: 56 vs. 39% (p = 0.024).
Conclusions
Although 30,000 IU/week of oral vitamin D3 is safe and effective in achieving adequate vitamin D levels, it was not associated with a decrease in AIMSS events based on the primary endpoint. Post-hoc analysis using a different tool suggests potential benefit of vitamin D3 in reducing AIMSS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Musculoskeletal (MS) pain is a frequent side effect among women taking adjuvant aromatase inhibitors (AIs) for hormone receptor positive early breast cancer [1,2,3]. Cross-sectional studies show that about half of the women receiving adjuvant AIs report new or worsening MS symptoms, with a quarter reporting severe symptoms [4]. As a result, considerable research activity has focused on preventing or treating the spectrum of side effects referred to collectively as aromatase inhibitor-associated musculoskeletal symptoms (AIMSS) [5].
Vitamin D deficiency is prevalent in women with breast cancer who also have MS symptoms and among women receiving adjuvant chemotherapy for breast cancer [6,7,8]. A pain syndrome similar to AIMSS has been described in subjects with extreme vitamin D deficiency and treatment with vitamin D results in rapid resolution of these symptoms [6, 9]. Estrogen can upregulate both the 1-α-hydroxylase enzyme required for conversion of 1,25(OH)2D from 25(OH)D and increase levels of vitamin D receptor [10, 11]. Thus, AI induced estrogen deprivation may unmask subclinical vitamin D deficiency, which also may increase the severity of AIMSS. Replacement with vitamin D3 may correct that subclinical deficiency.
Optimal serum 25-hydroxyvitaminD [25(OH)D] level, the best indicator of vitamin D stores in the body, is unknown. Levels less than 20 ng/ml are considered deficient for bone health; but higher levels may be necessary for optimal MS function. A level of more than 150 ng/ml is considered toxic [12, 13].
In a pilot study, we observed that 50,000 IU VitD3/week in women taking letrozole was associated with reduced disability from joint pain but that three women had 25(OH)D levels of >100 ng/ml [14]. Subsequently, we initiated the placebo-controlled VITAL trial (VITamin D treatment to prevent Arthralgia in women starting Letrozole) at the lower dose of 30,000 IU VitD3 weekly. With this dose of VitD3 we hypothesized that most participants with a starting 25(OH)D level of ≤40 ng/ml should experience an increase to 50–80 ng/ml but nobody should approach the toxic level of 150 ng/ml. Goals of the study were to examine the efficacy of VitD3 for preventing the worsening of AIMSS and to determine the efficacy of 30,000 IU VitD3 in achieving 25OHD levels of >40 ng/ml compared to placebo, and its safety.
Methods
Cohort
The study was conducted at the University of Kansas Medical Center (Kansas City) and the Cancer Center of Kansas (Wichita), under a protocol approved by local IRBs (NCT00867217). Subjects were postmenopausal women with stage I–III hormone receptor positive breast cancer scheduled to start treatment with an adjuvant AI and with a 25(OH)D level ≤40 ng/ml. Subjects were excluded if they had history of renal stones, hypercalcemia, or hyperparathyroidism. Informed consent was obtained from each participant. Upon study entry, participants were asked to stop any vitamin D and calcium supplements and were provided with a standard supplement.
Study schedule
Baseline assessments
Baseline assessments included history, physical exam, CBC, calcium and phosphorus, liver/renal functions, serum estradiol and 25(OH)D, handgrip test, and completion of various questionnaires (below). 25(OH)D levels using LC/MS/MS were performed at Quest Laboratories. Hand grip strength was measured using a Jamar dynamometer.
Questionnaires
-
Health assessment questionnaire II (HAQ-II) is commonly used in rheumatology. It consists of categorical (integer) values from 0 to 3 for ten separate functions; an increase of 0.25 in the mean score is considered clinically relevant worsening of symptoms (disability).
-
Categorical pain intensity scale (CPIS)—five descriptors (none, mild, moderate, severe, disabling) as reported by subject. Any step increase is considered relevant.
-
Brief pain inventory (BPI); consists of two separate scales, intensity and interference, with two sets of questions (integer values from 0 to 10) focused on intensity of pain and on Interference with a variety of normal activities, with an average score provided for both. A 1 point change is considered relevant.
-
Brief fatigue inventory (BFI); usual level of fatigue during the past 24 h is considered the most informative; integer values from 0 to 10. Any increase is considered as evidence of worsening.
-
Functional assessment of cancer therapy-breast (FACT-B) and menopause-specific quality of life (MENQOL).
Stratification, randomization, and treatment
Participants were stratified by site and use of adjuvant chemotherapy and randomized 1:1 to either 30,000 IU VitD3 (VitD3) or placebo weekly for 24 weeks (three capsules of 10,000 IU VitD3 or three capsules of matched placebo weekly). Only the study biostatistician and investigational pharmacists were aware of drug assignments. All subjects received 1200 mg of calcium plus 600 IU of vitamin D daily (“standard supplementation”) and Letrozole 2.5 mg PO daily. All study related drugs including standard supplements were provided without charge to participants. There were no dose adjustments.
Follow-up assessments
All baseline assessments were repeated at weeks 12 and 24. Since 25(OH)D measurement had the potential to reveal the study agent assignment, a serum specimen was sent to Quest Laboratories for assessment of 25(OH)D, and the results sent to a designated individual not involved with the study. If levels >100 ng/ml had been reported, the individual would have contacted the PI who would have taken the subject off-study. This did not occur. Once all study assessments had been completed, data audited, and the trial database locked, all 25(OH)D results were sent to the study biostatistician.
Statistical considerations
The planned accrual of 160 subjects was intended to provide at least 144 evaluable subjects (72/arm) if the drop-out rate was no greater than 10%. This would provide 88% power to detect a statistically significant reduction using 1-sided Fisher’s exact test at 5% type I error rate if the proportions of worsening AIMSS were 25% and 50% for women randomized to high dose vitamin D and placebo, respectively. These proportional reductions were estimated from our pilot study that compared women receiving VitD3 (50,000 IU/week) versus only standard supplementation [14]. All subjects that received study agent were considered evaluable for safety and for efficacy.
The protocol defined primary outcome was a worsening of AIMSS from baseline to 24 weeks, evidenced by any of the following three events: (1) an increase in the HAQ-II score of 0.25 or more; (2) an increase in CPIS score; or (3) discontinuation of letrozole specifically due to AIMSS. Secondary endpoints were changes in hand grip strength using a Jamar dynamometer; and increases in fatigue and menopause symptoms using BFI, FACT-B, and MENQOL.
For the primary endpoint, the difference in the incidence of worsening of AIMSS between individuals randomized to VitD3 vs. placebo was first examined by a 1-sided Fisher’s exact test. Multiple logistic regression analysis was then used to investigate whether changes in AIMSS from baseline to 24 weeks were due to variables other than assigned treatment.
For secondary categorical variables, Fisher’s exact test was used. For continuous variables, the Mann–Whitney test or Kruskal–Wallis test was used for comparison between groups; the Wilcoxon’s signed rank test was used for within-subject changes over time. Given the exploratory nature of the analyses dealing with AIMSS, no corrections were made for multiple comparisons and the two-sided type I error rate was maintained at 0.05.
Results
Characteristics of subjects
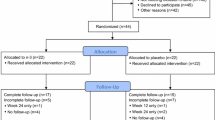
Accrual of 160 subjects was achieved between April 2009 and July 2010; with an exact 1:1 (80:80) randomization (Fig. 1). Distribution of demographics, stage, and adjuvant therapy was balanced between the two arms, as shown in Table 1.

Diagram of subject allocation, follow-up, and basis for analysis
Baseline serum 25(OH)D levels, AIMSS and quality of life assessments
Prior to enrollment, 49 of 160 subjects were taking vitamin D supplements. Baseline median 25(OH)D levels were 25.1 and 22.5 ng/ml for women randomized to placebo and VitD3, respectively (Table 1). As shown in Table 2, median baseline values for various quality of life assessments were similar for the two groups, except for several specific activities (mood, walking, relations, and enjoyment) in the brief fatigue inventory.
Study completion and evaluability
Of the 160 subjects enrolled and randomized (Fig. 1), one subject was determined to be not eligible because she consumed high dose VitD between screening assay and baseline visit. An additional 12 subjects did not complete the study for reasons unrelated to study agents and/or AIMSS (withdrawal of consent, never started study agent, personal decision, etc.). Additionally, all quality of life assessments were not available for all participants at all visits. Specifically, 72 and 70 complete sets of assessments were available for the vitD and placebo arms at week 12, and 73 and 65 at week 24. The number of missing values was not significantly different between arms at week 12 or 24 (2-sided p = 0.80 and 0.11 by Fisher’s exact test). Overall, sufficient information was available to allow assessment of protocol defined worsening of AIMSS for 147 evaluable subjects, with 77 in the placebo group and 70 in the VitD3 group. This formed the basis for all assessments of efficacy.
Effect of VitD3 on serum 25(OH)D levels
For women randomized to placebo (Table 3), median increase in 25(OH)D was 7.1 ng/ml between week 0 and 12, and then no further increase from 12 to 24 weeks. Only nine subjects achieved a level >40 ng/ml. In contrast, VitD3 (30,000 IU per week) increased 25(OH)D levels by a median of 32 ng/ml between week 0 and 12 (Table 3), with a slight further increase between weeks 12 and 24 (median 3.0 ng/ml). 91% achieved a level >40 ng/ml by 24 weeks; maximum value measured was 87 ng/ml. The differential effect of VitD3 is graphically displayed in Fig. 2.

Comparison of final (24 week) levels of 25(OH)D, plotted as a function of the baseline levels. There was no statistically significant difference between the two groups at baseline (p = 0.25); but there was for 12-week levels, 24-week levels, and change from baseline to 24-weeks (p < 0.001, Mann–Whitney test)
Adverse events
Excluding the adverse events related to letrozole, there were no adverse events attributed to vitamin D3. One subject in placebo group had mild hypercalcemia at 12 week assessment. She was taking a thiazide diuretic and had serum 25(OH)D level of 20 ng/ml at the time. There were no differences between the groups for off-study levels of urinary calcium, creatinine, or calcium:creatinine ratio.
Efficacy of vitamin D3 supplementation in preventing AIMSS
Frequency of subjects classified as exhibiting a worsening of AIMSS by each of the three individual measures that contribute to the protocol defined primary outcome is shown in Table 4. While for each there are numerical differences in favor of the VitD3 arm, none reached significance. For the protocol defined primary endpoint using the composite of HAQ-II, CPIS, and letrozole discontinuation due to AIMSS side effects, 51% of the women randomized to placebo vs. 37% of those randomized to VitD3 experienced onset or worsening of AIMSS (p = 0.069). Four other measures including hand grip strength showed a similar lack of difference between groups (Table 4), as did quality of life assessments using FACT-B and MENQOL (data not shown).
BPI-Intensity index, with greater dynamic range than the five category CPIS, did identify nine additional subjects exhibiting an increase in pain intensity. As an exploratory post hoc analysis when this was used instead of the CPIS in the composite, the difference between groups was statistically significant, with 56% (43/77) of women randomized to placebo vs. 39% (27/70) of those randomized to VitD3 classified as having worsening of AIMSS (p = 0.024).
Discussion
AIMSS is a symptom complex with variable phenotype consisting of varying degrees of joint and muscle pain, stiffness, and disability which may be difficult for a patient to describe and is not optimally defined with tools used in adjuvant trials. Trials of adjuvant AIs may therefore be underestimating the frequency of AIMSS due to a lack of tools specific for their assessment [15,16,17].
Estrogen deprivation is thought to be one of the underlying reasons for AIMSS. Joint stiffness and arthralgia are common after menopause [18], and especially among premenopausal women receiving gonadotropin-releasing hormone analogs such as luperolide. Estrogen has tissue specific effects on inflammatory cytokines and lack of estrogen may result in augmentation of inflammation and enhanced nociception from inflammation [19, 20]. An indirect evidence of this phenomenon comes from the observation that women on AIs complaining of joint pain have MRI findings of tenosynovitis suggesting local inflammation in the joints [21, 22]. A pain syndrome similar to AIMSS has been described in subjects with extreme vitamin D deficiency. More importantly, treatment with vitamin D results in rapid resolution of these symptoms [6, 9]. Vitamin D deficiency is prevalent in breast cancer patients who also have MS symptoms and among women receiving adjuvant chemotherapy [6, 7, 23].
The exact mechanism of a Vitamin D effect ameliorating AIMSS is not known but may be related to its anti-inflammatory properties [8]. The active hormone calcitriol is locally produced from vitamin D3 in macrophages and may have a role in limiting joint inflammation. Estrogen increases calcitriol, and estrogen deprivation from AIs is therefore expected to be pro-inflammatory by decrease in this active form of vitamin D [24, 25]. Higher doses of vitamin D3 would provide a substrate for increased local production of calcitriol serving to limit joint inflammation and pain resulting from AIs.
In our trial, supplementation with 30,000 IU of vitamin D3 weekly in women with early breast cancer who were receiving letrozole, was safe and extremely effective in replenishing vitamin D stores. No subject’s 25(OH)D level exceeded 87 ng/ml; yet the target level of >40 ng/ml was achieved in 91% of women. This extends our pilot trial experience [14] where 50,000 IU of vitamin D3 weekly for up to 24 weeks was quite tolerable and safe. Conversely, <10% of women had a level more than 40 ng/ml with standard supplementation of 600 IU vitamin D3 per day in the placebo group. The steep increase in 25(OH)D levels during the first 3 months followed by plateau suggests that 30,000 IU of vitamin D may be safely continued beyond 6 months if needed with minimal additional monitoring.
During the design of the trial, based on our phase II experience, our assumption was that no single measure of AIMSS worsening would be sufficient to detect a clinically meaningful change in AIMSS. This is particularly problematic in a trial designed to prevent development of AIMSS (rather than treat existing symptoms) since one does not know a priori what problems or complaints a woman might experience. For this reason, the protocol defined criteria for AIMSS worsening was any of three events (increase in HAQ-II score, increase in CPIS score, or discontinuation due to AI-related AEs). In general, these three measures tended to identify different women suffering different problems, as anticipated. However, the CPIS measure, which we had selected on the basis of our pilot study results, did not perform as well in this regard as it had in the pilot study. Despite achieving desired levels of 25(OH)D with supplementation, and observing a numerical decrease in worsening of AIMSS events (our primary, protocol defined endpoint) from 51 to 37%, the difference was not statistically significant. Thus, the trial was negative based on failure to meet its primary endpoint.
In contrast to CPIS, the BPI-Intensity index increased the number of subjects identified as having a worsening of AIMSS. The BPI provides four questions regarding severity of pain, with a broader range of possible responses (10 point scale) and is a more sensitive tool for pain assessment. Justification for use of the BPI comes from a phase II trial of women on anastrozole for at least 8 weeks who had existing AIMSS as well as serum 25(OH)D levels of 10–29 ng/ml, and were randomized to receive 50,000 IU weekly of vitamin D2 or placebo for 2–4 months in an attempt to reduce AIMSS [26]. At 2 months, scores for several measures of pain intensity in the BPI were more favorable in women randomized to vitamin D2 compared to placebo [26]. As an exploratory, post hoc analysis, when we replaced CPIS with BPI-Intensity in our composite index (i.e., HAQ-II + BPI-Intensity + letrozole discontinuation) to define an AIMSS event, the difference between groups was statistically significant (p = 0.024).
Based on the above encouraging result from an admittedly post hoc analysis, we proceeded to a further post hoc effort to develop a cumulative index (AIST—aromatase inhibitor symptom tool) from the VITAL trial that would capture as many AIMSS side effects as possible and would serve as a sensitive tool for future trials, designed specifically for AIMSS. For this purpose, all assessments were scored as change over the 24-week period of study agent intervention. For exploratory purposes, 2-sided statistical approaches were used. Of the seven measures in Table 4, no single assessment revealed statistically significant differences between the two randomized arms. However, the different tests did identify different subjects as having evidence of increasing discomfort/disability. Therefore, a stepwise approach was used to determine the utility of adding tests sequentially to provide evidence of “worsening” (Table 5). The first endpoint employed was discontinuation of AI therapy due to AEs. Even though this only contributed three subjects, it is obviously the most clinically relevant endpoint. It also avoided the need to censor subjects that dropped out prior to providing any of the 6-month objective assessments. Next, tests were added in order of the largest number of events that would be gained. BFI-Usual Activity (1 point increase) resulted in an additional 47 subjects with symptoms, followed by HAQ-II (0.25 increase) which identified 20 more subjects with symptoms and improved the discriminant ability to marginal statistical significance. When BPI-Interference was added, identifying 13 more subjects, the distinction between the two groups was definitely statistically significant. By this post hoc derived metric, evidence for worsening of AIMSS was observed in 71% of subjects randomized to placebo (plus the standard supplement of 600 IU of D3/day) versus only 40% of subjects randomized to high dose vitamin D3 plus the standard supplemental dose (p < 0.001). This indicates that a multi-component assessment may provide a robust means of demonstrating the beneficial effects of an intervention such as vitamin D3 on preventing the development or worsening of AIMSS. However, this new assessment tool needs to be validated in a prospective trial.
Our results indicate that six months of oral vitamin D3 at 30,000 IU weekly is safe in women starting an AI for adjuvant treatment of breast cancer and is very effective in increasing serum 25(OH)D levels. Although this intervention failed to show a benefit in preventing new or worsening AIMSS events in women starting adjuvant AIs based on the protocol defined primary endpoint, post hoc analysis using a more sensitive tool suggests benefit of vitamin D supplementation to prevent AIMSS. A new arthralgia assessment tool (AIST), specific for measuring AIMSS, needs to be validated in larger AI symptom intervention trials.
Abbreviations
- AIMSS:
-
Aromatase Inhibitor-Associated Musculoskeletal Symptoms
- AIST:
-
Aromatase Inhibitor Symptom Tool
- AIs:
-
Aromatase Inhibitors
- BFI:
-
Brief Fatigue Inventory
- BPI:
-
Brief Pain Inventory
- CPIS:
-
Categorical Pain Intensity Scale
- FACT-B:
-
Functional Assessment of Cancer Therapy-Breast
- HAQ-II:
-
Health Assessment Questionnaire II
- MENQOL:
-
Menopause-specific Quality of Life
- VITAL:
-
VITamin D treatment to prevent Arthralgia in women starting Letrozole
- VitD3:
-
30,000 IU vitamin D3 per week
References
Baum M, Budzar AU, Cuzick J, Forbes J, Houghton JH, Klijn JG, Sahmoud T, ATAC Trialists’ Group (2002) Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: first results of the ATAC randomised trial. Lancet 359:2131–2139
Coates AS, Keshaviah A, Thürlimann B, Mouridsen H, Mauriac L, Forbes JF, Paridaens R, Castiglione-Gertsch M, Gelber RD, Colleoni M, Láng I, Del Mastro L, Smith I, Chirgwin J, Nogaret JM, Pienkowski T, Wardley A, Jakobsen EH, Price KN, Goldhirsch A (2007) Five years of letrozole compared with tamoxifen as initial adjuvant therapy for postmenopausal women with endocrine-responsive early breast cancer: update of study BIG 1–98. J Clin Oncol 25:486–492
Coombes RC, Hall E, Gibson LJ, Paridaens R, Jassem J, Delozier T, Jones SE, Alvarez I, Bertelli G, Ortmann O, Coates AS, Bajetta E, Dodwell D, Coleman RE, Fallowfield LJ, Mickiewicz E, Andersen J, Lønning PE, Cocconi G, Stewart A, Stuart N, Snowdon CF, Carpentieri M, Massimini G, Bliss JM, van de Velde C, Intergroup Exemestane Study (2007) A randomized trial of exemestane after two to three years of tamoxifen therapy in postmenopausal women with primary breast cancer. New Engl J Med 350:1081–1092
Crew KD, Greenlee H, Capodice J, Raptis G, Brafman L, Fuentes D, Sierra A, Hershman DL (2007) Prevalence of joint symptoms in postmenopausal women taking aromatase inhibitors for early-stage breast cancer. J Clin Oncol 25:3877–3883
Roberts K, Rickett K, Greer R, Woodward N (2017) Management of aromatase inhibitor induced musculoskeletal symptoms in postmenopausal early Breast cancer: a systematic review and meta-analysis. Crit Rev Oncol Hematol 111:66–80
Plotnikoff GA, Quigley JM (2003) Prevalence of severe hypovitaminosis D in patients with persistent nonspecific musculoskeletal pain. Mayo Clin Proc 78:1463–1470
Taylor M, Rastelli A, Civitelli R et al. (2004) Incidence of 25-OH vitamin D deficiency in patients with a history of breast cancer who have musculoskeletal symptomatology. 27th Annual San Antonio Breast Cancer Symposium,San Antonio, TX, 8–11 Dec 2004 (abstract 3072)
Crew KD, Shane E, Cremers S, McMahon DJ, Irani D, Hershman DL (2009) High prevalence of vitamin D deficiency despite supplementation in premenopausal women with breast cancer undergoing adjuvant chemotherapy. J Clin Oncol 27:2151–2156
de Torrenté de la Jara G, Pécoud A, Favrat B (2004) Musculoskeletal pain in female asylum seekers and hypovitaminosis D3. BMJ 329:156–157
Caniggia A, Lorè F, di Cairano G, Nuti R (1987) Main endocrine modulators of vitamin D hydroxylases in human pathophysiology. J Steroid Biochem 27:815–824
Gilad LA, Bresler T, Gnainsky J, Smirnoff P, Schwartz B (2005) Regulation of vitamin D receptor expression via estrogen-induced activation of the ERK 1/2 signaling pathway in colon and breast cancer cells. J Endocrinol 185:577–592
Bischoff-Ferrari HA, Giovannucci E, Willett WC, Dietrich T, Dawson-Hughes B (2005) Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am J Clin Nutr 84:18–28
Holick MF (2007) Vitamin D deficiency. N Engl J Med 357(3):266–281
Khan QJ, Reddy PS, Kimler BF, Sharma P, Baxa SE, O’Dea AP, Klemp JR, Fabian CJ (2010) Effect of vitamin D supplementation on serum 25-hydroxy vitamin D levels, joint pain, and fatigue in women starting adjuvant letrozole treatment for breast cancer. Breast Cancer Res Treat 119:111–118
Fallowfield L, Cella D, Cuzick J, Francis S, Locker G, Howell A (2004) Quality of life of postmenopausal women in the Arimidex, Tamoxifen, Alone or in Combination (ATAC) adjuvant breast cancer trial. J Clin Oncol 22:4261–4271
Whelan TJ, Goss PE, Ingle JN, Pater JL, Tu D, Pritchard K, Liu S, Shepherd LE, Palmer M, Robert NJ, Martino S, Muss HB (2005) Assessment of quality of life in MA.17: a randomized, placebo-controlled trial of letrozole after 5 years of tamoxifen in postmenopausal women. J Clin Oncol 23:6931–6940
Fallowfield LJ, Bliss JM, Porter LS, Price MH, Snowdon CF, Jones SE, Coombes RC, Hall E (2006) Quality of life in the intergroup exemestane study: a randomized trial of exemestane versus continued tamoxifen after 2 to 3 years of tamoxifen in postmenopausal women with primary breast cancer. J Clin Oncol 24:910–917
Cecil RL, Archer BH (1925) Arthritis of the menopause. A study of fifty cases. JAMA 84:75–79
Felson DT, Cummings SR (2005) Aromatase inhibitors and the syndrome of arthralgias with estrogen deprivation. Arthritis Rheum 52:2594–2598
Vegeto E, Bonincontro C, Pollio G, Sala A, Viappiani S, Nardi F, Brusadelli A, Viviani B, Ciana P, Maggi A (2001) Estrogen prevents the lipopolysaccharide-induced inflammatory response in microglia. J Neurosci 21:1809–1818
Felson DT, Niu J, Clancy M, Aliabadi P, Sack B, Guermazi A, Hunter DJ, Amin S, Rogers G, Booth S (2007) Low levels of vitamin D and worsening of knee osteoarthritis: results of two longitudinal studies. Arthritis Rheum 56:129–136
Morales L, Pans S, Verschueren K, Van Calster B, Paridaens R, Westhovens R, Timmerman D, De Smet L, Vergote I, Christiaens MR, Neven P (2008) Prospective study to assess short-term intra-articular and tenosynovial changes in the aromatase inhibitor–associated arthralgia syndrome. J Clin Oncol 26:3147–3152
Hayes CE, Nashold FE, Spach KM, Pedersen LB (2003) The immunological functions of the vitamin D endocrine system. Cell Mol Biol 49:277–300
Cheema C, Grant BF, Marcus R (1989) Effects of estrogen on circulating “free” and total 1,25-dihydroxyvitamin D and on the parathyroid-vitamin D axis in postmenopausal women. J Clin Invest 83:537–542
Buchanan JR, Santen R, Cauffman S, Cavaliere A, Greer RB, Demers LM (1986) The effect of endogenous estrogen fluctuation on metabolism of 25-hydroxyvitamin D. Calcif Tissue Int 39:139–144
Rastelli AL, Taylor ME, Gao F, Armamento-Villareal R, Jamalabadi-Majidi S, Napoli N, Ellis MJ (2011) Vitamin D and aromatase inhibitor-induced musculoskeletal symptoms (AIMSS): a phase II, double-blind, placebo-controlled, randomized trial. Breast Cancer Res Treat 129:107–116
Acknowledgements
Letrozole and funding for this study were provided by Novartis Pharmaceutical Corporation (East Hanover, NJ). Vitamin D3 (10,000 IU capsules) and matched, blinded placebo were provided by BTR Group, Inc. (Pittsfield, IL). Neither company was the sponsor for the trial; nor were they involved in any aspect of design and conduct of the study; collection, management, analysis, or interpretation of the data; preparation, review, or approval of the manuscript; or decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The following authors are without financial interests or conflicts of interest related to this trial: Kimler, Reddy, Klemp, Nydegger, and Yeh. Within the past three years, Dr. Khan has served as a consultant to Novartis Pharmaceutical Company, Inc. and Pfizer. Drs. Khan and Sharma have received during the past three years, via their institution, funding for support of research and clinical trials from the following companies: AstraZeneca; Bristol-Myers Squibb; Celgene, Inc.; Novartis Pharmaceutical Company, Inc.; Genentech-Roche, GlaxoSmithKline, and Pfizer. Study agent but no funding has been provided by DSM and Pfizer for trials conducted by Dr. Fabian.
Ethical standards
The conduct of the trial complies with the current laws of the United States of America.
Rights and permissions
About this article

Cite this article
Khan, Q.J., Kimler, B.F., Reddy, P.S. et al. Randomized trial of vitamin D3 to prevent worsening of musculoskeletal symptoms in women with breast cancer receiving adjuvant letrozole. The VITAL trial. Breast Cancer Res Treat 166, 491–500 (2017). https://doi.org/10.1007/s10549-017-4429-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-017-4429-8