
Abstract
Purpose
Mutations in PALB2 have been associated with a predisposition to breast and pancreatic cancers. This study aims to characterize a novel PALB2 exon 13 duplication in a hereditary breast and ovarian cancer family.
Methods
The PALB2 exon 13 duplication in this family was evaluated using Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT™) and confirmed by multiplex ligation-dependent probe amplification (MLPA). The duplication breakpoints were determined by long-range PCR and DNA sequencing. The effects of this mutation on mRNA splicing were characterized using RT-PCR, cloning, and DNA sequencing.
Results
The 5′ and 3′ breakpoints were mapped to intron 12 and downstream of 3′UTR. The tandem duplication is mediated by Alu elements in these regions. This duplication disrupts normal mRNA splicing and presumably leads to a frameshift and premature protein truncation. This duplication segregates with ovarian and breast cancer in multiple members in this family.
Conclusions
Our results indicate that the PALB2 exon 13 duplication is a pathogenic variant. The presence of the PALB2 duplication in the proband affected with high-grade serous ovarian cancer suggests that PALB2 might be associated with a predisposition to ovarian cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
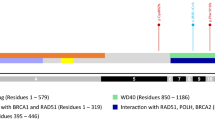
PALB2 (partner and localizer of BRCA2) was originally identified as a BRCA2-interacting protein that stabilizes BRCA2 in key nuclear complexes, which are crucial for its chromatin localization and recruitment to DNA damage sites [1, 2]. PALB2 serves as a central core of the BRCA1-PALB2-BRCA2 complex, which is essential for homologous recombination [3]. It associates with BRCA1 through its N-terminal coiled-coil domain, and the COOH terminus containing four WD40 domains is required for its interaction with BRCA2 [3–5]. Deletion of part of the PALB2 C-terminus containing the last WD domain abolishes its interaction with BRCA2 [2]. Deletion of the last 32 amino acids disrupts the protein interaction between PALB2 and BRCA2, leading to defective homologous recombination [6].
Germline heterozygous mutations in PALB2 have been linked to moderately increased risks for female breast cancer and pancreatic cancer [7]. PALB2 mutations occur in about 1–2 % of individuals with familial breast cancer [8–11] and in 3–4 % in breast cancer patients without BRCA1/2 mutations [12–14]. PALB2 mutations are present in 3–4 % of patients with familial pancreatic cancer [15, 16]. Some studies also suggest that PALB2 germline mutations confer increased risks of male breast cancer [8, 17], ovarian cancer [18, 19], and prostate cancer [20], although the spectrum of cancers and magnitude of cancer risks are still unclear.
Pennington KP et al. [21] hypothesize that patients with germline mutations in DNA homologous recombination genes will have a sensitivity to PARP inhibitors (PARPi). Inhibition of PARP is a potential synthetic lethal therapeutic approach to the treatment of patients with inherited mutations in genes such as BRCA1 and BRCA2 that are involved in DNA repair pathways [22–24]. Recently, Olaparib (AZD 2281), an oral PARP inhibitor, has been approved for the treatment of DNA repair-deficient high-grade ovarian tumors in BRCA1 or BRCA2 mutation carriers [25]. Prolonged responses to Olaparib were observed in patients harboring germline BRCA1/2 mutations with different tumor types including ovarian, breast, pancreatic, and prostate cancers [26, 27]. Like BRCA1 and BRCA2-deficient cells, cells with a genetic deficiency for PALB2 exhibit a defect in homologous repair [28] and display sensitivity to inhibition of PARP [28–31]. Since PALB2 protein participates in the same DNA repair pathway as BRCA2 [32], the synthetic lethal therapeutic strategy based on PARP inhibition could also be utilized for treatment of patients with germline mutations in PALB2. Determining PALB2 variant pathogenicity in patients with ovarian and related cancers is thus of significant clinical relevance, particularly for guiding targeted therapy regimens.
To date, most reported pathogenic PALB2 mutations detected in patients are truncating mutations (nonsense and small indel mutations) and mutations affecting the canonical splicing sites. Only a few PALB2 variants with large deletions or duplications have been identified, namely the deletions of exons 1–10 [5], 7–8, 9–10 [13], 7–11 [17], and 12–13 [33]; the duplication of exons 9–11 [13]. Here, we report a novel Alu-mediated duplication of PALB2 exon 13, which segregates with ovarian cancer and breast cancer in a large hereditary breast and ovarian cancer (HBOC) family. Our data indicate that this duplication disrupts normal splicing and leads to premature protein truncation. This study highlights the importance of functional studies in evaluating non-truncating variants including large duplications. It also indicates that PALB2 germline mutations may contribute to the development of ovarian cancer, in addition to breast and pancreatic cancers.
Materials and methods
Subject
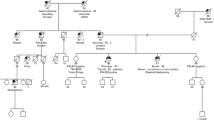
A 53-year-old female of Norwegian/German/Italian ancestry presented to the Clinical Genetics Service (CGS) at Memorial Sloan Kettering Cancer Center (MSKCC) following the diagnosis of stage IV, high-grade serous ovarian cancer involving bilateral ovaries and fallopian tubes. Given the high-grade serous pathology of her tumor and the reported maternal family history of breast and ovarian cancer, the patient was offered genetic testing for inherited mutations in known breast/ovarian cancer predisposition genes. A four-generation pedigree (Fig. 1) demonstrated that one of the proband’s three sisters was diagnosed with breast cancer (ductal carcinoma in situ, DCIS) at 42 years of age. A maternal aunt was diagnosed with bilateral breast cancer at 54 and 56 years of age (of note, this aunt had been evaluated for inherited mutations in BRCA1 and BRCA2 via full gene sequencing and was not found to carry a mutation). The proband’s maternal grandmother was diagnosed with colon cancer at 67 years of age. Her maternal great-aunt was diagnosed with ovarian cancer at her 50s (medical records were not available).

Patient pedigree. The patient (indicated with the arrow) is a 56-year-old Norwegian/German/Italian woman who was diagnosed with stage IV, high-grade serous ovarian cancer involving bilateral ovaries and fallopian tubes. One of the proband’s three sisters was diagnosed with breast cancer (ductal carcinoma in situ, DCIS) at 42 years of age. A maternal aunt was diagnosed with bilateral breast cancer at 54 and 56 years of age. The proband’s paternal great-aunt was diagnosed with ovarian cancer (medical records were not available)
Given the strong family history of breast and ovarian cancer, the proband opted to proceed with testing via a commercially available hereditary cancer multi-gene panel (sequencing and large rearrangement analysis was performed for the following genes: APC, ATM, BARD1, BMPR1A, BRCA1, BRCA2, BRIP1, CDH1, CDK4, CDKN2A, CHEK2, EPCAM (large rearrangement only), MLH1, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, PTEN, RAD51C, RAD51D, SMAD4, STK11, and TP53). The patient provided written informed consent for genetic testing as part of a study approved by the Institutional Review Board of MSKCC (protocol #96-051 “Clinical Significance of Germline BRCA Mutations”). The proband was subsequently found to carry a duplication of the last exon of the PALB2 gene (duplication exon 13). No other mutations or variants of uncertain clinical significance were identified in the remaining 24 genes analyzed. Additional studies performed at the commercial laboratory indicated that the PALB2 rearrangement was a tandem head-to-tail duplication of exon 13. Given the potentially important clinical significance of this variant, the patient and her family agreed to provide additional blood samples to help further characterize the variant at the MSKCC. Peripheral blood samples were collected using the EDTA Blood tube and PAXgene Blood RNA tube and submitted to the Diagnostics Molecular Genetics Laboratory at MSKCC for further analysis. Control RNAs were extracted from unrelated individuals seen at MSKCC who do not carry the PALB2 large duplication variant.
Duplication analysis
The PALB2 gene copy number was determined for the proband’s family members via the Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) test [34]. Duplication of PALB2 exon 13 was confirmed by multiplex ligation-dependent probe amplification (MLPA) using the SALSA MLPA P260 PALB2-RAD50-RAD51C-RAD51D probe mix (MRC Holland, Amsterdam, The Netherlands).
Long-range PCR and breakpoint determination
Long-range (LR) PCR was performed on genomic DNA from the patient to confirm the large DNA duplication in PALB2 detected by the commercial laboratory. LR-PCR was performed using a forward primer in intron 12 and a reverse primer in 3′-untranslated region (UTR) of PALB2 and the TaKaRa LA PCR kit (TaKaRa, Clontech) following the manufacturer’s instructions. The following thermal cycling conditions were used to perform LR-PCR: initial denaturation at 94 °C for 2 min, 30 cycles at 98 °C for 10 s, 68 °C for 6 min, and a final elongation at 68 °C for 10 min.
The breakpoint was determined by LR-PCR with the primer located at the end of exon 13 and the reverse primer located at the beginning of exon 12. The duplication breakpoint was more specifically identified using a primer walking strategy with a series of forward primers in the 3′-UTR and reverse primers in intron 12. The duplication junction was amplified with a specific primer followed by direct sequencing to confirm the breakpoint. Sequencing reactions were performed with the BigDye Terminator v3.1 cycle sequencing kit (Applied Biosystems) on an ABI 3730XL sequencer according to the manufacturer’s instructions. The sequences were aligned against the wild-type PALB2 nucleotide sequence (NM_024675; transcript ID, ENST00000261584) using LALIGN (ExPASy: SIB bioinformatics resource portal).
In silico analysis
The intron 12 sequence plus 2000 bps downstream of the 3′-UTR were analyzed using RepeatMasker (http://www.repeatmasker.org) to identify interspersed repeats. DNA sequence regions with Alu repeat signatures of interest were aligned using LALIGN to determine the similarity of the two Alu repeats.
cDNA analysis
The PALB2 exon 13 duplication identified through commercial testing was confirmed prior to transcript analysis. Total RNA was extracted using the PAXgene BloodRNA Kit (PreAnalytiX, Qiagen, Valencia, CA) and was subsequently used for cDNA synthesis (Superscript III First-Strand Synthesis SuperMix, Invitrogen Life Technologies, Carlsbad, CA). Control RNA was extracted from another individual who did not carry the PALB2 exon 13 duplication. RT-PCR was performed using the JumpStart REDTaq Ready Mix (Sigma), with control cDNA or the patient’s cDNA in the presence of M13-tagged forward and reverse primers (Forward, I12F: 5′- GTAAAACGACGGCCAGTCTGTGCCAAAGAGAGTGAGTC -3′; Reverse, 3′R: 5′- CAGGAAACAGCTATGAC CTGTCTGGACATAAACAAGCAA -3′). Each PCR reaction contained 12.5 µl 2 × JumpStart REDTaq Ready Mix, 2 µl 10 µM primers (1 µl for each), 2 µl cDNA, and water to make a final volume of 25 µl. Cycling conditions used were 96 °C for 5 min, 94 °C for 30 s (35×), 58 °C for 45 s (35×), and 72 °C for 60 s (35×) with a final extension at 72 °C for 5 min (1×).
Cloning
The RT-PCR products were cloned into pCR4 TOPO vectors (Invitrogen, Carlsbad, CA), following the manufacturer’s procedures (Invitrogen, Carlsbad, CA). DNA from colonies was amplified using the I12F and 3′R primers covering cDNA regions of exon 13. The PCR products were visualized by QIAxcel (QIAGEN), purified by ExoSAP-IT (Affymetrix), and then subjected to direct DNA sequencing analysis using primers M13F and M13R (BigDye Terminator v3.1 Cycle Sequencing Kit and 3730 DNA Analyzer, Applied Biosystems, Foster City, CA).
Results
Segregation analysis
The pattern of ovarian and breast cancers in the maternal kindred (Fig. 1) was consistent with autosomal dominant inheritance of a deleterious PALB2 mutation. Given our suspicion based on family history, we wanted to identify whether the PALB2 duplication segregated with the breast cancer in the family. Our family studies revealed that the proband’s sister with breast DCIS and her maternal aunt with bilateral breast cancer also carry the PALB2 duplication exon 13, as determined by the MSK-IMPACT assay (we reviewed PALB2 copy number only) and confirmed by MLPA analysis (Fig. 2). Therefore, the PALB2 duplication initially identified in the proband with high-grade serous ovarian cancer segregates with the breast cancers in this family.

PALB2 Exon 13 duplication testing. a PALB2 Exon 13 duplication detected by the MSK-IMPACT. b PALB2 Exon 13 confirmation by MLPA
Identification of duplication breakpoints
Given that the PALB2 duplication was tracking with the cancers in the family, and the possibility of targeted therapeutic options becoming available for our patient should she be found to carry a deleterious mutation in a homologous recombination repair gene, we decided to investigate the biological effects of the exon 13 duplication. We first confirmed this rearrangement by amplifying exon 13 using LR-PCR. We observed an extra band of approximately 4–6 kb in size, which was absent in the negative control sample (Fig. 3a). One primer pair specific to the patient sample was designed to obtain adequate length of PCR product to determine the breakpoints. The 5′ breakpoint located in intron 12 was mapped using a few primers. The right breakpoint was determined by sequencing the LR-PCR product using three forward sequencing primers about 600 bp apart, specific to the downstream region of 3′-UTR. Combination of the forward and reverse sequencing primers amplified a fragment of approximately 300–400 bp size. A sequence analysis of this fragment revealed the duplication conjunctions located 1089 bp downstream of exon 12, in intron 12, and 989 bp downstream of the PALB2 3′-UTR (Fig. 4), resulting in a duplicated region of 4594 bp.

Breakpoint determination. a PALB2 exon 13 duplication confirmation by long-range PCR. An extra band of approximately 4–6 kb in size was detected in the proband’s blood DNA, which is absent in the negative control sample. b Localization of breakpoint. Electropherogram showing the breakpoint sequence. c Sequence alignment in the breakpoint region showing the 32-bp perfect identity between the intron 12 and 3′- UTR of PALB2

An Alu-mediated mechanism appears to be responsible for exon 13 duplication. A sequence analysis of the LR-PCR product allowed us to identify the duplication conjunctions which are located 1089 bp downstream of exon 12, in intron 12, and 989 bp downstream of the PALB2 3′-UTR
Analysis of intron 12 and the region downstream of the PALB2 3′-UTR by RepeatMasker revealed that there are eight Alu repeats in intron 12 and five Alu elements within the region 2000 bp downstream of the 3′-UTR. Both 5′ and 3′ breakpoints occurred within Alu elements, named AluSg in intron 12 and AluSc downstream of the 3′-UTR. These elements are characterized by a 32-bp region of perfect identity (Fig. 3c), implicating homologous recombination as the underlying mechanism of the tandem duplication (Fig. 4).
PALB2 exon 13 duplication disrupts normal splicing and presumably leads to premature protein truncation
The effect of PALB2 exon 13 on RNA splicing was evaluated by amplifying regions of PALB2 from cDNA derived from the patient. PCR was designed to generate a fragment that spanned part of exon 12 and the entire coding region of exon 13, which is likely affected by the duplication. An additional band was identified in the patient, patient’s 48-year-old sister, aunt, and 52-year-old sister. It is absent in her 57-year-old sister (lane 5, Fig. 5a). This band represents an aberrant RNA splicing product attributable to the exon 13 duplication. Further RT-PCR, cloning, and sequencing results revealed that the duplication leads to a duplication of the first 53 bps at the beginning of exon 13 (Fig. 5b). This insertion causes a frameshift that creates a premature stop codon and leads to the loss of 41 amino acids within the WD domain of the C-terminus of the PALB2 protein, which is critical for its interaction with BRCA2 [6, 14, 35].

PALB2 Exon 13 duplication disrupts normal splicing and leads to frameshift. a RT-PCR products run on QIAxcel. An extra band was observed in the patient, patient’s 48-year-old sister, aunt, and 52-year-old sister. b Electropherogram showing the inserted sequence from the upper band in the patient of figure (a). The insertion leads to a new stop codon as indicated by the red box
Family expansion testing
Prior to meeting with the CGS, all three of the proband’s sisters had opted to undergo risk-reducing bilateral salpingo-oophorectomy based on the family history of ovarian cancer. Prior to genetic testing, all three sisters expressed that they would not regret having had risk-reducing surgery should they be shown to not share the familial PALB2 duplication that is expected to be responsible for the ovarian cancer, particularly since the proband was not found to carry another mutation that could explain her personal history of cancer. Subsequent to the molecular studies being performed to determine the pathogenicity of the PALB2 duplication in this family, we offered genetic testing to the proband’s unaffected sisters through MSK-IMPACT and confirmed by both RT-PCR (Fig. 5a, 52-year-old and 57-year-old sisters) and MLPA analysis (data not shown). One of the unaffected sisters was found to carry the PALB2 duplication and the other unaffected sister does not carry the familial duplication. Since we have confirmed that the PALB2 duplication is pathogenic, we have spoken to the maternal aunt informing her of the amended results and recommended that her three sons, all of whom have daughters, meet with a local genetic counselor for an individualized risk assessment and their own genetic testing. At the time of submission, genetic testing results were pending for these family members.
Discussion
This study identified the exact breakpoints of the PALB2 exon 13 duplication and demonstrated a large duplication involving AluSg in intron 12 and AluSc8 in the downstream region of the PALB2 3′-UTR through a mechanism of Alu-mediated non-allelic homologous recombination (NAHR). Alu-mediated HR inversions, duplications, deletions, and other alterations have been implicated not only in copy number and structural variations within the human genome but also in numerous human genetic disorders [36, 37]. Similar to the BRCA1 gene known to bear Alu-mediated rearrangements [38, 39], analysis of PALB2 also shows a high density of Alu elements which favor the occurrence of non-allelic homologous recombination. Therefore, Alu-mediated NAHR could be responsible for other exonic deletions or duplications in PALB2. To date, six large deletions and duplications have been found in the PALB2 gene: three of which were mediated by Alu sequences and the rest were not yet characterized [5, 13, 17, 33].
We need to be cautious when interpreting truncating variants downstream of the most 3′ truncating variants established as pathogenic in the literature [40]. In our case, the large duplication disrupts normal splicing, introduces a premature stop codon at amino acid position 1151, and putatively produces a truncated PALB2 protein of 1150 amino acids instead of 1186 amino acids. PALB2 pathogenic truncating mutations affecting the very C-terminus of the protein have been reported to be associated with breast cancer and/or Fanconi anemia [14, 41]. Since the premature truncation caused by the reported PALB2 exon 13 duplication presumably removes the 36 amino acids at the C-terminus of the PALB2 protein, which is critical for its normal function, we speculate that the duplication would contribute to the ovarian cancer in the proband and the breast cancers that occurred in the family. Our co-segregation data further supports a role for this duplication in cancer predisposition in this family.
To date, PALB2 gene mutations have not been extensively studied in patients with ovarian cancer and its role in ovarian cancer predisposition is not well-established. Two out of 339 patients with ovarian cancer (0.6 %) were found to carry a PALB2 gene mutation (c.509_510delGA) in a study from central Poland [42]. One pathogenic PALB2 mutation (c.172_175delTTGT) was identified in 1/253 (0.4 %) ovarian cancer patients from the Volga-Ural region [43]. PALB2 promoter hypermethylation was detected in 4 of 53 sporadic ovarian tumor cell lines [44]. In our study, the proband who carries this PALB2 duplication was diagnosed with stage IV high-grade serous ovarian cancer. Her maternal aunt and one sister carrying the same alternation were diagnosed with breast cancer. It is known that germline loss-of-function mutations in PALB2 confer a hereditary predisposition to breast cancer [12, 45, 46] and pancreatic cancer [16, 33, 47]. Our results suggest that PALB2 deleterious mutations may also predispose to ovarian cancer. The presented results support the recently published suggestion that genetic screening for PALB2 protein-truncating alterations and large genome rearrangement mutations in the clinical setting may be considered for hereditary breast/ovarian cancer families [12, 32]. It is worthy to note that the proband’s mother, who was 80 years of age and cancer-free during these analyses, is an obligate carrier. This fits in with our understanding that the PALB2 gene is a moderate risk gene with reduced penetrance compared to that of a BRCA1/2 mutation carrier.
For the proband, identification of a pathologic germline PALB2 mutation identified her as a potential candidate for treatment with a DNA damage repair inhibitor such as a PARP inhibitor [48]. Had this variant not been identified (since she was BRCA-negative), PARPi may not have been a therapeutic option for treatment of her ovarian cancer. For the proband’s unaffected sister and any other female family members who do not carry the PALB2 duplication, these results provide reassurance regarding their breast cancer risk. Since the PALB2 gene has been shown to be associated with increased risks of developing breast cancer and the duplication segregates with the breast cancer diagnoses in this family, this is convincing data that the duplication accounts for the family history of breast cancer. Thus, unaffected relatives who do not carry the PALB2 duplication can follow general population guidelines for breast cancer surveillance. Had a cancer predisposition marker not been identified; this sister and other unaffected female family members would be subjected to unnecessary breast surveillance by way of annual magnetic resonance imaging (MRI) based on an unexplained significant family history of breast cancer. In addition, these negative results provide reassurance for any non-carrier’schildren in terms of future cancer risks. For the proband’s unaffected sister and any other female relatives who are found to carry the PALB2 duplication, knowing this result provides them with information so they can proceed with appropriate cancer surveillance. Various groups, including the National Comprehensive Cancer Network, recommend that females with PALB2 mutations undergo increased breast surveillance, including annual mammograms and breast MRI, with some suggesting that surveillance begin at age 30 [7]. Based on currently available data, risk-reducing BSO is not typically recommended for PALB2 mutation carriers, although given this family’s history of ovarian cancer, surgery could be supported. Further analyses of a larger population of PALB2 mutation carriers, including classification of novel PALB2 variants of unclear clinical significance, are highly desirable for precise evaluation of ovarian cancer penetrance to improve clinical management in individuals and families with PALB2 mutations.
References
Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, Livingston DM (2006) Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell 22(6):719–729. doi:10.1016/j.molcel.2006.05.022
Sy SM, Huen MS, Chen J (2009) PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci USA 106(17):7155–7160. doi:10.1073/pnas.0811159106
Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, Yu X (2009) PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol 19(6):524–529. doi:10.1016/j.cub.2009.02.018
Tischkowitz M, Xia B, Sabbaghian N, Reis-Filho JS, Hamel N, Li G, van Beers EH, Li L, Khalil T, Quenneville LA, Omeroglu A, Poll A, Lepage P, Wong N, Nederlof PM, Ashworth A, Tonin PN, Narod SA, Livingston DM, Foulkes WD (2007) Analysis of PALB2/FANCN-associated breast cancer families. Proc Natl Acad Sci USA 104(16):6788–6793. doi:10.1073/pnas.0701724104
Xia B, Dorsman JC, Ameziane N, de Vries Y, Rooimans MA, Sheng Q, Pals G, Errami A, Gluckman E, Llera J, Wang W, Livingston DM, Joenje H, de Winter JP (2007) Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat Genet 39(2):159–161. doi:10.1038/ng1942
Sy SM, Huen MS, Zhu Y, Chen J (2009) PALB2 regulates recombinational repair through chromatin association and oligomerization. J Biol Chem 284(27):18302–18310. doi:10.1074/jbc.M109.016717
Tung N, Domchek SM, Stadler Z, Nathanson KL, Couch F, Garber JE, Offit K, Robson ME (2016) Counselling framework for moderate-penetrance cancer-susceptibility mutations. Nat Rev Clin Oncol. doi:10.1038/nrclinonc.2016.90
Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, Reid S, Spanova K, Barfoot R, Chagtai T, Jayatilake H, McGuffog L, Hanks S, Evans DG, Eccles D, Breast Cancer Susceptibility C, Easton DF, Stratton MR (2007) PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet 39(2):165–167. doi:10.1038/ng1959
Hellebrand H, Sutter C, Honisch E, Gross E, Wappenschmidt B, Schem C, Deissler H, Ditsch N, Gress V, Kiechle M, Bartram CR, Schmutzler RK, Niederacher D, Arnold N, Meindl A (2011) Germline mutations in the PALB2 gene are population specific and occur with low frequencies in familial breast cancer. Hum Mutat 32(6):E2176–E2188. doi:10.1002/humu.21478
Southey MC, Teo ZL, Dowty JG, Odefrey FA, Park DJ, Tischkowitz M, Sabbaghian N, Apicella C, Byrnes GB, Winship I, Baglietto L, Giles GG, Goldgar DE, Foulkes WD, Hopper JL, kConFab for the Beast Cancer Family R (2010) A PALB2 mutation associated with high risk of breast cancer. Breast Cancer Res BCR 12(6):R109. doi:10.1186/bcr2796
Fernandes PH, Saam J, Peterson J, Hughes E, Kaldate R, Cummings S, Theisen A, Chen S, Trost J, Roa BB (2014) Comprehensive sequencing of PALB2 in patients with breast cancer suggests PALB2 mutations explain a subset of hereditary breast cancer. Cancer 120(7):963–967. doi:10.1002/cncr.28504
Casadei S, Norquist BM, Walsh T, Stray S, Mandell JB, Lee MK, Stamatoyannopoulos JA, King MC (2011) Contribution of inherited mutations in the BRCA2-interacting protein PALB2 to familial breast cancer. Cancer Res 71(6):2222–2229. doi:10.1158/0008-5472.CAN-10-3958
Janatova M, Kleibl Z, Stribrna J, Panczak A, Vesela K, Zimovjanova M, Kleiblova P, Dundr P, Soukupova J, Pohlreich P (2013) The PALB2 gene is a strong candidate for clinical testing in BRCA1- and BRCA2-negative hereditary breast cancer. Cancer Epidemiol Biomark Prev 22(12):2323–2332. doi:10.1158/1055-9965.EPI-13-0745-T
Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, Neveling K, Kelly P, Seal S, Freund M, Wurm M, Batish SD, Lach FP, Yetgin S, Neitzel H, Ariffin H, Tischkowitz M, Mathew CG, Auerbach AD, Rahman N (2007) Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet 39(2):162–164. doi:10.1038/ng1947
Hofstatter EW, Domchek SM, Miron A, Garber J, Wang M, Componeschi K, Boghossian L, Miron PL, Nathanson KL, Tung N (2011) PALB2 mutations in familial breast and pancreatic cancer. Fam Cancer 10(2):225–231. doi:10.1007/s10689-011-9426-1
Slater EP, Langer P, Niemczyk E, Strauch K, Butler J, Habbe N, Neoptolemos JP, Greenhalf W, Bartsch DK (2010) PALB2 mutations in European familial pancreatic cancer families. Clin Genet 78(5):490–494. doi:10.1111/j.1399-0004.2010.01425.x
Blanco A, de la Hoya M, Balmana J, Ramon y Cajal T, Teule A, Miramar MD, Esteban E, Infante M, Benitez J, Torres A, Tejada MI, Brunet J, Grana B, Balbin M, Perez-Segura P, Osorio A, Velasco EA, Chirivella I, Calvo MT, Feliubadalo L, Lasa A, Diez O, Carracedo A, Caldes T, Vega A (2012) Detection of a large rearrangement in PALB2 in Spanish breast cancer families with male breast cancer. Breast Cancer Res Treat 132(1):307–315. doi:10.1007/s10549-011-1842-2
Norquist BM, Harrell MI, Brady MF, Walsh T, Lee MK, Gulsuner S, Bernards SS, Casadei S, Yi Q, Burger RA, Chan JK, Davidson SA, Mannel RS, DiSilvestro PA, Lankes HA, Ramirez NC, King MC, Swisher EM, Birrer MJ (2016) Inherited mutations in women with ovarian carcinoma. JAMA Oncol 2(4):482–490. doi:10.1001/jamaoncol.2015.5495
Ramus SJ, Song H, Dicks E, Tyrer JP, Rosenthal AN, Intermaggio MP, Fraser L, Gentry-Maharaj A, Hayward J, Philpott S, Anderson C, Edlund CK, Conti D, Harrington P, Barrowdale D, Bowtell DD, Alsop K, Mitchell G, Group AS, Cicek MS, Cunningham JM, Fridley BL, Alsop J, Jimenez-Linan M, Poblete S, Lele S, Sucheston-Campbell L, Moysich KB, Sieh W, McGuire V, Lester J, Bogdanova N, Durst M, Hillemanns P, Ovarian Cancer Association C, Odunsi K, Whittemore AS, Karlan BY, Dork T, Goode EL, Menon U, Jacobs IJ, Antoniou AC, Pharoah PD, Gayther SA (2015) Germline Mutations in the BRIP1, BARD1, PALB2, and NBN genes in women with ovarian cancer. J Natl Cancer Inst. doi:10.1093/jnci/djv214
Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, Garofalo A, Gulati R, Carreira S, Eeles R, Elemento O, Rubin MA, Robinson D, Lonigro R, Hussain M, Chinnaiyan A, Vinson J, Filipenko J, Garraway L, Taplin ME, AlDubayan S, Han GC, Beightol M, Morrissey C, Nghiem B, Cheng HH, Montgomery B, Walsh T, Casadei S, Berger M, Zhang L, Zehir A, Vijai J, Scher HI, Sawyers C, Schultz N, Kantoff PW, Solit D, Robson M, Van Allen EM, Offit K, de Bono J, Nelson PS (2016) Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med 375(5):443–453. doi:10.1056/NEJMoa1603144
Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil CC, Rendi MH, Thornton A, Norquist BM, Casadei S, Nord AS, Agnew KJ, Pritchard CC, Scroggins S, Garcia RL, King MC, Swisher EM (2014) Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res 20(3):764–775. doi:10.1158/1078-0432.CCR-13-2287
Ashworth A (2008) A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol 26(22):3785–3790. doi:10.1200/JCO.2008.16.0812
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JH, de Bono JS (2009) Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361(2):123–134. doi:10.1056/NEJMoa0900212
Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, Wardley A, Mitchell G, Earl H, Wickens M, Carmichael J (2010) Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 376(9737):235–244. doi:10.1016/S0140-6736(10)60892-6
Kim G, Ison G, McKee AE, Zhang H, Tang S, Gwise T, Sridhara R, Lee E, Tzou A, Philip R, Chiu HJ, Ricks TK, Palmby T, Russell AM, Ladouceur G, Pfuma E, Li H, Zhao L, Liu Q, Venugopal R, Ibrahim A, Pazdur R (2015) FDA approval summary: Olaparib monotherapy in patients with deleterious germline BRCA-Mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin Cancer Res 21(19):4257–4261. doi:10.1158/1078-0432.CCR-15-0887
Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, De Greve J, Lubinski J, Shanley S, Messiou C, A’Hern R, Tutt A, Ashworth A, Stone J, Carmichael J, Schellens JH, de Bono JS, Kaye SB (2010) Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol 28(15):2512–2519. doi:10.1200/JCO.2009.26.9589
Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, Mitchell G, Fried G, Stemmer SM, Hubert A, Rosengarten O, Steiner M, Loman N, Bowen K, Fielding A, Domchek SM (2015) Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol 33(3):244–250. doi:10.1200/JCO.2014.56.2728
Buisson R, Dion-Cote AM, Coulombe Y, Launay H, Cai H, Stasiak AZ, Stasiak A, Xia B, Masson JY (2010) Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat Struct Mol Biol 17(10):1247–1254. doi:10.1038/nsmb.1915
Ghosh S, Sur S, Yerram SR, Rago C, Bhunia AK, Hossain MZ, Paun BC, Ren YR, Iacobuzio-Donahue CA, Azad NA, Kern SE (2014) Hypersensitivities for acetaldehyde and other agents among cancer cells null for clinically relevant Fanconi anemia genes. Am J Pathol 184(1):260–270. doi:10.1016/j.ajpath.2013.09.023
Park JY, Singh TR, Nassar N, Zhang F, Freund M, Hanenberg H, Meetei AR, Andreassen PR (2014) Breast cancer-associated missense mutants of the PALB2 WD40 domain, which directly binds RAD51C, RAD51 and BRCA2, disrupt DNA repair. Oncogene 33(40):4803–4812. doi:10.1038/onc.2013.421
Bleuyard JY, Buisson R, Masson JY, Esashi F (2012) ChAM, a novel motif that mediates PALB2 intrinsic chromatin binding and facilitates DNA repair. EMBO Rep 13(2):135–141. doi:10.1038/embor.2011.243
Southey MC, Teo ZL, Winship I (2013) PALB2 and breast cancer: ready for clinical translation! Appl Clin Genet 6:43–52. doi:10.2147/TACG.S34116
Tischkowitz MD, Sabbaghian N, Hamel N, Borgida A, Rosner C, Taherian N, Srivastava A, Holter S, Rothenmund H, Ghadirian P, Foulkes WD, Gallinger S (2009) Analysis of the gene coding for the BRCA2-interacting protein PALB2 in familial and sporadic pancreatic cancer. Gastroenterology 137(3):1183–1186. doi:10.1053/j.gastro.2009.06.055
Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, Chandramohan R, Liu ZY, Won HH, Scott SN, Brannon AR, O’Reilly C, Sadowska J, Casanova J, Yannes A, Hechtman JF, Yao J, Song W, Ross DS, Oultache A, Dogan S, Borsu L, Hameed M, Nafa K, Arcila ME, Ladanyi M, Berger MF (2015) Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn 17(3):251–264. doi:10.1016/j.jmoldx.2014.12.006
Oliver AW, Swift S, Lord CJ, Ashworth A, Pearl LH (2009) Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep 10(9):990–996. doi:10.1038/embor.2009.126
Sasaki M, Lange J, Keeney S (2010) Genome destabilization by homologous recombination in the germ line. Nat Rev Mol Cell Biol 11(3):182–195. doi:10.1038/nrm2849
Konkel MK, Batzer MA (2010) A mobile threat to genome stability: the impact of non-LTR retrotransposons upon the human genome. Semin Cancer Biol 20(4):211–221. doi:10.1016/j.semcancer.2010.03.001
Mazoyer S (2005) Genomic rearrangements in the BRCA1 and BRCA2 genes. Hum Mutat 25(5):415–422. doi:10.1002/humu.20169
Ticha I, Kleibl Z, Stribrna J, Kotlas J, Zimovjanova M, Mateju M, Zikan M, Pohlreich P (2010) Screening for genomic rearrangements in BRCA1 and BRCA2 genes in Czech high-risk breast/ovarian cancer patients: high proportion of population specific alterations in BRCA1 gene. Breast Cancer Res Treat 124(2):337–347. doi:10.1007/s10549-010-0745-y
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5):405–424. doi:10.1038/gim.2015.30
Peterlongo P, Catucci I, Pasquini G, Verderio P, Peissel B, Barile M, Varesco L, Riboni M, Fortuzzi S, Manoukian S, Radice P (2011) PALB2 germline mutations in familial breast cancer cases with personal and family history of pancreatic cancer. Breast Cancer Res Treat 126(3):825–828. doi:10.1007/s10549-010-1305-1
Dansonka-Mieszkowska A, Kluska A, Moes J, Dabrowska M, Nowakowska D, Niwinska A, Derlatka P, Cendrowski K, Kupryjanczyk J (2010) A novel germline PALB2 deletion in Polish breast and ovarian cancer patients. BMC Med Genet 11:20. doi:10.1186/1471-2350-11-20
Prokofyeva D, Bogdanova N, Bermisheva M, Zinnatullina G, Hillemanns P, Khusnutdinova E, Dork T (2012) Rare occurrence of PALB2 mutations in ovarian cancer patients from the Volga-Ural region. Clin Genet 82(1):100–101. doi:10.1111/j.1399-0004.2011.01824.x
Potapova A, Hoffman AM, Godwin AK, Al-Saleem T, Cairns P (2008) Promoter hypermethylation of the PALB2 susceptibility gene in inherited and sporadic breast and ovarian cancer. Cancer Res 68(4):998–1002. doi:10.1158/0008-5472.CAN-07-2418
Ding YC, Steele L, Kuan CJ, Greilac S, Neuhausen SL (2011) Mutations in BRCA2 and PALB2 in male breast cancer cases from the United States. Breast Cancer Res Treat 126(3):771–778. doi:10.1007/s10549-010-1195-2
Antoniou AC, Casadei S, Heikkinen T, Barrowdale D, Pylkas K, Roberts J, Lee A, Subramanian D, De Leeneer K, Fostira F, Tomiak E, Neuhausen SL, Teo ZL, Khan S, Aittomaki K, Moilanen JS, Turnbull C, Seal S, Mannermaa A, Kallioniemi A, Lindeman GJ, Buys SS, Andrulis IL, Radice P, Tondini C, Manoukian S, Toland AE, Miron P, Weitzel JN, Domchek SM, Poppe B, Claes KB, Yannoukakos D, Concannon P, Bernstein JL, James PA, Easton DF, Goldgar DE, Hopper JL, Rahman N, Peterlongo P, Nevanlinna H, King MC, Couch FJ, Southey MC, Winqvist R, Foulkes WD, Tischkowitz M (2014) Breast-cancer risk in families with mutations in PALB2. N Engl J Med 371(6):497–506. doi:10.1056/NEJMoa1400382
Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, Lin JC, Palmisano E, Brune K, Jaffee EM, Iacobuzio-Donahue CA, Maitra A, Parmigiani G, Kern SE, Velculescu VE, Kinzler KW, Vogelstein B, Eshleman JR, Goggins M, Klein AP (2009) Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science 324(5924):217. doi:10.1126/science.1171202
Grisham RN, Hyman DM, Iyer G (2014) Targeted therapies for treatment of recurrent ovarian cancer. Clin Adv Hematol Oncol 12(3):158–162
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
David M. Hyman has received research funding from LOXO, PUMA, and AstraZeneca. The rest authors declare no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Ciyu Yang and Angela G. Arnold have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Yang, C., Arnold, A.G., Trottier, M. et al. Characterization of a novel germline PALB2 duplication in a hereditary breast and ovarian cancer family. Breast Cancer Res Treat 160, 447–456 (2016). https://doi.org/10.1007/s10549-016-4021-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-016-4021-7