
Abstract
Purpose
Multiple aspects of the tumor microenvironment (TME) impact breast cancer, yet the genetic modifiers of the TME are largely unknown, including those that modify tumor vascular formation and function.
Methods
To discover host TME modifiers, we developed a system called the Consomic/Congenic Xenograft Model (CXM). In CXM, human breast cancer cells are orthotopically implanted into genetically engineered consomic xenograft host strains that are derived from two parental strains with different susceptibilities to breast cancer. Because the genetic backgrounds of the xenograft host strains differ, whereas the inoculated tumor cells are the same, any phenotypic variation is due to TME-specific modifier(s) on the substituted chromosome (consomic) or subchromosomal region (congenic). Here, we assessed TME modifiers of growth, angiogenesis, and vascular function of tumors implanted in the SSIL2Rγ and SS.BN3IL2Rγ CXM strains.
Results
Breast cancer xenografts implanted in SS.BN3IL2Rγ (consomic) had significant tumor growth inhibition compared with SSIL2Rγ (parental control), despite a paradoxical increase in the density of blood vessels in the SS.BN3IL2Rγ tumors. We hypothesized that decreased growth of SS.BN3IL2Rγ tumors might be due to nonproductive angiogenesis. To test this possibility, SSIL2Rγ and SS.BN3IL2Rγ tumor vascular function was examined by dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI), micro-computed tomography (micro-CT), and ex vivo analysis of primary blood endothelial cells, all of which revealed altered vascular function in SS.BN3IL2Rγ tumors compared with SSIL2Rγ. Gene expression analysis also showed a dysregulated vascular signaling network in SS.BN3IL2Rγ tumors, among which DLL4 was differentially expressed and co-localized to a host TME modifier locus (Chr3: 95–131 Mb) that was identified by congenic mapping.
Conclusions
Collectively, these data suggest that host genetic modifier(s) on RNO3 induce nonproductive angiogenesis that inhibits tumor growth through the DLL4 pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multiple nonmalignant cell types in the TME impact breast cancer risk and progression [1,2,3,4], yet the underlying heritable mechanisms that alter TME cell function and influence breast cancer outcomes are largely unknown. We developed CXM as the first strategy for mapping host TME modifiers [5]. In CXM, human breast cancer cells are orthotopically implanted into consomic and congenic xenograft host strains, which are derived from two parental strains with different susceptibilities to breast cancer (see Fig. 1a for schematic). Because the strain backgrounds are different, whereas the inoculated tumor cells are the same, any phenotypic variation is due to TME-specific modifier(s) that are localized to the substituted chromosome (consomic) or subchromosomal region (congenic), and can be elucidated by network-based expression analysis.

Schematic of the SS.BN3IL2Rγ and SSIL2Rγ strains and tumor growth curves. a In CXM, the host genetic background is altered by one chromosome inherited from a different genetic background (black bar), enabling any phenotypic differences to be linked to the substituted chromosome (e.g., RNO3 in this case). b The growth of 231Luc+ tumors was monitored by caliper measurement at 13, 20, and 27 days post-implantation in SSIL2Rγ (n = 5) and SS.BN3IL2Rγ (n = 7) rats. c The growth of HCC1806-RR tumors was monitored by caliper measurement at 10, 17, and 24 days post-implantation in SSIL2Rγ (n = 8) and SS.BN3IL2Rγ (n = 6) rats. Data are presented as mean tumor volume ± SEM. *p < 0.05 as determined by a repeated measures ANOVA
In the present study, we found that multiple breast cancer xenograft lines that were implanted into the consomic SS.BN3IL2Rγ CXM strain had significant tumor growth inhibition compared with the parental SSIL2Rγ strain, despite a paradoxical increase in tumor blood vessel density in SS.BN3IL2Rγ. Multiple clinical breast cancer studies have also correlated tumor vascular perfusion kinetics (i.e., tumor vascular function) with histopathologic prognostic factors [6], responses to neoadjuvant chemotherapy [7,8,9,10], and patient overall survival [11], leading us to hypothesize that TME modifier(s) on rat chromosome 3 (RNO3) might decrease tumor vascular function, which overrides the phenotypic effects of increased vascular density. Here, we characterized the growth and vasculature of triple-negative breast cancer (TNBC) tumors implanted in SS.BN3IL2Rγ and SSIL2Rγ rats using micro-CT, DCE-MRI, and in vitro morphometric analysis of primary blood vascular endothelial cells (EC). Additionally, species-specific RNAseq (SSRS) and quantitative immunofluorescent imaging were used to identify the potential TME modifier(s) that might alter tumor growth and vascular function. We found that SS.BN3IL2Rγ tumors form a more tortuous and denser vascular network and have altered vascular function, which was similar to qualitative perfusion imaging in patients that had lower grade breast cancers [6] and improved 5-year overall survival [11]. The changes in the SS.BN3IL2Rγ tumors also coincided with altered expression of DLL4, which was previously linked with nonproductive angiogenesis [12]. Moreover, DLL4 was co-localized within a host TME modifier locus (Chr3: 95–131 Mb) that was identified by congenic mapping and correlated with the phenotypic differences that were observed at the consomic level. Collectively, these data suggest that germline host TME modifier(s) of the pathways regulating tumor angiogenesis (e.g., the DLL4 pathway) might induce a switch from productive to nonproductive angiogenesis that inhibits breast cancer.
Materials and methods
Animals
All rats were provided food and water ad libitum and were housed at the Medical College of Wisconsin (MCW) Animal Resource Center. All procedures were approved by the MCW IACUC committee. The generation of the SSIL2Rγ and SS.BN3IL2Rγ rats has been described elsewhere [5]. Congenic strains were generated by crossing SS/Mcwi and the SS.BN3 consomic strain, followed by intercrossing the F1 progeny and F2 generation to capture different regions of RNO3 by marker-assisted selection, as described previously [13, 14]. Three new congenic strains were generated: line AIL2Rγ [SS.BN-(D3Rat93-D3Mit3)/Mcwi], line BIL2Rγ [SS.BN-(D3Rat26-D3Mgh18)/Mcwi], and line CIL2Rγ [SS.BN-(D3Rat222-D3Rat218)/Mcwi].
Tumor implantation
The TNBC orthotopic models of firefly luciferase-tagged MDA-MB-231 (231Luc+) cells and Renilla-tagged HCC-1806 (HCC1806-RR) were described previously [5, 15, 16]. Briefly, 231Luc+ cells (6 × 106) or HCC1806-RR cells (4 × 106) were suspended in 50% Matrigel and implanted into the #3 mammary fat pad (MFP) of female SSIL2Rγ and SS.BN3IL2Rγ rats. Tumor volume was determined by caliper measurements using the formula: volume = Dd2π/6, where D equals larger diameter and d equals smaller diameter.
DCE-MRI
The MRI study was performed on a 9.4T Bruker AVANCE Scanner fitted with a volume coil. The rats were anesthetized with 1.5% isoflurane and immobilized with a fiberglass bite-bar. Temperature was monitored and maintained at 37 ± 1.5 °C throughout the experiment. A RARE (rapid acquisition rapid echo) imaging sequence (TE/TR = 8/4 ms; matrix = 256 × 256; FOV = 3.5 cm, slice = 17.5 mm) was used to acquire sagittal scout images. A dynamically acquired T1-weighted spin-echo imaging sequence was acquired during the rapid injection of a gadolinium (0.1 mmol/kg Omniscan, Nycomed Amersham) contrast agent for a total of 5 min. Acquisition parameters included a TE/TR = 11/500 ms, matrix = 256 × 256, FOV = 3.5 cm, slice 1 mm, and phase repetition time of 6 s. Four coronal slices were chosen based on the RARE images and the tumor inoculation site. Pre- and post-T1-weighted spin-echo image were also acquired (TE/TR = 11/500 ms; matrix = 256 × 256; FOV = 3.5 cm; slice 2 mm) to delineate enhancing tumor. The DCE data were processed to generate initial area under the curve (IAUC), contrast washout, and initial slope images using the IB-DCE software plugin (Imaging Biometrics, Elm Grove, WI). Tumor and contralateral normal flank regions of interest (ROI) were drawn manually on the T1 + Gd images. Parameters were extracted from these ROIs for direct comparison between groups. The IAUC values were normalized by the contralateral flank values.
Micro-CT
Rats were completely perfused with saline containing 5 U/mL of heparin, followed by systemic injection of Microfil casting agent (Flow Tech, Inc., Carver, MA) and tissue processing, as described previously [17]. Micro-CT data were acquired on a Triumph SPEC/CT scanner (Gamma Medica-Ideas, Nortridge, CA) at a nominal resolution of 27 µm (65 kVp, 170 µA, 230-ms integration time, 2048 proj/180°, 28.9-mm-diameter field of view, 592 × 592 reconstruction matrix, cone-beam reconstruction. Gaussian smoothing and global thresh-holding procedures were applied to the grayscale data to contour the tumor (sigma = 2, support = 3, threshold = 30% of max grayscale value) and extract the tumor vascular network (sigma = 0.8, support = 1, threshold = 200% of max grayscale value).
Tube formation assay
Primary blood vascular endothelial cells (EC) were isolated using magnetic bead-conjugated anti-Pecam-1 antibodies (Cell Biologics; Chicago, IL) and cultured in DMEM containing 10% FBS and PenStrep. Four-chamber slides (Nunc Lab-Tek) were coated with 250 µL of matrigel (CB40234B, BD Biosciences) and allowed to solidify at 37 °C for 1 h, followed by seeding of 25,000 EC per well. After 24 h, images were acquired using a Nikon TS-100 Microscope equipped with a Flex camera (Nikon). Morphometric analysis of tube-like structures was performed using Pipeline [18].
Proliferation assay
EC were seeded in DMEM containing 1% FBS (control) or 10% FBS in 24-well plates at the density of 10,000 cells/well and incubated for 72 h. At 72 h post-seeding, EC were trypsinized and enumerated using an automated cell counter.
Species-specific RNAseq
The SSRS method is outlined in Supplemental Fig. 1. Briefly, total RNA was extracted by Trizol from whole xenograft tumors that were excised from SSIL2Rγ (n = 4) and SS.BN3IL2Rγ (n = 4) rats, followed by library preparation using Illumina’s TruSeq RNA library kit and sequencing on an Illumina HiSeq 2000. All raw sequencing data can be accessed from the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA278049). A joint transcriptome reference file was produced by concatenating all the RNA sequences from human build GRCh38 and rat build Rnor5.0 and appending the coding sequence of firefly luciferase (GenBank accession U47295). Alignment and joint transcript abundance estimation, using the joint transcriptome reference, was performed for each separate xenograft sample with bowtie2 [19] and eXpress (version 1.5.1) [20]. Default parameters were used with the exception of a bowtie2 offset of 1, trading index size for increased alignment speed. An estimated counts table was compiled from all transcripts and samples and analyzed using the DESeq2 (version 1.4.5) [21] library for R. The rat and human (plus luciferase) transcripts were analyzed separately to allow for normalization of different relative contributions of malignant tumor cells and the nonmalignant host stroma. Differential expression values were estimated using individual transcript data and also at the gene level by first summing estimate counts across all alternative transcripts for each gene. This yielded transcript- and gene-level-estimated fold-changes and FDR-adjusted p values for human and rat transcripts separately (Supplemental Tables 1–4).
Immunofluorescent staining and measurement of mean fluorescent intensity
Immunofluorescent staining was performed using antibodies against CD31 (R&D Systems; AF3628 or BD; 555,025) and DLL4 (R&D Systems; AF1389), as described previously [5, 22]. Images were acquired at ×200 and 400 magnification on a Nikon E400 microscope equipped with a Spot Insight camera (Nikon Instruments).
Near infrared imaging of tumor vascular function
A bifurcated optical fiber bundle is used to deliver a 785 nm wavelength (power 75 mW, diode laser, Thorlab Inc.,) from two positions to uniformly excite the entire rat. A 16-bit deep cooled charge-coupled device (CCD) camera (PI-XAM, Princeton Instruments) is used to image the rats through a computer-controlled WinView/32 software: first, the rat image with a 785-nm laser exposure and neutral density filter (OD = 4 at 785 nm) for 25-ms exposure to focus and align. For molecular imaging, a combination of 785-nm notch filter and 830 ± 10-nm bandpass filters was used on emission side and a series of 1500 frames having 256 × 256 pixels size (50-ms exposure time per frame and gain 10) were acquired in 5 min 43 s. Indocyanine green (ICG) was injected intravenously at 2 s after the image acquisition starts using a programmable syringe pump and the frames before ICG injection was used for the background correction.
Statistical analysis
Sigma Plot 11.0 was used to perform unpaired Student’s t test. Chi-square, ANOVA, Fisher’s exact, and Kolmogorov–Smirnov tests and empirical cumulative distribution plots for differential expression significance values were performed with R.
Results
Host TME modifier(s) of TNBC growth on RNO3 also elicit nonproductive angiogenesis
To assess tumor growth, 231Luc+ or HCC1806-RR cells were orthotopically implanted in the MFP of SSIL2Rγ and SS.BN3IL2Rγ CXM rats and tumor volumes were assessed by caliper measurement. Compared with 231Luc+ tumors implanted in SSIL2Rγ rats at 27 days post-implant (3860 ± 754 mm3, n = 5), SS.BN3IL2Rγ tumors (1863 ± 395 mm3, n = 7) were twofold smaller (p < 0.05) (Fig. 1b). Likewise, HCC1806-RR showed significant growth inhibition in SS.BN3IL2Rγ rats (5904 ± 700 mm3, n = 6) compared with SSIL2Rγ rats (8893 ± 984 mm3, n = 8) (Fig. 1c). These data demonstrate that TME-specific modifier(s) residing on RNO3 inhibit breast cancer growth [5].
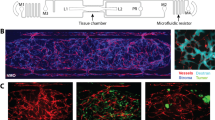
Tumor growth requires vascular supply [23] and changes in vascular function are correlated with breast tumor grade [6] and overall survival [11], leading us to examine whether the TME-specific modifier(s) on RNO3 altered the tumor vasculature. Compared with SSIL2Rγ tumors, the blood vasculature of TNBC tumors implanted in the SS.BN3IL2Rγ genetic background was denser and more tortuous (Fig. 2a–e). Likewise, tumor blood vessel function differed between 231Luc+ tumors implanted in SS.BN3IL2Rγ and SSIL2Rγ, as determined by DCE-MRI (Fig. 2f, g). Compared with SSIL2Rγ tumors, the nIAUC was significantly higher in SS.BN3IL2Rγ tumors (p < 0.05) (Fig. 2h), demonstrating that SS.BN3IL2Rγ tumors have increased total blood volume compared with SSIL2Rγ tumors. The increased tumor blood volume might be explained by the higher density of tortuous blood vessels in tumors implanted in SS.BN3IL2Rγ compared with SSIL2Rγ, as determined histologically (Fig. 2a–d) and by micro-CT (Fig. 2e). Moreover, the rate of washout of gadolinium contrast agent from SS.BN3IL2Rγ tumors was significantly lower than SSIL2Rγ (p < 0.05, Fig. 2i), indicating that outward blood flow is slower in SS.BN3IL2Rγ tumors and subsequent blood pooling could result in the higher peak total blood volume. Collectively, these data suggest that SS.BN3IL2Rγ tumors have more disordered vasculature that functions differently than the SSIL2Rγ parental strain and possibly contribute to delayed tumor growth in the SS.BN3IL2Rγ rat.

Analysis of blood vasculature of TNBC tumors implanted in SS.BN3IL2Rγ and SSIL2Rγ rats. a Immunofluorescent staining of CD31+ blood vessels of 231Luc+ implanted in SSIL2Rγ (n = 5) and SS.BN3IL2Rγ (n = 5) rats. b Quantification of CD31+ tumor blood vessels per 200× field (n = 3 fields per tumor). Data are presented as mean ± SEM. c Immunofluorescent staining of CD31+ blood vessels of HCC1806-RR implanted in SSIL2Rγ (n = 8) and SS.BN3IL2Rγ (n = 6) rats. d Quantification of CD31+ tumor blood vessels per 200× field (n = 3 fields per tumor). Data are presented as mean ± SEM. e Micro-CT reconstructions of the vascular networks of 231Luc+ tumors implanted in SSIL2Rγ and SS.BN3IL2Rγ rats. f Representative T1 + Gd and nIAUC images from 231Luc+ tumors implanted in SS.BN3IL2Rγ and SSIL2Rγ rats. g Plot of dynamic flow curves (n = 6 rats per group). h Bar charts showing the comparison of nIAUC and i washout of Gd contrast agent from tumor regions of interest. Data are presented as mean normalized values ± SEM. *p < 0.05 as determined by Student unpaired t test
One possible explanation of the altered vasculature in SS.BN3IL2Rγ tumors is that genetic modifier(s) on the BN-derived RNO3 intrinsically alter EC function at the cellular level. To explore this possibility, we used an in vitro model of primary blood vascular EC derived from SS.BN3 consomic and SS rats to examine proliferative capacity and tube formation. As shown in Fig. 3a, b, isolated EC showed cobblestone morphology and expression of multiple vascular EC markers. The proliferative capacity of the SS.BN3-derived EC was significantly higher (p < 0.05) compared with SS-derived EC (Fig. 3c). Likewise, the density of tube-like structures formed by SS.BN3-derived EC was significantly higher (p < 0.001) than SS-derived EC (Fig. 3d, e), despite the same number of EC being seeded per well. Further morphometric analysis revealed that SS.BN3-derived EC formed tube-like structures with significantly more branch points (Fig. 3f) and thinner tubes (Fig. 3g) compared with SS-derived EC. Collectively, these data suggest that the altered vasculature in SS.BN3IL2Rγ tumors could be due to TME modifier(s) on RNO3 that directly alter EC function.

Functional analysis of blood vascular endothelial cells (EC) derived from SS and SS.BN3 rats. a Morphology of EC derived from SS and SS.BN3 rats was identical between strains and resembled a cobblestone morphology that is characteristic of EC. Images were acquired at 200× and the scale bar represents 100 µm. b Purified ECs express blood endothelium markers. (+) indicates positive control RNA and (−) indicates a no template control. c Proliferative response of EC to serum. Data are presented as mean ± SEM (n = 4 per group). *p < 0.05 and **p < 0.01, as determined by ANOVA followed by the Holm–Sidak multiple comparison test. d Brightfield images of EC tube formation for SS (n = 8) and SSBN3 (n = 8). Images were acquired at 400× and the scale bar represents 500 µm. e Quantification of the total area of tube-like structures per 40× field. f Quantification of EC tube branch points per 40× fields. g Quantification of EC tube thickness per 400× field. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001, as determined by Student unpaired t test
Host TME modifier(s) on RNO3 alter a vascular gene network
Species-specific RNAseq (SSRS) was used to identify host TME gene networks that were differentially expressed (DE; FDR < 0.1) in 231Luc+ tumors implanted in SSIL2Rγ (n = 4) and SS.BN3IL2Rγ (n = 4). This revealed 541 DE genes and 489 DE transcript isoforms in the host TME of SS.BN3IL2Rγ tumors compared with SSIL2Rγ (Supplemental Tables 1, 2). DE genes and transcript isoforms were significantly enriched on RNO3 compared to the genome-wide average (p < 0.001; Fig. 4a–d), as were the distributions of p values for RNO3 versus the rest of the genome (Fig. 4e, f).

Localization of differentially expressed genes and transcripts in the TME of SS.BN3IL2Rγ (n = 4) and SSIL2Rγ (n = 4) rats, as determined by SSRS. a Chromosomal distribution of the 541 differentially expressed genes in the TME of SS.BN3IL2Rγ rats compared with the SSIL2Rγ. b Differentially expressed genes from panel (a) presented as differentially expressed genes per chromosome normalized to total number of genes per chromosome. c Chromosomal distribution of the 489 differentially expressed transcript isoforms in the TME of SS.BN3IL2Rγ rats compared with the SSIL2Rγ. d Differentially expressed transcript isoforms from panel (c) presented as differentially expressed transcript isoforms per chromosome normalized to the total number of transcript isoforms per chromosome. ***p < 0.001, as determined by Fisher’s exact test. Distributions of adjusted p values for differentially expressed genes (e) and differentially expressed transcripts (f) on RNO3 versus the rest of the rat genome were tested by a two-sample Kolmogorov–Smirnov test
Gene ontology (GO) analysis of the merged list of DE genes and transcript isoforms using ingenuity pathway analysis (IPA) revealed that the most enriched GO cluster was related to vascular development and angiogenesis (Supplemental Tables 5, 6). Of the DE candidates in the vascular development and angiogenesis GO, only DLL4 was previously linked with altered vascular branching that leads to nonproductive angiogenesis and tumor growth inhibition [12, 24,25,26,27,28], which matches the phenotypes observed in the SS.BN3IL2Rγ CXM model (Figs. 1, 2, 3, 4). Expression of DLL4 protein on tumor blood vessels was also reduced in 231Luc+ and HCC1806-RR tumors implanted in SS.BN3IL2Rγ rats compared with SSIL2Rγ tumors (Fig. 5c, d). Compared with SSIL2Rγ tumors, the density DLL4+ blood vessels decreased 50–60% in 231Luc+ and HCC1806-RR tumors implanted in SS.BN3IL2Rγ rats (Fig. 5e, f), despite a nearly twofold increase in density of total CD31+ tumor blood vessels (Fig. 2a–d). Additionally, blockade of the DLL4 pathway in SS-derived EC by the γ-secretase inhibitor, DAPT [24], enhanced in vitro tube formation (124 ± 6 branchpoints; p < 0.05) compared with control-treated EC (106 ± 5 branchpoints) (Supplemental Fig. 3), suggesting that inhibition of the DLL4 pathway increases vascular branching, as has been demonstrated previously [12, 24,25,26,27,28].

DLL4 is a candidate TME modifier that is downregulated in the vasculature of SS.BN3IL2Rγ tumors compared with SSIL2Rγ. a Heatmap of the 43 differentially expressed mRNAs (FDR < 0.1) that make up the blood vasculature GO cluster. b Heatmap of the six differentially expressed mRNAs in the blood vasculature GO cluster that reside on RNO3. c, d Expression of DLL4 protein (green) on CD31+ blood vessels (red) was assessed by dual immunofluorescent staining of 231Luc+ (c) and HCC1806-RR (d) tumors implanted in SSIL2Rγ and SS.BN3IL2Rγ rats. Note that DLL4 is co-localized to CD31+ tumor blood vessels and is downregulated in SS.BN3IL2Rγ tumors compared to SSIL2Rγ. e, f Quantification of the percentage of DLL4+ blood vessels in 231Luc+ (e) and HCC1806-RR (f) tumors implanted in SSIL2Rγ and SS.BN3IL2Rγ rats (n = 5–8 tumors per group). Data are presented as mean ± SEM. *p < 0.05 and ***p < 0.001, as determined by Student unpaired t test
DLL4 co-localizes within a host TME modifier locus of tumor growth and angiogenesis
We next attempted to test the molecular role of DLL4 in our model by shRNA-mediated knockdown; however, we were unable to identify shRNA reagents that reproducibly downregulated DLL4 expression in the rat. Thus, we instead mapped the host TME modifier(s) (e.g., DLL4) on RNO3 using three SS.BN3IL2Rγ congenic xenograft host strains (line AIL2Rγ, line BIL2Rγ, and line CIL2Rγ). As shown in Fig. 6, 231Luc+ or HCC1806-RR cells were orthotopically implanted into the MFP of SSIL2Rγ, SS.BN3IL2Rγ, line AIL2Rγ, line BIL2Rγ, and line CIL2Rγ. We found that 231Luc+ tumors implanted SSIL2Rγ (6680 ± 719 mm3, n = 10) were significantly larger than SS.BN3IL2Rγ consomic (2714 ± 149 mm3, p < 0.05, n = 26) and line AIL2Rγ (3630 ± 289 mm3, p < 0.05, n = 16) tumors at 24 days post-implantation, whereas the volumes of 231Luc+ tumors implanted in line BIL2Rγ (4518 ± 278 mm3, n = 13) and line CIL2Rγ (4614 ± 335 mm3, n = 13) did not significantly differ from SSIL2Rγ (Fig. 6). Similarly, HCC1806-RR tumor growth was significantly inhibited in the SS.BN3IL2Rγ consomic and line AIL2Rγ compared with SSIL2Rγ, line BIL2Rγ, and line CIL2Rγ (Fig. 6). By exclusion mapping, these data suggest that host TME modifier(s) of TNBC growth reside within a 36 Mb locus on RNO3 (Chr3: 95–131 Mb), a region that contains DLL4 (Fig. 5a–c).

Schematic representation of the SS.BN3IL2Rγ congenic strains that were generated by introgressing segments of BN chromosome 3 (black) into the genetic background of the parental SSIL2Rγ strain (white) by marker-assisted breeding. Thin black bars represent confidence intervals, which are chromosomal regions that could be BN or SS. The position of DLL4 (chr3: 111,135,011–111,146,746 bp) and the 36 Mb host TME modifier locus (chr3: 95,176,874–131,051,652 bp). Tumor volumes were measured at 24 days post-implantation of the 231Luc+ and HCC1806-RR TNBC cell lines. Data are presented as mean ± SEM. *p < 0.05, as determined by 1-way ANOVA followed by Holm–Sidak post hoc test
We next assessed whether the host TME modifier locus on RNO3 (Chr3: 95–131 Mb) altered the tumor vascular density and function, and vascular-specific DLL4 expression. Compared with the CD31+ vessel density of SS.BN3IL2Rγ tumors (80 ± 9 vessels/field), the 231Luc+ tumors implanted in SSIL2Rγ (54 ± 5 vessels/field; p < 0.01) and line CIL2Rγ (47 ± 3 vessels/field; p < 0.001) had significantly decreased tumor vascular density (Fig. 7a). Likewise, HCC1806-RR tumor vascular density was significantly higher in the SS.BN3IL2Rγ consomic compared with SSIL2Rγ and line CIL2Rγ (Fig. 7a). For both TNBC lines, the density of CD31+/DLL4+ double-positive vessels in SS.BN3IL2Rγ tumors was significantly less (p < 0.05) than SSIL2Rγ and line CIL2Rγ tumors, despite significantly increased density of CD31+ blood vessels in SS.BN3IL2Rγ tumors (Fig. 7a). Finally, analysis of tumor vascular function showed a delayed washout of ICG dye in 231Luc+ tumors implanted in SS.BN3IL2Rγ compared with SSIL2Rγ and line CIL2Rγ (Fig. 7b). Collectively, these data suggest that the host TME modifier(s) of tumor growth inhibition and nonproductive angiogenesis reside within the 36 Mb locus on RNO3 (Chr3: 95–131 Mb), which co-localizes with differential expression of DLL4.

Congenic mapping of vascular density and function, which coincide with altered vascular-specific DLL4 protein expression. a Analysis of the mean density of CD31+ blood vessels per 200× field (n = 3 images per tumor) and the percentage of CD31+/DLL4+ double-positive blood vessels per 400× field (n = 3 images per tumor). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001, as determined by 1-way ANOVA followed by Holm–Sidak post hoc test. b Tumor vascular function was assessed by near infrared imaging of circulating ICG dye in 231Luc+ tumors at 10 days post-implantation
Discussion
Perfusion MRI analysis of tumor vascular function can be used to predict more aggressive histopathologic features and outcomes in breast cancer patients, such as tumor grade [6], response to neoadjuvant chemotherapy [7,8,9,10], and overall survival [11]. However, the genetic and molecular mechanism(s) underlying the tumor vascular changes and the prognostic factors remain poorly understood. Here, we present a novel experimental model with key vascular phenotypes that recapitulate perfusion characteristics that may correlate with the aggressiveness of human breast cancers. Compared with the SS.BN3IL2Rγ strain, tumor growth in the SSIL2Rγ rat was significantly more aggressive and this coincided with a rapid washout on DCE-MRI that has been associated with higher grade of tumors [6] and worse overall survival prognosis in human patients [11]. These findings, combined with morphometric and functional analyses of the blood vasculature (Figs. 2, 3), indicate that host genetic modifier(s) on RNO3 alter vascular structure and function in the SS.BN3IL2Rγ CXM rat strain. These vascular alterations were also detected at the molecular level by SSRS and immunofluorescent imaging, which revealed an altered vascular gene network that was highlighted by dysregulation of DLL4 in the TME of SS.BN3IL2Rγ tumors (Fig. 5). By congenic mapping, we localized a host TME modifier locus (Chr3: 95–131 Mb) that contains DLL4 and was correlated with the tumor growth and vascular phenotypes that were observed at the consomic level (Figs. 6, 7). Collectively, these data suggest that DLL4 is likely a host TME modifier of breast cancer in the SS.BN3IL2Rγ CXM strain, albeit further congenic mapping or gene-editing is necessary to exclude other potential candidate(s) in the region and isolate the causative variant(s).
It is notable that the DLL4 pathway is a master regulator of angiogenic vascular patterning [12, 24,25,26,27,28,29,30,31] and inhibition of DLL4 attenuates tumor growth and progression by eliciting nonproductive angiogenesis (i.e., a higher density of poorly functioning vasculature) [12, 24,25,26,27,28], which is similar to the phenotypes observed in the SS.BN3IL2Rγ tumors. During angiogenesis, VEGF upregulates DLL4 expression on the vascular tip cells and DLL4 then activates NOTCH1 signaling in the adjacent endothelial cells to suppress aberrant tip cell formation and excessive vascular branching [25, 29,30,31]. In the SS.BN3IL2Rγ model, we observed a TME-specific decrease in expression of DLL4 that coincided with nonfunctional angiogenesis and decreased growth of SS.BN3IL2Rγ tumors. Congenic mapping further localized the host TME locus on RNO3 (Chr3: 95–131 Mb) that contains DLL4 and modified DLL4 expression (Fig. 7), which coincided with altered tumor growth (Fig. 6) and altered tumor vascular density and function (Fig. 7). Although these data due not exclude the possibility of other TME modifier(s) within the TME locus on RNO3 (Chr3: 95–131 Mb), the downregulation of DLL4 and the observations in SS.BN3IL2Rγ tumors that phenocopy the inhibition of DLL4 [12, 24,25,26,27,28] suggest that the DLL4 pathway is a plausible candidate on RNO3. However, these findings do not exclude additional host TME modifier(s) on RNO3 that might interact with the DLL4 pathway (e.g., upstream or downstream mediators) or function independently of the DLL4 pathway. The next step to verifying the causative DLL4 allele(s) or discovering new candidates will be to further map the TME modifier(s) using smaller subcongenic strains or gene-editing approaches.
Tumor angiogenesis is a highly coordinated process that is necessary for growth and progression of breast cancer [23, 32]. A denser tumor vasculature is largely correlated with increased tumor growth and hematogenous metastasis, which is thought to be due to enhanced supply of oxygen and nutrients to the tumor and by providing more routes for metastatic dissemination [33]. Although many initial studies focused on the prognostic value of tumor blood vessel density [33, 34], it is increasingly evident that functionality of the tumor vasculature is also an important predictor of patient outcomes (e.g., tumor grade [6], response to neoadjuvant chemotherapy [7,8,9,10], and overall survival [11]). A rapid accumulation and washout of Gd+ that is associated with higher grade of tumors [6] and worse overall survival prognosis in human patients [11] is thought to reflect a denser network of tumor vessels that facilitate tumor growth and metastasis. However, our data suggest that the predictive values of tumor vascular density and function might not always coincide. Although the vascular density was increased in SS.BN3IL2Rγ tumors compared with SSIL2Rγ tumors (Fig. 2a–e) [5], this correlated negatively with tumor growth (Fig. 1) and the DCE-MRI washout curves that were indicative of less aggressive breast cancer (Fig. 2). Collectively, these data suggest that germline genetic variants in the host TME can modify the pathways that regulate tumor vascular density and function (e.g., the DLL4 pathway), which ultimately impacts tumor growth and disease progression. Identifying these host TME modifiers will likely improve the accuracy of breast cancer patient prognosis and potentially provide novel therapeutic targets for treating breast cancer patients.
Abbreviations
- TME:
-
Tumor microenvironment
- CXM:
-
Consomic Xenograft Model
- DCE-MRI:
-
Dynamic contrast-enhanced magnetic resonance imaging
- SSRS:
-
Species-specific RNAseq
- PBST:
-
Phosphate-buffered saline plus Tween-20
- RNO3:
-
Rat chromosome 3
- TNBC:
-
Triple-negative breast cancer
- micro-CT:
-
Micro-computed tomography
- EC:
-
Endothelial cell
- DMEM:
-
Dulbecco’s modifier Eagle’s medium
- MFP:
-
Mammary fat pad
- RARE:
-
Rapid acquisition rapid echo
- IAUC:
-
Initial area under the curve
- ROI:
-
Region of interest
- FDR:
-
False discovery rate
References
Quail DF, Joyce JA (2013) Microenvironmental regulation of tumor progression and metastasis. Nat Med 19(11):1423–1437
Polyak K, Kalluri R (2010) The role of the microenvironment in mammary gland development and cancer. Cold Spring Harbor Perspect Biol 2(11):a003244
Cook LM, Hurst DR, Welch DR (2011) Metastasis suppressors and the tumor microenvironment. Semin Cancer Biol 21(2):113–122
McAllister SS, Weinberg RA (2010) Tumor-host interactions: a far-reaching relationship. J Clin Oncol 28(26):4022–4028
Flister MJ, Endres BT, Rudemiller N, Sarkis AB, Santarriaga S, Lemke A, Roy I, Geurts AM, Moreno C, Ran S et al (2014) CXM—a new tool for mapping breast cancer risk in the tumor microenvironment. Cancer Res 74(22):6419–6429
Han M, Kim TH, Kang DK, Kim KS, Yim H (2012) Prognostic role of MRI enhancement features in patients with breast cancer: value of adjacent vessel sign and increased ipsilateral whole-breast vascularity. AJR 199(4):921–928
Craciunescu OI, Blackwell KL, Jones EL, Macfall JR, Yu D, Vujaskovic Z, Wong TZ, Liotcheva V, Rosen EL, Prosnitz LR et al (2009) DCE-MRI parameters have potential to predict response of locally advanced breast cancer patients to neoadjuvant chemotherapy and hyperthermia: a pilot study. Int J Hyperth 25(6):405–415
Ah-See ML, Makris A, Taylor NJ, Harrison M, Richman PI, Burcombe RJ, Stirling JJ, d’Arcy JA, Collins DJ, Pittam MR et al (2008) Early changes in functional dynamic magnetic resonance imaging predict for pathologic response to neoadjuvant chemotherapy in primary breast cancer. Clin Cancer Res 14(20):6580–6589
Jia WR, Tang L, Wang DB, Chai WM, Fei XC, He JR, Chen M, Wang WP (2016) Three-dimensional contrast-enhanced ultrasound in response assessment for breast cancer: a comparison with dynamic contrast-enhanced magnetic resonance imaging and pathology. Sci Rep 6:33832
Sun YS, He YJ, Li J, Li YL, Li XT, Lu AP, Fan ZQ, Cao K, Ouyang T (2016) Predictive value of DCE-MRI for early evaluation of pathological complete response to neoadjuvant chemotherapy in resectable primary breast cancer: a single-center prospective study. Breast 30:80–86
Heldahl MG, Bathen TF, Rydland J, Kvistad KA, Lundgren S, Gribbestad IS, Goa PE (2010) Prognostic value of pretreatment dynamic contrast-enhanced MR imaging in breast cancer patients receiving neoadjuvant chemotherapy: overall survival predicted from combined time course and volume analysis. Acta Radiol 51(6):604–612
Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, Lin HC, Yancopoulos GD, Thurston G (2006) Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 444(7122):1032–1037
Flister MJ, Hoffman MJ, Reddy P, Jacob HJ, Moreno C (2013) Congenic mapping and sequence analysis of the Renin locus. Hypertension 61(4):850–856
Hoffman MJ, Flister MJ, Nunez L, Xiao B, Greene AS, Jacob HJ, Moreno C (2013) Female-specific hypertension Loci on rat chromosome 13. Hypertension 62(3):557–563
Volk LD, Flister MJ, Bivens CM, Stutzman A, Desai N, Trieu V, Ran S (2008) Nab-paclitaxel efficacy in the orthotopic model of human breast cancer is significantly enhanced by concurrent anti-vascular endothelial growth factor A therapy. Neoplasia 10(6):613–623
Volk-Draper LD, Rajput S, Hall KL, Wilber A, Ran S (2012) Novel model for basaloid triple-negative breast cancer: behavior in vivo and response to therapy. Neoplasia 14(10):926–942
Walker EJ, Shen F, Young WL, Su H (2011) Cerebrovascular casting of the adult mouse for 3D imaging and morphological analysis. J Vis Exp 57:e2958–e2958
Prisco AR, Bukowy JD, Hoffmann BR, Karcher JR, Exner EC, Greene AS (2014) Automated quantification reveals hyperglycemia inhibits endothelial angiogenic function. PLoS ONE 9(4):e94599
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4):357–359
Roberts A, Pachter L (2013) Streaming fragment assignment for real-time analysis of sequencing experiments. Nat Methods 10(1):71–73
Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11(10):R106
Flister MJ, Volk LD, Ran S (2011) Characterization of Prox1 and VEGFR-3 expression and lymphatic phenotype in normal organs of mice lacking p50 subunit of NF-kappaB. Microcirculation 18(2):85–101
Hanahan D, Folkman J (1996) Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86(3):353–364
Scehnet JS, Jiang W, Kumar SR, Krasnoperov V, Trindade A, Benedito R, Djokovic D, Borges C, Ley EJ, Duarte A et al (2007) Inhibition of Dll4-mediated signaling induces proliferation of immature vessels and results in poor tissue perfusion. Blood 109(11):4753–4760
Hellstrom M, Phng LK, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, Alva J, Nilsson AK, Karlsson L, Gaiano N et al (2007) Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 445(7129):776–780
Xu Z, Wang Z, Jia X, Wang L, Chen Z, Wang S, Wang M, Zhang J, Wu M (2016) MMGZ01, an anti-DLL4 monoclonal antibody, promotes nonfunctional vessels and inhibits breast tumor growth. Cancer Lett 372(1):118–127
Ridgway J, Zhang G, Wu Y, Stawicki S, Liang WC, Chanthery Y, Kowalski J, Watts RJ, Callahan C, Kasman I et al (2006) Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 444(7122):1083–1087
Hoey T, Yen WC, Axelrod F, Basi J, Donigian L, Dylla S, Fitch-Bruhns M, Lazetic S, Park IK, Sato A et al (2009) DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell 5(2):168–177
Suchting S, Freitas C, le Noble F, Benedito R, Breant C, Duarte A, Eichmann A (2007) The notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc Natl Acad Sci USA 104(9):3225–3230
Siekmann AF, Lawson ND (2007) Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature 445(7129):781–784
Lobov IB, Renard RA, Papadopoulos N, Gale NW, Thurston G, Yancopoulos GD, Wiegand SJ (2007) Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc Natl Acad Sci USA 104(9):3219–3224
Bergers G, Benjamin LE (2003) Tumorigenesis and the angiogenic switch. Nat Rev Cancer 3(6):401–410
Nico B, Benagiano V, Mangieri D, Maruotti N, Vacca A, Ribatti D (2008) Evaluation of microvascular density in tumors: pro and contra. Histol Histopathol 23(5):601–607
Weidner N, Semple JP, Welch WR, Folkman J (1991) Tumor angiogenesis and metastasis–correlation in invasive breast carcinoma. N Engl J Med 324(1):1–8
Acknowledgements
We thank M. Tschannen, R. Schilling, E. Schneider, A. Zappa, and Y. Liu for excellent technical support and the Center for Imaging Research in the Medical College of Wisconsin Department of Radiology, and the Biomedical Imaging Shared Resource supported by the MCW Cancer Center.
Funding
This work was supported by a seed grant from the Wisconsin Breast Cancer Showhouse and the MCW Cancer Center, the Mary Kay Foundation (Grant No. 024.16), and the NCI (R01CA193343) to M.J.F. Support was also received from the National Center for Research Resources, the National Center for Advancing Translational Sciences, and the Office of the Director of the NIH via the Clinical & Translational Science Institute (#8KL2TR000056), the Wisconsin Breast Cancer Showhouse and the MCW Cancer Center, the Rosenberg Translational Research Award, and an institutional research Grant (#86-004-26) from the American Cancer Society to C.B.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interests to declare.
Ethical approval
All applicable international, national, and institutional guidelines for the care and use of animals were followed. The Institutional Animal Care and Use Committee (IACUC) of the Medical College of Wisconsin approved all animal studies. All procedures involving animals were conducted in accordance with the National Institutes of Health guidelines concerning the use and care of experimental animals.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Flister, M.J., Tsaih, SW., Stoddard, A. et al. Host genetic modifiers of nonproductive angiogenesis inhibit breast cancer. Breast Cancer Res Treat 165, 53–64 (2017). https://doi.org/10.1007/s10549-017-4311-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-017-4311-8