
Abstract
Bone metastases from breast cancer are common, causing significant morbidity. Preclinical data of dasatinib, an oral small molecule inhibitor of multiple oncogenic tyrosine kinases, suggested efficacy in tumor control and palliation of bone metastases in metastatic breast cancer (MBC). This clinical trial aimed to determine whether treatment with either of 2 dose schedules of dasatinib results in a progression-free survival (PFS) >50 % at 24 weeks in bone metastasis predominant MBC, to evaluate the toxicity of the 2 dosing regimens, and explore whether treatment results in decreased serum bone turnover markers and patient-reported “worst pain.” Subjects with bone metastasis predominant MBC were randomly assigned to either 100 mg of dasatinib once daily, or 70 mg twice daily, with treatment continued until time of disease progression or intolerable toxicity. Planned accrual was 40 patients in each arm. The primary trial endpoint was PFS, defined as time from registration to progression or death due to any cause. Median PFS for all eligible patients (79) was 12.6 weeks (95 % CI 9.1–16.7). Neither cohort met the threshold for further clinical interest. There were no significant differences in PFS by randomized treatment arm (p = 0.85). Toxicity was similar in both cohorts, with no clear trend in serum biomarkers of bone turnover or patient-reported pain. Dasatinib was ineffective in controlling bone-predominant MBC in a patient population, unselected by molecular markers. Further study of dasatinib in breast cancer should not be pursued unless performed in molecularly determined patient subsets, or rational combinations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Breast cancer is a common cause of morbidity and mortality in the United States and worldwide. Despite optimal use of adjuvant anthracycline and taxane chemotherapy, breast cancer recurrence occurs in up to 30 % of high-risk or node-positive patients within 8 years of diagnosis, resulting in 21 % breast cancer mortality [21]. In the United States, approximately 40,000 deaths are attributable to breast cancer each year, predominantly related to distant metastatic disease. One of the most common locations of metastatic relapse in breast cancer, regardless of cancer subtype, is in bone [23], and these are clinically important because skeletal related events from bony metastases lead to significant morbidity and mortality.
Dasatinib is an orally available tyrosine kinase inhibitor (TKI) that inhibits multiple oncogenic tyrosine kinases including BCR-ABL, SRC family kinases (SFKs), platelet derived growth factor (PDGF), and c-KIT. It is approved for use in Philadelphia chromosome-positive (Ph+) chronic myelogenous leukemia (CML) and acute lymphoblastic leukemia (ALL), and is also relevant in gastrointestinal stromal tumors (GIST; off-label use). SFK’s have a role in multiple pathways of signal transduction, many of which are relevant to breast cancer, includes the following: (1) osteoclast proliferation, survival, and resorptive function; (2) transduction of cell proliferative and antiapoptotic signals; (3) angiogenesis and vascular permeability; and (4) cell motility and migration, cell–cell adhesion, anchorage independence, and other cytoskeletal events which characterize the invasive and metastatic phenotype. [12, 19, 22] Substantial preclinical data support the role of SFKs in human breast cancer, including the finding of elevated c-Src in breast tumors, as well as preclinical studies show that 7/23 breast cancer cell lines were sensitive to dasatinib (IC50 <1 μM). Also, c-Src plays a role in mitogenic signaling via ER and PgR (in the presence or absence of ligand), as well as through EGFR, HER2, and p130Cas, which are found to be upregulated in breast cancer models of antiestrogen resistance. These preclinical effects of dasatinib suggest that it has potential for tumor control and palliation of bone metastases in patients with bone-predominant breast cancer metastasis.
Materials and methods
Study design
The study consisted of two parallel Phase II trials of two different doses and schedules of dasatinib. The original approved dose of dasatinib for CML was 70 mg twice daily (BID), and at the time of initiation of this study, exploration of dosing at 100 mg daily (QD) was just beginning (now standard). Therefore, this trial tested both 70 mg BID and 100 mg QD dosing in the bone metastasis predominant breast cancer population. Planned accrual was forty patients in each arm over 78 weeks (1.5 years) with an additional 24 weeks of follow-up.
The study was performed within SWOG, a cooperative group within the National Clinical Trials Network. The participating sites obtained institutional review board’s approval. Informed, written consent was obtained from all patients prior to enrollment, and the study was registered on ClinicalTrials.gov Identifier: NCT00410813. Subjects were randomized to either Arm 1: dasatinib, 100 mg by mouth QD, or Arm 2: dasatinib, 70 mg by mouth BID. Randomization was stratified by use of trastuzumab at the time of registration. Treatment was given continuously until disease progression, unacceptable toxicity, symptomatic deterioration, or treatment delay in excess of 4 weeks.
The primary trial endpoint was progression-free survival (PFS), defined as time from registration to progression or death due to any cause. The progression was determined locally without the central review. Secondary trial endpoints included the following: (1) Response Evaluation Criteria in Solid Tumors (RECIST) response (in those with measurable disease); (2) mucin-1 (MUC-1) antigen response; (3) circulating tumor cell (CTC) response; (4) incidence of Grade 3–4 toxicity; (5) change in serum bone turnover markers; and (6) change in the primary measure “worst pain” on the Brief Pain Inventory (BPI) at 8, 16, and 24 weeks. [8]
Patient population
Eligible patients were women or men with bone-predominant metastatic breast cancer. Bone-predominant breast cancer was defined as the presence of one or more bone metastases with or without nonbone (visceral or soft tissue) disease. The number of documented bone lesions was greater than or equal to the number of RECIST measurable visceral target lesions, and visceral disease was not causing symptoms to reduce the performance status. Eligible patients satisfied either criterion a or b: a. Measurable disease by RECIST criteria; b. Nonmeasurable disease only, with rising serum CA 15-3, CA 27-29, CEA, or CA-125 documented by two measurements taken at least 14 days apart with the more recent measurement within 42 days prior to registration. The second serum marker value was greater than the institution’s upper limit of normal and had at least a 20 % increase over the earlier measurement.
Patients may have had 0 or 1 prior cytotoxic chemotherapy regimens for metastatic disease. Patients whose tumors were ER and/or PgR positive must have experienced progression of disease on at least one hormonal therapy in the metastatic setting.
Patients may have had previously treated and currently asymptomatic brain or CNS metastasis with radiation completed at least 8 weeks prior to registration. Patients could not receive concurrent antineoplastic therapy for breast cancer while on protocol treatment; one exception was that patients with HER2-positive breast cancer, who were on trastuzumab for at least 12 weeks could continue to take trastuzumab concurrently with dasatinib.
Patients who were on bisphosphonates must not have had a dose of the bisphosphonate within 3 weeks prior to the enrollment. Patients and their physicians agreed to hold bisphosphonates for the duration of study treatment in order to limit interaction between concurrent bisphosphonate administration and markers of bone metabolism. Concurrent therapy with RANKL inhibitors was not allowed.
Endpoint evaluations
Monitoring of toxicity occurred at a clinic visit on Weeks 4, 8, 16, 24, and every 8 weeks thereafter, with reporting of serious adverse events (SAE) by the NCI Common Terminology Criteria for Adverse Events (CTCAE Version 4.0). CTCAE Version 3.0 was used for routine toxicity reporting. Efficacy was evaluated using CT scans, bone scans, and other assessments as required by RECIST 1.0 criteria. Each individual site was responsible for response and progression assessment; central review was not performed.
Blood biomarker evaluation
Serum biomarkers were tested centrally, and were not used for treatment decision making. Blood was drawn at the sites at baseline and Weeks 4, 8, 16, and 24 after registration and shipped to the SWOG Solid Tumor Tissue bank for serum separation and storage. In addition, sites obtained the whole blood via one Cell Save™ tube at the same timepoints, which was shipped to Janssen Diagnostics laboratories for CTC enumeration with CellSearch® (Janssen Diagnostics, Raritan, New Jersey).
CA 15-3 and CEA measurements were performed in the University of Michigan clinical laboratories, using standard laboratory methods. Serum bone biomarker expression was analyzed using ELISA kits according to the manufacturers’ instructions for expression of vascular endothelial growth factor (R&D Systems Inc., Minneapolis, MN), interleukin-6 (R&D Systems Inc.), dickkopf 1 (R&D Systems Inc.), bone alkaline phosphatase (IDS Inc., Gaithersburg, MD), tartrate-resistant acid phosphatase 5b (IDS Inc.), osteocalcin (IDS Inc.), soluble receptor activator of nuclear factor kappa-B ligand (ALPCO, Salem, NH), osteoprotegerin (ALPCO), and N-terminal telopeptide (Alere Inc., Waltham, MA). Serum samples were analyzed in duplicate.
Statistical methods
Analysis of PFS
Estimates of PFS in this patient population selected for bone-dominant disease are difficult to obtain. We considered historical trials of second- and third-line hormonal therapy, assuming that these trials may include a preponderance of patients with “bone-predominant” disease and estimated that between 40–50 % of such patients treated with aromatase inhibitors or fulvestrant as second- or third-line hormonal therapy will have stable disease at 24 weeks [6, 7]. Therefore, the PFS target was chosen to be 50 % at 24 weeks, and the null hypothesis of PFS was 30 % at 24 weeks.
Planned accrual was forty patients in each arm over 78 weeks (1.5 years) with an additional 24 weeks of follow-up. This design had power of 0.90 for each arm given a Type I error rate of 0.025 (1-sided). If PFS was favorable for both arms, the arm with the greatest tolerability would be the dose/schedule chosen to proceed to a Phase III study. A comparison of efficacy between the two arms was a secondary analysis conducted using a log-rank test for PFS and OS with estimation of the hazard ratio from Cox regression.
Secondary trial endpoints
Only patients with measurable disease at baseline were evaluated for RECIST response. Patients analyzed for overall MUC-1 antigen response, defined as previously described, [24] included those whose initial baseline MUC-1 antigen level was >2 X ULN. Patients analyzed for CTC response rate included those who had elevated CTCs (≥5 cells/7.5 ml) at baseline. CTC response was defined as the percentage of patients with initially elevated CTCs, whose CTC level drops to <5. The RECIST, MUC-1, and CTC response rates were estimated with exact 95 % two-sided confidence intervals using standard methods based on the binomial distribution. The analysis of changes in exploratory serum biomarkers associated with bone was descriptive in nature and based on all patients who started trial therapy.
The toxicity was recorded at clinic visits scheduled at Weeks 4, 8, 16, 24, and then every 8 weeks thereafter. With a minimum of 25 patients per treatment arm, any toxicity occurring in >10 % of patients had a 93 % probability of being observed.
The Brief Pain Inventory (BPI) [8] is a patient-reported measure of pain including severity and impact of pain on daily functioning. The BPI was administered at randomization and weeks 8, 16, and 24 in either English or Spanish. Here we consider worst pain as a measure of severity and the average interference score. Additional exploratory analyses from baseline to Week 8, Week 16, and Week 24 were also performed.
Results
Consort diagram
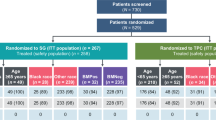
This study was opened to accrual on March 15, 2007 and closed on December 15, 2010, after reaching full accrual with 85 registrations. Six patients were found to be ineligible for the following reasons: no progression on hormonal therapy (2 patients); prior chemotherapy less than three weeks prior to registration; pleural effusion at baseline; no 20 % increase in CA 27–29; and not bone-predominant disease. (Fig. 1)

Flow diagram of the progress through the randomized trial
Description of enrolled population
Patients in the two arms were similar, as shown in Table 1. The median age was 60 years, 88.6 % of participants had hormone receptor positive (HR+) tumors, and 6.3 % had HER2-positive tumors. As per eligibility, all HR+ patients must have received at least one line of endocrine therapy in the metastatic setting, and only 0–1 prior lines of chemotherapy in the metastatic setting.
Analysis of PFS
All patients have had progression of disease or have died so there was no censoring of the primary endpoint. The overall median PFS was 10.3 weeks (95 % CI 8.4–16.7) in Arm 1 and 15.3 weeks (95 % CI 8.7–20.1) in Arm 2 (Fig. 2). Neither Arm met the a priori criterion of interest (50 % PFS at 24 weeks): Arm 1 29 % (95 % CI 16–43 %); Arm 2 34 % (95 % CI 20–49 %). There were no apparent differences in PFS between the two arms (log-rank p = 0.85), with HR for Arm 2 versus Arm 1 = 1.04 (95 % CI 0.66–1.65). Combining the Arm 1 and Arm 2 together, we observed an overall median PFS = 12.6 weeks (95 % CI 9.1–16.7).

Kaplan–Meier estimates of progression-free and overall survival in the patient population, by arm of enrollment
Deaths have been recorded for 64 of 79 patients. Median overall survival was observed at 99.7 weeks (95 % CI 79.3–137.3), and there were no apparent differences between the randomized arms (Fig. 2): log-rank p = 0.61; HR = 0.88 (95 % CI 0.53–1.44).
RECIST response
Of 25 patients with measurable disease, only one patient had a partial response, so this outcome was not evaluated further.
Toxicity
Toxicities are summarized in Table 2. Among 79 patients evaluated for toxicity, three (one on Arm 1; two on Arm 2) reported Grade 4 toxicities. The toxicities included thrombocytopenia, pulmonary hypertension, and hypokalemia. Twenty-seven additional patients (9 on Arm 1; 18 on Arm 2) experienced Grade 3 toxicities as maximum degree. Seventeen patients discontinued protocol treatment early due to adverse events.
Blood biomarkers
CTC response evaluation was limited to those patients who had measurements performed at baseline and at a subsequent time point. Due to diminishing numbers of samples collected at later time points, we reported only the analysis of those who had CTCs enumerated at baseline and 4 weeks. In total, 60 patients had CTCs measured at baseline, (33 < 5 cells/7.5 mg; 27 ≥ 5 cells/7.5 mg) and 51 had measurements at week 4 (31 < 5 cells/7.5 mg; 20 ≥ 5 cells/7.5 mg). Only 41 patients had measurements at both time points. Within this subset, 4 of 17 patients had reduction in CTCs from high to low (CTC response rate of 24 % (95 % CI 7–50 %). As shown in other trials, CTC >5 cells/7.5 ml was negatively prognostic for both PFS and OS. For those with elevated CTC’s at baseline, the PFS HR = 2.27 (95 % CI 1.29–3.98) and OS HR = 2.27 (95 % CI 1.22–4.21). Of 38 individuals with high CA15-3 at baseline, 37 remained high at 4 weeks (mRR = 3 %). All 26 individuals with high CEA at baseline remained high at 4 weeks.
The analysis of serum biomarkers associated with bone focused on baseline, 4 week, and 8 week biomarkers. Observations at 16 and 24 weeks had very small numbers (n = 21 and 12, respectively) because of patient dropout due to disease progression and the estimated mean was very unstable, so are not included. Figure 3 depicts the mean values (and 95 % CI) of the markers at baseline, 4, and 8 weeks. No significant trend in biomarker levels over the three time points was observed. The baseline marker was dichotomized at the median and PFS compared for low or high values at baseline. There was a trend toward worse prognosis in patients with a baseline IL-6 level above the mean, but this did not reach statistical significance (data not shown).

Bone turnover markers (box plots) at 0, 4, and 8 weeks; combined Arm 1 and Arm 2
Brief pain inventory
All 79 patients completed the Brief Pain Inventory at baseline, but the numbers were reduced at 8, 16, and 24 weeks (n = 68; 37; and 41, respectively). Pain severity and pain interference did not differ significantly between the two treatment arms at 8, 16, or 24 weeks. Pain severity did not differ across the time points. Mean pain severity was 3.37, 3.82, 3.06, and 3.73 at 0, 8, 16, and 24 weeks based on a 10-point pain severity score (10 highest). Overall interference was significantly worse at 8 weeks compared to baseline (p = 0.005), but not at weeks 16 or 24. Mean interference levels were 2.05, 2.71, 1.71, and 2.34 at 0, 8, 16, and 24 weeks on a 10-point scale. Therefore, any interference due to pain induced by the start of treatment was transitory.
Discussion
Our original hypothesis was that dasatinib, a SFK would alter the course of bone-predominant breast cancer by inhibiting osteoclasts, reducing tumor cell invasiveness, reducing cellular proliferation, and causing apoptosis. However, our trial results did not support this hypothesis.
As the measurement of response to the treatment in a bone metastasis predominant population can be challenging, our trial design included multiple methods of evaluation for anticancer activity: including progression-free survival; RECIST response rate [26]; and serial measurement of MUC-1 antigens [24, 28], CTC’s [10, 25, 32], and patient-reported measures of pain utilizing the Brief Pain Inventory. [8] Although diminishing numbers of patients at later time points resulted in few measurements after 16 weeks, none of the measures pointed toward a significant benefit from treatment. Given that multiple surrogate measures for efficacy were negative, it is unlikely that the study falsely missed a positive signal.
In addition, based on our exploratory analysis of serum markers of bone turnover, we could not confirm significant activity of dasatinib in modulating either bone resorption or bone deposition in this patient population. Dasatinib was predicted to have significant effect on osteoclast proliferation and survival through its effects on RANKL [16]. We studied a panel of collagen markers of bone turnover based on abundant evidence that these are raised in the blood and urine of a high percentage of patients with progressive skeletal metastases. [4, 9, 11, 14, 17, 29–31, 33] Although we observed a baseline elevation in bone metabolism marker levels in these patients with bone metastases [15, 29], treatment with dasatinib did not have a discernable effect on these markers. Antiresorptive agents, such as the bisphosphonate class of compounds and denosumab, a RANKL inhibitor, elicit a rapid and sustained suppression of these bone collagen breakdown markers in patients with skeletal metastases who are responding favorably to this form of therapy. [5, 13, 27, 31] A Phase II clinical trial of dasatinib in combination with zoledronic acid has suggested that a subset of patients with low grade, HR(+), and high baseline NTX levels may be more likely to experience a response to this combination regimen, which may warrant additional study [18].
Dasatinib has been evaluated as a single agent in other solid tumors, with low-reported RECIST response rates of 5–6 %. [2] In breast cancer specifically, TKI’s have had limited activity when used as single agents in patients unselected for molecular aberrations. In contrast, use of TKI’s can be effective when used in molecularly targeted breast cancer populations; lapatinib has demonstrated efficacy in patients with HER2 positive breast cancer [1], and neratinib has been suggested to have activity not only in HER2-positive breast cancer, but also in patients with tumors that harbor HER2 mutations [3]. Interestingly, in our trial there was a single patient with HER2-positive disease. This patient experienced a long progression-free survival for about 20 months. Although this patient’s response evaluation is confounded by the concurrent use of trastuzumab, it is possible that dasatinib had some additive activity with the monoclonal antibody therapy. This hypothesis is supported by some preclinical models and is currently being explored in clinical trials of combined therapy [20].
In summary, further exploration of dasatinib as a single agent in bone-predominant metastatic breast cancer, unselected by additional predictive factors, is not warranted. Future evaluation of dasatinib in breast cancer could be pursued in molecularly determined patient subsets or in rational combinations.
References
Amir E, Ocana A, Seruga B, Freedman O, Clemons M (2010) Lapatinib and HER2 status: results of a meta-analysis of randomized phase III trials in metastatic breast cancer. Cancer Treat Rev 36:410–415. doi:10.1016/j.ctrv.2009.12.012
Araujo J, Logothetis C (2010) Dasatinib: a potent SRC inhibitor in clinical development for the treatment of solid tumors. Cancer Treat Rev 36:492–500. doi:10.1016/j.ctrv.2010.02.015
Ben-Baruch NE, Bose R, Kavuri SM, Ma CX, Ellis MJ (2015) HER2-Mutated Breast cancer responds to treatment with single-agent neratinib, a second-generation HER2/EGFR tyrosine kinase inhibitor. J Natl Compr Canc Netw 13:1061–1064
Blomqvist C, Risteli L, Risteli J, Virkkunen P, Sarna S, Elomaa I (1996) Markers of type I collagen degradation and synthesis in the monitoring of treatment response in bone metastases from breast carcinoma. Br J Cancer 73:1074–1079
Body JJ, Lipton A, Gralow J, Steger GG, Gao G, Yeh H, Fizazi K (2010) Effects of denosumab in patients with bone metastases with and without previous bisphosphonate exposure. J Bone Miner Res 25:440–446. doi:10.1359/jbmr.090810
Buzdar A, Douma J, Davidson N, Elledge R, Morgan M, Smith R, Porter L, Nabholtz J, Xiang X, Brady C (2001) Phase III, multicenter, double-blind, randomized study of letrozole, an aromatase inhibitor, for advanced breast cancer versus megestrol acetate. J Clin Oncol 19:3357–3366
Buzdar A, Jonat W, Howell A, Jones SE, Blomqvist C, Vogel CL, Eiermann W, Wolter JM, Azab M, Webster A, Plourde PV (1996) Anastrozole, a potent and selective aromatase inhibitor, versus megestrol acetate in postmenopausal women with advanced breast cancer: results of overview analysis of two phase III trials. Arimidex Study Group. J Clin Oncol 14:2000–2011
Cleeland CS (2006) The measurement of pain from metastatic bone disease: capturing the patient’s experience. Clin Cancer Res 12:6236s–6242s. doi:10.1158/1078-0432.ccr-06-0988
Costa L, Demers LM, Gouveia-Oliveira A, Schaller J, Costa EB, de Moura MC, Lipton A (2002) Prospective evaluation of the peptide-bound collagen type I cross-links N-telopeptide and C-telopeptide in predicting bone metastases status. J Clin Oncol 20:850–856
Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LW, Hayes DF (2004) Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med 351:781–791. doi:10.1056/NEJMoa040766
Demers LM, Costa L, Chinchilli VM, Gaydos L, Curley E, Lipton A (1995) Biochemical markers of bone turnover in patients with metastatic bone disease. Clin Chem 41:1489–1494
Frame MC (2002) Src in cancer: deregulation and consequences for cell behaviour. Biochim Biophys Acta 1602:114–130
Hortobagyi GN, Theriault RL, Porter L, Blayney D, Lipton A, Sinoff C, Wheeler H, Simeone JF, Seaman J, Knight RD (1996) Efficacy of pamidronate in reducing skeletal complications in patients with breast cancer and lytic bone metastases. Protocol 19 Aredia Breast Cancer Study Group. N Engl J Med 335:1785–1791. doi:10.1056/nejm199612123352401
Houze P, Bellik B, Extra JM, Bouro F, Bousquet B (1999) Urinary carboxyterminal telopeptide of collagen I as a potential marker of bone metastases chemotherapy monitoring in breast cancer. Clin Chim Acta 281:77–88
Jablonka F, Schindler F, Lajolo PP, Pinczowski H, Fonseca FL, Barbieri A, Massonetto LH, Katto FT, Del Giglio A (2009) Serum cross-linked n-telopeptides of type 1 collagen (NTx) in patients with solid tumors. Sao Paulo Med J 127:19–22
Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, Khoo W, Wakeham A, Dunstan CR, Lacey DL, Mak TW, Boyle WJ, Penninger JM (1999) OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 397:315–323. doi:10.1038/16852
Lipton A, Costa L, Ali SM, Demers LM (2001) Bone markers in the management of metastatic bone disease. Cancer Treat Rev 27:181–185. doi:10.1053/ctrv.2000.0212
Mitri ZI, Nanda R, Blackwell KL, Costelloe C, Hood I, Brewster AM, Ibrahim NK, HigginbothamKoenig K, Hortobagyi GN, Van Poznak CH, Rimawi MF, Moulder SL (2015) TBCRC-010: Phase I/II study of dasatinib in combination with zoledronic acid (ZA) for the treatment of breast cancer bone metastasis (MBC-bone). NCT00566618. J Clin Oncol 33:11080
Miyazaki T, Sanjay A, Neff L, Tanaka S, Horne WC, Baron R (2004) Src kinase activity is essential for osteoclast function. J Biol Chem 279:17660–17666. doi:10.1074/jbc.M311032200
Montero JC, Seoane S, Ocana A, Pandiella A (2011) Inhibition of SRC family kinases and receptor tyrosine kinases by dasatinib: possible combinations in solid tumors. Clin Cancer Res 17:5546–5552. doi:10.1158/1078-0432.ccr-10-2616
Peto R, Davies C, Godwin J, Gray R, Pan HC, Clarke M, Cutter D, Darby S, McGale P, Taylor C, Wang YC, Bergh J, Di Leo A, Albain K, Swain S, Piccart M, Pritchard K (2012) Comparisons between different polychemotherapy regimens for early breast cancer: meta-analyses of long-term outcome among 100,000 women in 123 randomised trials. Lancet 379:432–444. doi:10.1016/s0140-6736(11)61625-5
Playford MP, Schaller MD (2004) The interplay between Src and integrins in normal and tumor biology. Oncogene 23:7928–7946. doi:10.1038/sj.onc.1208080
Savci-Heijink CD, Halfwerk H, Hooijer GK, Horlings HM, Wesseling J, van de Vijver MJ (2015) Retrospective analysis of metastatic behaviour of breast cancer subtypes. Breast Cancer Res Treat 150:547–557. doi:10.1007/s10549-015-3352-0
Schott AF, Barlow WE, Albain KS, Chew HK, Wade JL 3rd, Lanier KS, Lew DL, Hayes DF, Gralow JR, Livingston RB, Hortobagyi GN (2012) Phase II trial of simple oral therapy with capecitabine and cyclophosphamide in patients with metastatic breast cancer: SWOG S0430. Oncologist 17:179–187. doi:10.1634/theoncologist.2011-0235
Smerage JB, Barlow WE, Hortobagyi GN, Winer EP, Leyland-Jones B, Srkalovic G, Tejwani S, Schott AF, O’Rourke MA, Lew DL, Doyle GV, Gralow JR, Livingston RB, Hayes DF (2014) Circulating tumor cells and response to chemotherapy in metastatic breast cancer: SWOG S0500. J Clin Oncol 32:3483–3489. doi:10.1200/jco.2014.56.2561
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European organization for research and treatment of cancer, national cancer institute of the United States, National cancer institute of Canada. J Natl Cancer Inst 92:205–216
Theriault RL, Lipton A, Hortobagyi GN, Leff R, Gluck S, Stewart JF, Costello S, Kennedy I, Simeone J, Seaman JJ, Knight RD, Mellars K, Heffernan M, Reitsma DJ (1999) Pamidronate reduces skeletal morbidity in women with advanced breast cancer and lytic bone lesions: a randomized, placebo-controlled trial. protocol 18 Aredia breast cancer study group. J Clin Oncol 17:846–854
Van Poznak C, Somerfield MR, Bast RC, Cristofanilli M, Goetz MP, Gonzalez-Angulo AM, Hicks DG, Hill EG, Liu MC, Lucas W, Mayer IA, Mennel RG, Symmans WF, Hayes DF, Harris LN (2015) Use of biomarkers to guide decisions on systemic therapy for women with metastatic breast cancer: American society of clinical oncology clinical practice guideline. J Clin Oncol 33:2695–2704. doi:10.1200/jco.2015.61.1459
Vinholes J, Coleman R, Lacombe D, Rose C, Tubiana-Hulin M, Bastit P, Wildiers J, Michel J, Leonard R, Nortier J, Mignolet F, Ford J (1999) Assessment of bone response to systemic therapy in an EORTC trial: preliminary experience with the use of collagen cross-link excretion. European organization for research and treatment of cancer. Br J Cancer 80:221–228. doi:10.1038/sj.bjc.6690506
Vinholes J, Guo CY, Purohit OP, Eastell R, Coleman RE (1997) Evaluation of new bone resorption markers in a randomized comparison of pamidronate or clodronate for hypercalcemia of malignancy. J Clin Oncol 15:131–138
Walls J, Assiri A, Howell A, Rogers E, Ratcliffe WA, Eastell R, Bundred NJ (1999) Measurement of urinary collagen cross-links indicate response to therapy in patients with breast cancer and bone metastases. Br J Cancer 80:1265–1270. doi:10.1038/sj.bjc.6690496
Witzig TE, Bossy B, Kimlinger T, Roche PC, Ingle JN, Grant C, Donohue J, Suman VJ, Harrington D, Torre-Bueno J, Bauer KD (2002) Detection of circulating cytokeratin-positive cells in the blood of breast cancer patients using immunomagnetic enrichment and digital microscopy. Clin Cancer Res 8:1085–1091
Yoshida K, Sumi S, Arai K, Koga F, Umeda H, Hosoya Y, Honda M, Yano M, Moriguchi H, Kitahara S (1997) Serum concentration of type I collagen metabolites as a quantitative marker of bone metastases in patients with prostate carcinoma. Cancer 80:1760–1767
Funding
This work was supported by the National Institutes of Health/National Cancer Institute/National Clinical Trials Network grants CA180888, CA180819, CA180801, CA180834, CA180846; National Institutes of Health/National Cancer Institute Community Oncology Research Program grants CA189971, CA189830, CA189872, CA189954, CA189952, CA189858, CA189856, CA189817, CA190002; National Institutes of Health/National Cancer Institute legacy Grants CA35158, CA35119, CA11083, CA76448, CA04919, CA46282, CA76447, CA22433, CA16385; and in part by the Bristol-Myers Squibb and Janssen Diagnostics Corporation. The content is solely the responsibility of the authors and does not necessarily represent the views of the National Institute of Health or Bristol-Myers Squibb and Janssen Diagnostics Corporation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
William E. Barlow, Evan T. Keller, Gabriel N. Hortobagyi, Jill M. Keller, Danika L. Lew, Carol M. Moinpour, Philip A. Dy, Anne F. Schott declare no conflict of interest; Catherine H. Van Poznak reports sponsorship of clinical trial to institution by the Bayer Pharmaceuticals; Daniel F. Hayes reports Stock Ownership: Oncimmune LLC, De Soto, KS, USA—stock options (7/20/09), Inbiomotion, Barcelona, Spain—stock options (10/22/12); Lecture/Honorarium: Visiting Consultant for Lilly Oncology, Indianapolis, IN (11/7/14); Sponsored Clinical Research—Principle or co-Investigator: Merrimack Pharmaceuticals, Inc. (Parexel Intl Corp) (01/24/15-02/02/20), Eli Lilly Company (06/19/15-04/30/19), Janssen R&D, LLC (Johnson & Johnson) (12/23/08-04/28/18), Puma Biotechnology, Inc., (subcontract Wash Univ St. Louis to Univ Mich) (07/19/13-07/31/18), Pfizer (07/22/13-07/14/18), Astra Zeneca (11/01/14-10/31/16), Astra Zeneca (02/06/15-02/05/16; Royalties from licensed technology: Janssen R&D, LLC (Johnson & Johnson) (08/01/14); Patents: Title: A method for predicting progression-free and overall survival at each follow-up timepoint during therapy of metastatic breast cancer patients using circulating tumor cells. Filed 14 Mar 2005 with the European Patent Office, the Netherlands. Application No./Patent No. 05725638.0-1223-US2005008602. Applicant/Proprietor: Immunicon Corporation. Dr. Daniel F. Hayes is designated as inventor/co-inventor; Title: Diagnosis and Treatment of Breast Cancer. Patent No.: US 8,790,878 B2. Date of Patent: Jul. 29, 2014. Applicant Proprietor: University of Michigan. Dr. Daniel F. Hayes is designated as inventor/co-inventor; Title: Circulating Tumor Cell Capturing Techniques and Devices. Patent No.: US 8,951,484 B2. Date of Patent: Feb. 10, 2015. Applicant Proprietor: University of Michigan. Dr. Daniel F. Hayes is designated as inventor/co-inventor.
Rights and permissions
About this article

Cite this article
Schott, A.F., Barlow, W.E., Van Poznak, C.H. et al. Phase II studies of two different schedules of dasatinib in bone metastasis predominant metastatic breast cancer: SWOG S0622. Breast Cancer Res Treat 159, 87–95 (2016). https://doi.org/10.1007/s10549-016-3911-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-016-3911-z