
Abstract
Hyperphenylalaninemia (HPA) caused by hepatic phenylalanine hydroxylase (PAH) deficiency has severe consequences on brain monoamine neurotransmitter metabolism. We have studied monoamine neurotransmitter status and the effect of tetrahydrobiopterin (BH4) treatment in Pahenu1/enu2 (ENU1/2) mice, a model of partial PAH deficiency. These mice exhibit elevated blood L-phenylalanine (L-Phe) concentrations similar to that of mild hyperphenylalaninemia (HPA), but brain levels of L-Phe are still ~5-fold elevated compared to wild-type. We found that brain L-tyrosine, L-tryptophan, BH4 cofactor and catecholamine concentrations, and brain tyrosine hydroxylase (TH) activity were normal in these mice but that brain serotonin, 5-hydroxyindolacetic acid (5HIAA) and 3-methoxy-4-hydroxyphenylglycol (MHPG) content, and brain TH protein, as well as tryptophan hydroxylase type 2 (TPH2) protein levels and activity were reduced in comparison to wild-type mice. Parenteral L-Phe loading conditions did not lead to significant changes in brain neurometabolite concentrations. Remarkably, enteral BH4 treatment, which normalized brain L-Phe levels in ENU1/2 mice, lead to only partial recovery of brain serotonin and 5HIAA concentrations. Furthermore, indirect evidence indicated that the GTP cyclohydrolase I (GTPCH) feedback regulatory protein (GFRP) complex may be a sensor for brain L-Phe elevation to ameliorate the toxic effects of HPA. We conclude that BH4 treatment of HPA toward systemic L-Phe lowering reverses elevated brain L-Phe content but the recovery of TPH2 protein and activity as well as serotonin levels is suboptimal, indicating that patients with mild HPA and mood problems (depression or anxiety) treated with the current diet may benefit from supplementation with BH4 and 5-OH-tryptophan.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Classical hyperphenylalaninemia (HPA), caused by autosomal recessive defects in hepatic phenylalanine hydroxylase (PAH, EC 1.14.16.1), leads to reduced L-phenylalanine (L-Phe) degradation and thus systemic HPA or phenylketonuria (PKU, OMIM 261600) with severe consequences on brain monoamine neurotransmitter metabolism (Blau et al 2010; Donlon et al 2015). The severity of PKU is classified by the level of HPA with normal peripheral blood L-Phe concentrations ranging from 50 to 110 μmol/L and, based on a commonly accepted classification, mild HPA ranging from 120 to 600 μmol/L, mild PKU ranging from 600 to 1200 μmol/L, and classic PKU for concentrations above 1200 μmol/L (Blau et al 2010). This differentiation is mainly determined by the molecular defect in the PAH gene that is primarily expressed in liver but also in kidneys (Lichter-Konecki et al 1999; Heintz et al 2013). Thus, a mutation-associated decrease in protein stability appears as the key determinant for phenotypic outcome in PKU (Pey et al 2007; Gersting et al 2008; Wettstein et al 2015). However, other factors may modulate the outcome, including interallelic complementation, as a consistent genotype-phenotype correlation based on the combination of recessive genomic mutations is still missing (Heintz et al 2013; Wettstein et al 2015; Shen et al 2016). Furthermore, an elevated blood L-Phe concentration does not elicit any known adverse effects in peripheral organs, including liver, despite the fact that an amyloidosis-like etiology for PKU has been suggested in brain tissue based on the L-Phe assembly at high concentrations into toxic fibrils (Adler-Abramovich et al 2012). Supplementation with the PAH cofactor tetrahydrobiopterin (BH4) is an approved therapeutic approach for about 20–30% of PKU patients, notably those with mild HPA and mild PKU (Kure et al 1999; Heintz et al 2013). Investigating the corrective effects of BH4 supplementation is relevant to further understand the molecular mechanisms in BH4-responsive PKU and for patient management. Extensive characterization of the hepatic PAH reaction, also exploiting mouse models, has previously revealed that the most relevant mechanisms at the molecular level are the increased intracellular cofactor concentration (kinetic or Michaelis-Menten effect) and the pharmacological chaperone effect on mild PKU mutations (Erlandsen et al 2004; Lagler et al 2010; Sarkissian et al 2012; Heintz et al 2013).
Yet, despite intensive investigations, there are many aspects of HPA or PKU that are unclear, notably the molecular mechanisms for neurotoxic effect of HPA and the pathophysiology to explain the neurological damage of PKU patients remains elusive. Evidence for an inverse relationship between blood L-Phe and monoamine neurotransmitter metabolites has been found in human autopsies and cerebral fluid (CSF) samples, but also in the Pahenu2/enu2 (hereafter designated ENU2/2) mouse model of PKU with blood L-Phe levels >1200 μmo/L (McKean 1972; Curtius et al 1981; Guttler and Lou 1986; Lykkelund et al 1988; Puglisi-Allegra et al 2000; Pascucci et al 2002). Other brain changes and pathologies in patients or in the PKU mouse model ENU2/2 are reduced brain concentrations of large neutral amino acids, hypomyelination, gliosis of central nervous system white matter, and reduction of cerebral protein synthesis (Pietz et al 1999; Smith and Kang 2000; Joseph and Dyer 2003; Hoeksma et al 2009) (for an overview see (de Groot et al 2010)). Most studies point to the neuronal pathology being mainly associated with alteration of relevant (brain) enzymatic activities, such as tyrosine hydroxylase (TH) and the brain specific tryptophan hydroxylase type 2 (TPH2), responsible for monoamine neurotransmitter biosynthesis (catecholamines and serotonin, respectively). The inverse relationship between blood L-Phe and monoamine neurotransmitters and their metabolite concentrations has in fact been associated with the saturation of the blood-brain-barrier transport of large neutral amino acids by L-Phe, which would lead to a reduction in the other amino acids, and notably L-Tyr, the substrate of TH, has been emphasized (van Spronsen et al 2009). In addition, the high concentration of L-Phe itself inhibits TH activity without altering TH gene expression but reduces the amount of TH protein (Joseph and Dyer 2003; Oh et al 2005; Ogawa and Ichinose 2006; Park et al 2009; Winn et al 2017). THP2 activity is also inhibited by elevated L-Phe levels, in fact more strongly than TH activity (Ogawa and Ichinose 2006). Earlier studies on the effect of brain metabolites following L-Phe-restricted diet in mice reported a partial but not complete recovery of monoamine neurotransmitters metabolites, which may be explained by the lack of 5-OH-Trp and L-Dopa supplementation together with the L-Phe restricted diet (Pascucci et al 2009; Pascucci et al 2012). Actually, a high level of circulating L-Phe was reported to correlate with neurological deterioration at any age despite dietary treatment during childhood (Joseph and Dyer 2003).
However, little is known on the neurometabolic changes in mild HPA and mild PKU. The ENU1/2 mouse was originally described as a heteroallelic orthologous model that exhibits partial liver PAH deficiency and blood L-Phe concentrations characteristic of mild HPA (< 600 μmol/L) (Sarkissian et al 2000). Thus, the mouse represents a good model for a large number of patients, which are compound heterozygous for a severe and a mild mutation. Besides, we have previously reported on the hepatic molecular changes and BH4-cofactor responsiveness of the ENU1/2 genetic mouse model with blood L-Phe concentrations <600 μmol/L, characteristic of mild HPA (Sarkissian et al 2012). Here we present brain analyses from this ENU1/2 mouse compared with wild-type mice. Our results point toward a more critically compromised serotonin metabolism in the brain of mild HPA mice models compared to the disturbance in catecholamine metabolism.
Materials, methods, and animal husbandry
Animal experiments and molecular analysis of brain
Animal experiments were carried out in accordance with the guidelines and policies of the Canadian Council on Animal Care, or the State Veterinary Office of Zurich and Swiss law on animal protection, the Swiss Federal Act on Animal Protection (1978), and the Swiss Animal Protection Ordinance (1981). All animal studies presented here received approval from the Animal Care Committee Review Board at McGill University in Canada or the State of Zurich. The studies were performed with ENU1/2 mutant or wild-type mice all on C57BL/6 background. For all mice experiments, including wild-type controls, we used littermates, and the age of mice was between 10 and 20 weeks (fertile adults). To determine blood L-Phe levels, mice were fasted for 4–6 h prior to blood withdrawal from tail vein. Mice on L-Phe administration received 1.1 mg/g [L-Phe/body weight], which was administered on day 1 via a subcutaneous (s.c.) bolus on the animal’s lower back. Blood samples were collected from the tail vein of all animals, 1 h prior to, and 30 min and 1 h (upon termination) post-time of L-Phe administration. Blood samples were collected on filter cards and amino acid quantification was done using a standard tandem mass spectrometry method (Rebuffat et al 2010). Whole mouse brains were removed from killed animals and immediately shock-frozen in liquid nitrogen. Before preparing tissue extracts, the entire brains were pulverized in metal nuts that were pre-cooled in liquid nitrogen and by drilling bolts from both sides.
Mice on BH4 treatment received enteral doses, once daily via a 22-gauge gavage needle, directly to the stomach as described in Sarkissian et al 2012. Briefly, BH4 (50 mg/kg/day), suspended in a solution of 11 mg/kg/day ascorbic acid and 5% dimethyl sulfoxide (DMSO), was administered to ENU1/2 mice under standard dietary condition. The control ENU1/2 animals received the buffer solution (11 mg/kg ascorbic acid and 5% DMSO only) and were otherwise treated as above.
Preparation of brain extracts for determination of BH4 and monoamine neurotransmitters, enzyme activity measurements, and immuno-quantification
Tissue extracts were prepared by homogenization in either 4 × volumes (immunodetection and TH or TPH enzyme activities; see below) or 10 × volumes (for BH4 and neurotransmitter metabolites and GTPCH/GFPR assay) of 50 mM Tris-HCl, pH 7.5, 100 mM KCl, 1 mM EDTA, 1 mM DTT, 1 μM leupeptin, 1 μM pepstatin, and 200 μM phenylmethylsulfonyl fluoride with a Tissue Lyser II (100-240 V, 50/60 Hz; Qiagen), with 5 mm beads (Qiagen) in two consecutive shaking steps with 20 Hz at 4 °C for 90 s. Extracts were clarified by centrifugation at 15,000 g for 20 min at 4 °C. Supernatants were stored in liquid nitrogen prior to use, with the exception of GTPCH activity which was measured immediately after producing the lysate. Protein concentration was measured using the M-TP Mikroprotein kit from Beckman Coulter Synchron LX-System (Beckman Coulter Inc., Brea, CA; kit-no. 445860). Measurements of pterin, monoamine neurotransmitters, and their metabolites were performed as reported (Elzaouk et al 2003; Blau and Thöny 2008). Note that DOPAC concentrations were in our samples below or at limit of detection of the method we used and therefore were not reported.
mRNA quantification
Total RNA was isolated from brain powder with the QIAamp RNA Blood Mini Kit from QIAGEN and translated to cDNA with the Reverse Transcription system from Promega. Quantification of mouse Gch1-mRNA, Gchfr-mRNA, Th-mRNA, Tph1-mRNA, and Tph2-mRNA by RT-qPCR was performed as described using the commercially available ABI assays (Calvo et al 2010; Sarkissian et al 2012). Numbers are Mm00447546_m1 for Th-mRNA, NCBI RefSeq NM_009377.1; Mm00493798_m1 for Tph1-mRNA, NCBI RefSeq NM_001136084.1 and NM 009414.2; Mm00557717_m1 for Tph2-mRNA, NCBI RefSeq NM_173391.2; for control, the murine Gapdh gene was used, ABI assay ID: Mm99999915_g1; NCBI RefSeq NM_008084.2). Values were normalized relative to Gapdh-mRNA of the control group as described (Livak and Schmittgen 2001).
Immunodetection
Brain extracts were prepared by homogenization in four volumes of 50 mM Tris-HCl, pH 7.5, 100 mM KCl, 1 mM EDTA, 1 mM DTT, 200 μM PMSF, and protease inhibitor cocktail (Roche). Extracts were clarified by centrifugation at 20,000×g for 15 min at 4 °C. Free amino acids and contaminants of low molecular weight were removed from the supernatants using Zebra Desalt Spin columns (Thermo Scientific), and then the supernatants were stored in liquid nitrogen prior to use. Protein concentration was measured by the Bradford method or by Direct Detect Spectrometer (Merck Millipore). Quantification of TH and TPH2 protein was performed by Western blot analyses after SDS-PAGE (10% acrylamide) with 10–20 μg total protein in each lane, and transfer to nitrocellulose membrane and immunostaining using affinity-purified polyclonal rabbit anti-rat TH (Fisher Scientific) in a 1:1000 dilution or rabbit polyclonal anti-mouse TPH2 (Santa Cruz biotechnology) in a 1:300 dilution, used as the primary antibodies. Secondary antibody was goat anti-rabbit IgG (H + L) horseradish peroxidase conjugate (Bio-Rad) in a 1:2000 dilution. As a loading control, a polyclonal antibody against glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Abcam) was used for immunostaining of the membrane. Purified recombinant human TH and TPH2 (10 ng/lane) were used as standard for immune-quantification. Detection was performed by enhanced chemiluminescence, and immune-quantification in a Molecular Image (Bio-Rad) using Image Lab 3.01 software (Calvo et al 2010).
Enzyme assays
Tyrosine hydroxylase (TH) and tryptophan hydroxylase (TPH) activities were assayed at 30 °C for the brain extract as described (Reinhard Jr et al 1986) using an incubation mixture containing 100 mM Na-HEPES, pH 7.0, 50 μM L-[3,5-3H]-tyrosine or 50 μM L-[3,5-3H]-tryptophan, 0.05 mg/ml catalase, and 10 μM ferrous ammonium sulphate. The enzyme was pre-incubated for 1 min in this mixture, and reaction started by addition of 500 μM BH4 and 5 mM DTT. The reaction was stopped after 30 min by addition of a slurry of activated charcoal in 1 M HCl. After centrifugation at 10,000×g, the supernatant was counted in a scintillation counter. Tryptophan hydroxylase (TPH) activity was assayed at 30 °C for the brain extract as described (Calvo et al 2010) using an incubation mixture containing 100 mM Na-HEPES, pH 7.0, 50 μM L-[3,5-3H]-tryptophan, 0.05 mg/ml catalase, and 20 μM ferrous ammonium sulphate. The enzyme was pre-incubated for 1 min in this mixture, and reaction started by addition of 500 μM BH4 and 5 mM DTT. The reaction was stopped after 20–30 min by addition of a slurry of activated charcoal in 1 M HCl. After centrifugation at 10,000×g, the supernatant was counted in a scintillation counter.
GTP cyclohydrolase 1 (GTPCH) activity was measured as described (Blau and Thöny 2008). To inhibit GTPCH activity in extracts of mouse brain, 2,4-diamino-6-hydroxypyrimidine (DAHP; Enzo Life Sciences) was added as indicated to a fixed amount of extract or homogenate, containing between 50 and 100 μg of protein. For DAHP titration, a 10 mM solution of DAHP was prepared in homogenization buffer (50 mM Tris-HCl, pH 7.5, 100 mM KCl, 1 mM EDTA, 1 mM DTT).
Statistical methods
For statistical analyses, two-way analyses of variance (ANOVA) was used with either genotype and sex or sex and treatment as between-subjects factors. We considered p < 0.05 as statistically significant. Statistical analyses were performed using GraphPad Prism™ (version 6, San Diego) software.
Results
Phenylalanine, tyrosine, and BH4 in brain of ENU1/2 mice
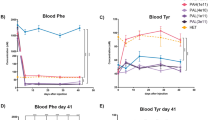
We previously reported on the mechanism of BH4-responsiveness in the ENU1/2 mouse model of mild/cofactor-responsive HPA where we found that (oral) BH4 supplementation increased hepaticL-Phe hydroxylation through a kinetic rather than chaperone stabilizing effect (Sarkissian et al 2012). Here we present brain analysis of ENU1/2 treated and untreated in the same manner. Treatment of ENU1/2 mice included BH4 loading (50 mg/kg/day for 10 days (Sarkissian et al 2012)). For comparison, L-Phe levels in blood in untreated and treated wild-type and ENU1/2 mice are presented in Fig. 1a. L-Phe level in brain of wild-type mice, whether they were under fasting or L-Phe loading conditions, was 0.53 ± 0.02 nmol/mg protein, and ENU1/2 mice had fourfold elevated brain L-Phe levels (1.98 ± 0.31 nmol/mg) when untreated, and roughly sixfold elevated (3.23 ± 0.77 nmol/mg protein) under L-Phe loading (see values normalized vs. wild-type control in Fig. 1b). The elevation of L-Phe in non L-Phe treated ENU1/2 compared to wild-type mice completely normalized upon BH4 treatment in blood (Fig. 1a) and, remarkably, in brain (Fig. 1b). We observed lower brain L-Phe levels in ENU1/2 mice when treated with BH4 than in wild-type, independent of whether wild-type mice were treated with cofactor or not (p = 0.016), which is in agreement with what we observed in liver (Sarkissian et al 2012). On the other hand, we found statistically no significant differences in brain L-Tyr concentrations under any conditions studied (Fig. 1b).

Blood L-Phe and L-Phe and L-Tyr content in brain of wild-type and ENU1/2 mice; effect of L-Phe administration and BH4 treatment. a Blood L-Phe values (from dried blood spots) are shown for comparison (“+ L-Phe” corresponds to 1 h after a single s.c. dose of L-Phe of 1.1 mg/kg; n = 10 for wild-type untreated and n = 5 for all other groups). b Brain L-Phe and L-Tyr content, normalized to wild-type control. We measured 0.53 ± 0.02 nmol L-Phe/mg protein and 0.32 ± 0.04 nmol L-Tyr/mg protein in untreated wild-type mice, which were defined as “1”; n.a., not available (n = 5 for all groups). For statistical analyses, two-way ANOVA test was used; **, p < 0.005
As depicted in Fig. 2, brain BH4 levels were unchanged in wild-type (defined as 100%) and in ENU1/2 mice, even under oral L-Phe loading conditions. On the other hand BH4 treatment resulted in an elevation of the cofactor in brain of wild-type animals, as previously shown (Thony et al 2008). We also observed a trend for higher cofactor levels in BH4-treated ENU1/2 but differences were not significant, most probably due to the higher inter-individual variability that we measured in these HPA mice (Fig. 2), and the number of mice we had could be too low for the high biological variance. The concentration of brain neopterin, the oxidized and dephosphorylated intermediate product of GTPCH, remained below 1 pmol/mg in all animals and under all conditions (data not shown).

Brain BH4 levels in wild-type and ENU1/2; effect of L-Phe administration and BH4 treatment. BH4 levels are shown as % of wild-type controls. Every group represents determinations from four to ten individual mice. Note that the absolute values of brain BH4 concentrations were determined in two different groups: wild-type compared to ENU1/2: 17.6 ± 2.6 pmol/mg in wild-type (defined as 100 ± 21%), compared to 17.2 ± 2.6 pmol/mg in wild-type L-Phe treated (97 ± 20%), 17.4 ± 2.6 pmol/mg in ENU1/2 untreated (99 ± 21%), and 18.8 ± 3.8 pmol/mg in ENU1/2L-Phe treated (107 ± 27%). Data on BH4 supplemented mice were obtained after loading with 50 mg/kg/day for 10–12 days, with the exception of wild-type mice that were loaded with 20 mg/kg/day (which leads to a 61% increase of brain BH4 over control; n = 4 to 10 mice per group). For statistical analyses, two-way ANOVA test was used; **, p < 0.005
Effect of HPA and BH4 supplementation on monoamine neurotransmitter and metabolites in brain of ENU1/2 mice
The metabolites of the brain serotonin and catecholamine pathways in untreated and L-Phe and/or BH4-treated ENU1/2 mice, compared to wild-type, are summarized in Fig. 3. Note that the neurochemical analyses were done on whole brain rather than on specific brain areas. We found reduced serotonin, 5HIAA, and MHPG levels in ENU1/2 mice compared to wild-type, while catecholamine neurotransmitters were not affected (Fig. 3a). L-Phe administration did not lead to any remarkable change in neurotransmitter levels (Fig. 3a). Furthermore, the BH4 treatment in ENU1/2 did not result in substantial alteration of brain neurotransmitter metabolites, though a trend for increased levels was observed, notably for serotonin and 5HIAA (Fig. 3b), most probably as a result of the normalization of brain L-Phe upon BH4 supplementation (Fig. 1b).

Biogenic amine metabolites in the brain of wild-type and ENU1/2; effect of L-Phe administration and BH4 treatment. The neurotransmitter metabolites, as shown in the insert, are depicted relative to wild-type values, which are defined as 1. a Comparison of wild-type to ENU1/2: “+ L-Phe” corresponds to 1 h after a single s.c. dose of L-Phe of 1.1 mg/kg. b Data on BH4 supplemented ENU1/2 mice were obtained after loading with 50 mg/kg/day for 10–12 days. For statistical analyses, two-way ANOVA test was used (n = 5 to 8 for all groups); *, p < 0.05; **, p < 0.005. Abbreviations: L-Dopa, L-3,4-dihydroxyphenylalanine; 3OMD, 3-o-methyldopa; HVA, homovanillic acid; 5HTP, 5-hydroxytryptophan; 5HIAA, 5-hydroxyindolacetic acid; MHPG, 3-methoxy-4-hydroxyphenylglycol
HPA compromises TH and TPH2 levels and activity
As discussed in the Introduction, previous studies have indicated that total TH can be reduced and inhibited under conditions of severe HPA, which might be the mechanism underlying catecholamine neurotransmitter depletion in this disease (Joseph and Dyer 2003; Oh et al 2005; Ogawa and Ichinose 2006; Park et al 2009). We thus characterized the effect of HPA on the levels of TH and TPH activity, and of the levels of neuronal TH and TPH2 proteins in brain extracts of ENU1/2 and wild-type mice. We measured a trend for decreased TH protein and activity between WT and ENU1/2, both treated and untreated with BH4, with significant differences only measured for TH protein (Fig. 4). Furthermore, significant differences were measured for TPH2 protein level and activity, which was reduced in the HPA mouse model while Th-, Tph1, and Tph2-mRNA levels were not altered (Table 1). Besides, BH4 treatment did not elicit any corrective effect on TPH2 protein and activity levels (Fig. 4).

TH and TPH2 protein and activity in the brain of wild-type and ENU1/2 mice; effect of BH4 treatment. TH protein is measured as the density of the ~60 immunoquantified kDa band and TPH2, as the density of the ~61 kDa band, both relative to the intensity of the GAPDH band in whole brain extracts, whereas the activity was measured as indicated in Materials, methods, and animal husbandry. All values are related to those in untreated wild-type (defined as 1). For statistical analyses, two-way ANOVA test was used; *, p < 0.05 (n = 5 to 8 for all groups)
HPA induces up-regulation of GTPCH-GFPR expression
In liver, the de novo biosynthesis of BH4 is thought to be regulated by the GTP cyclohydrolase I (GTPCH) feedback regulatory protein (GFRP) complex (Harada et al 1993; Werner et al 2011), and at least in serotonin neurons, BH4 biosynthesis was reported to be regulated by the GFRP (Kapatos et al 1999). We thus analyzed the relative gene expression levels of Gch1-mRNA and Gchfr-mRNA, encoding GTPCH and GFRP, respectively, in ENU1/2 in brain and, for comparison, in liver of the same animals (Table 2). As previously reported, we found no change of Gchfr gene expression (relative to Gapdh-mRNA) in liver of ENU1/2 mice (Sarkissian et al 2012). In whole brain extracts of mice, relative Gchfr-mRNA (as well as Gch1-mRNA) expression was between 50 to 100-fold lower compared to liver in all animals, confirming that GFRP has a much lower overall expression level in the brain compared to liver (see Discussion). However, in brain, we found that Gchfr-mRNA, but not Gch1–mRNA, expression was 3–4-fold elevated in ENU1/2 mice independent of L-Phe loading (Table 2). At the same time, BH4 loading in wild-type and ENU1/2 mice lowered Gchfr-mRNA expression by maximally a factor of 2 (not shown). Using a GTPCH enzyme activity assay with whole brain extracts, we did not see any difference between ENU1/2 and wild-type mice. We thus examined a putative up-regulation of the GTPCH activity via the GTPCH-GFRP complex in whole brain extracts of ENU1/2 mice. Although our antibody against GFRP detected the 9.7 kDa protein in crude liver extracts upon Western blot analysis (SZ2309 in Fig. S2 in (Sarkissian et al 2012)), we were not able to identify GFRP in extracts from whole mouse brain (not shown). Again this might reflect the relatively low expression level of GFRP in these whole brain extracts. An alternative way to assess the amount (and activity) of GFPR is to measure the response of GTPCH enzymatic activity in the presence of 2,4-diamino-6-hydroxypyrimidine (DAHP). DAHP is a specific inhibitor of GTPCH activity that was found to interact via the GFRP in the GTPCH-GFRP complex (Kolinsky and Gross 2004). We thus performed GTPCH activity assays in the presence of increasing amounts of DAHP with crude brain extracts from either wild-type or ENU1/2 mice. The corresponding titration curves are shown in Fig. 5 where we found no significantly increased GTPCH activity in ENU1/2 mice compared to wild-type mice with normal L-Phe levels. Such behavior could possibly be interpreted as a moderately elevated expression of GFRP — as a consequence of the up-regulated Gchfr–mRNA expression — requiring more DAHP to inhibit GTPCH activity. Since homozygous ENU2/2 (PKU) mice exhibit much higher systemic L-Phe levels, we performed the same assay with brain extracts from mutant and control mice and found a significant difference, i.e., elevated GTPCH activity in brain lysates of ENU2/2 mice (see Suppl. Fig. S1). In summary, this assay showed indirectly increased expression of the GFRP in response to high L-Phe levels at least in ENU2/2 (PKU) mice, and that neuronal up-regulation of GTPCH activity may operate via the GTPCH-GFRP complex.

Inhibition of GPTCH activity by 2,4,-diamino-6-hydroxypyrimidine (DAHP) in crude brain extracts from either wild-type or ENU1/2 mice. Activity was assayed as the quantity of 7,8,-dihydroneopterin triphosphate produced by 80 μg of brain extract from either wild-type or ENU1/2 mice in the presence of increasing amounts of added DAHP. Assays were performed in triplicates (n = 3 mice per group)
Discussion
We have previously investigated the mechanism of BH4-responsiveness in the ENU1/2 mouse model of mild/cofactor-responsive HPA, where (oral) BH4 supplementation increased hepatic L-Phe hydroxylation through a kinetic rather than chaperon stabilizing effect (Sarkissian et al 2012). There is to our knowledge no previous brain investigation in ENU1/2 mice, except for blood L-Phe values that were published to exhibit twofold elevation compared to wild-type (Sarkissian et al 2000). The here presented analysis was performed at 1.5 h after the L-Phe loading although the kinetics of potential changes in brain neurotransmitter metabolites following a L-Phe challenge are to our knowledge not known. Our present whole brain analysis of BH4-treated and untreated ENU1/2 mice lead to major unprecedented observations. ENU1/2 show a significant elevation of L-Phe in brain even in the absence of L-Phe administration. Thus, ENU1/2 mice presented a fourfold elevation of brain L-Phe under fasting conditions and sixfold elevation upon L-Phe administration (Fig. 1b), concomitant with abnormal neurotransmitter metabolites in brain (serotonin, 5HIAA, and MHPG; Fig. 3), while plasma L-Phe levels were only mildly or insignificantly elevated, at least under fasting conditions (Fig. 1). A less dramatic but also a non-linear correlation between plasma and brain L-Phe values were described in HPA patients using proton magnetic resonance spectroscopy (Moller et al 2000).
Our results also indicate a reduction of TH and TPH2, serotonin and its metabolites in ENU1/2 compared to wild-type mice. It is largely established that classic, untreated PKU leads to a decrease in monoamine neurotransmitters, notably explained by a reduction of L-Tyr and L-Trp in brain (Blau et al 2010) and by the competitive inhibition of neuronal TH and TPH2 activity by L-Phe (Ogawa and Ichinose 2006). Furthermore, a direct effect of classic PKU on brain TH protein levels has also been shown (Joseph and Dyer 2003; Oh et al 2005; Park et al 2009). However, less is known about the effects of mild HPA upon TPH2 protein and activity levels, and upon the dopaminergic and serotonergic pathways. In this work, using the ENU1/2 model of mild HPA, we observed that their fourfold elevation of brain Phe was associated with a significant decrease in TPH2 protein and activity, and reduced levels of serotonin and its metabolites. BH4-responsive HPA in these mice — as demonstrated by a decrease in blood-Phe levels — has been described previously (Sarkissian et al 2012), but here we show complete normalization of brain L-Phe levels following BH4-treatment. A trend was also observed for increased monoamine neurotransmitters in ENU1/2 mice, indicating the importance of treating patients with mild forms of HPA and PKU. Despite the modest increase in blood Phe levels in ENU1/2 compared with wild-type mice, L-Phe is effectively elevated in brain, most probably through the action of the blood-brain barrier large neutral amino acid (LNAA) transporter, which shows high affinity for LNAA and causes the competition among LNAA for entering the brain (Cansev and Wurtman 2007). However, the dominant L-Phe transport does not seem to largely affect the transport of other LNAA than L-Phe in ENU1/2 mice, as seen by the comparative L-Tyr values (Fig. 1b), further supporting that the reduced serotonin levels in this mice model is related to the inhibitory effect of L-Phe on TPH2 (Ogawa and Ichinose 2006; Winn et al 2017). We have previously shown that BH4 supplementation increases the cofactor levels in liver, where it stimulates PAH activity (Sarkissian et al 2012). As seen in this work, this stimulating effect leads to normalization of blood and brain L-Phe values. Despite this effective L-Phe reduction, TPH2 protein and activity levels were not normalized following BH4 treatment, and recovery of brain serotonin content was suboptimal, pointing to a possible HPA-associated constitutive deficiency affecting the serotonergic system in ENU1/2 mice. Thus, our results advocate for combined 5-OH-Trp supplementation and BH4 treatment in patients with mild BH4-responsive HPA/PKU who exhibit concomitant mood problems such as depression or anxiety.
Another important finding of the present work was the moderately increased expression in brain of GFRP in response to elevated L-Phe levels. Neuronal up-regulation of GTPCH activity is thought to operate via the GTPCH-GFRP complex, as has been described in liver (Harada et al 1993). The hepatic GTPCH-GFRP complex is a known sensor for BH4 and L-Phe, thereby regulating the de novo biosynthesis of the BH4-cofactor via feedback inhibition or stimulation, respectively (Werner et al 2011). Gchfr-mRNA expression was reported to also be abundantly present in (rat) brain, specifically in serotonin neurons but undetectable in dopaminergic neurons (Kapatos et al 1999). However, regulation of the GPTCH-GFRP complex in the brain has not been fully elucidated. Park et al reported in 2009 gene expression profiling in brain of hyperphenylalaninemic ENU2/2 mice but up-regulation of Gchfr-mRNA was not detected, although a very limited number of mice were investigated in that study (see Discussion in (Park et al 2009)). Nevertheless, modulation of GTPCH activity by regulated gene expression and/or post-translational regulation involving, among others, interaction via GFRP in specific brain sub-regions or neuronal cell types has been postulated by several authors (for an overview see (Werner et al 2011)). Although the detailed mechanism and location of this regulation needs further analyses, we suggest that the GTPCH-GFRP complex is also up-regulated in the brain as it is in the liver in response to L-Phe elevation (Harada et al 1993). Elevated L-Phe would thus result in elevated GTPCH enzyme activity and BH4-cofactor biosynthesis in the brain which may have a positive effect on brain TH and THP2 activity to synthesize L-Dopa and serotonin, respectively. Further studies are required to support this hypothesis.
Abbreviations
- PAH:
-
phenylalanine hydroxylase
- LNAA:
-
large neutral amino acid
- BH4 :
-
6R–L-erythro-5,6,7,8-tetrahydrobiopterin
- GTPCH:
-
GTP cyclohydrolase I
- GFRP:
-
GTPCH feedback regulatory protein (gene Gchfr)
- HPA:
-
hyperphenylalaninemia
- PKU:
-
phenylketonuria
- L-Phe:
-
L-phenylalanine
- TH:
-
tyrosine hydroxylase
- TPH:
-
tryptophan hydroxylase
- L-Dopa:
-
L-3,4-dihydroxyphenylalanine
- 3OMD:
-
3-o-methyldopa
- HVA:
-
homovanillic acid
- 5HTP:
-
5-hydroxytryptophan
- 5HIAA:
-
5-hydroxyindolacetic acid
- MHPG:
-
3-methoxy-4-hydroxyphenylglycol
References
Adler-Abramovich L, Vaks L, Carny O et al (2012) Phenylalanine assembly into toxic fibrils suggests amyloid etiology in phenylketonuria. Nat Chem Biol 8:701–706
Blau N, Thöny B (2008) Pterins and related enzymes. In: Blau N, Duran M, Gibson KM (eds) Laboratory guide to the methods in biochemical genetics. Springer, Berlin, p 665–701
Blau N, van Spronsen FJ, Levy HL (2010) Phenylketonuria. Lancet 376:1417–1427
Calvo AC, Scherer T, Pey AL et al (2010) Effect of pharmacological chaperones on brain tyrosine hydroxylase and tryptophan hydroxylase 2. J Neurochem 114:853–863
Cansev M, Wurtman RJ (2007) Aromatic amino acids in the brain. In: Lajtha A (ed) Handbook of neurochemistry and molecular neurobiology. Springer, Berlin, pp 60–97
Curtius HC, Niederwieser A, Viscontini M et al (1981) Serotonin and dopamine synthesis in phenylketonuria. Adv Exp Med Biol 133:277–291
de Groot MJ, Hoeksma M, Blau N, Reijngoud DJ, van Spronsen FJ (2010) Pathogenesis of cognitive dysfunction in phenylketonuria: review of hypotheses. Mol Genet Metab 99 Suppl 1:S86–S89
Donlon J, Sarkissian CN, Levy HL, Scriver CR (2015) Hyperphenylalaninemia: phenylalanine hydroxylase deficiency. Chapter 77. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Stylianos E, Antonarakis MD, Ballabio A, Gibson M, Mitchell G (eds) The metabolic and molecular bases of inherited disease. McGraw-Hill, New York http://ommbid.mhmedical.com/content.aspx?bookid=971§ionid=62673211
Elzaouk L, Leimbacher W, Turri M et al (2003) Dwarfism and low insulin-like growth factor-1 due to dopamine depletion in pts−/− mice rescued by feeding neurotransmitter precursors and H4-biopterin. J Biol Chem 278:28303–28311
Erlandsen H, Pey AL, Gamez A et al (2004) Correction of kinetic and stability defects by tetrahydrobiopterin in phenylketonuria patients with certain phenylalanine hydroxylase mutations. Proc Natl Acad Sci U S A 101:16903–16908
Gersting SW, Kemter KF, Staudigl M et al (2008) Loss of function in phenylketonuria is caused by impaired molecular motions and conformational instability. Am J Hum Genet 83:5–17
Guttler F, Lou H (1986) Dietary problems of phenylketonuria: effect on CNS transmitters and their possible role in behaviour and neuropsychological function. J Inherit Metab Dis 9 Suppl 2:169–177
Harada T, Kagamiyama H, Hatakeyama K (1993) Feedback regulation mechanism for the control of GTP cyclohydrolase I activity. Science 260:1507–1510
Heintz C, Cotton RG, Blau N (2013b) Tetrahydrobiopterin, its mode of action on phenylalanine hydroxylase, and importance of genotypes for pharmacological therapy of phenylketonuria. Hum Mutat 34:927-36
Hoeksma M, Reijngoud DJ, Pruim J, de Valk HW, Paans AM, van Spronsen FJ (2009) Phenylketonuria: high plasma phenylalanine decreases cerebral protein synthesis. Mol Genet Metab 96:177–182
Joseph B, Dyer CA (2003) Relationship between myelin production and dopamine synthesis in the PKU mouse brain. J Neurochem 86:615–626
Kapatos G, Hirayama K, Shimoji M, Milstien S (1999) GTP cyclohydrolase I feedback regulatory protein is expressed in serotonin neurons and regulates tetrahydrobiopterin biosynthesis. J Neurochem 72:669–675
Kolinsky MA, Gross SS (2004) The mechanism of potent GTP cyclohydrolase I inhibition by 2,4-diamino-6-hydroxypyrimidine: requirement of the GTP cyclohydrolase I feedback regulatory protein. J Biol Chem 279:40677–40682
Kure S, Hou DC, Ohura T et al (1999) Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. J Pediatr 135:375–378
Lagler FB, Gersting SW, Zsifkovits C et al (2010) New insights into tetrahydrobiopterin pharmacodynamics from Pah enu1/2, a mouse model for compound heterozygous tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. Biochem Pharmacol 80:1563–1571
Lichter-Konecki U, Hipke CM, Konecki DS (1999) Human phenylalanine hydroxylase gene expression in kidney and other nonhepatic tissues. Mol Genet Metab 67:308–316
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408
Lykkelund C, Nielsen JB, Lou HC et al (1988) Increased neurotransmitter biosynthesis in phenylketonuria induced by phenylalanine restriction or by supplementation of unrestricted diet with large amounts of tyrosine. Eur J Pediatr 148:238–245
McKean CM (1972) The effects of high phenylalanine concentrations on serotonin and catecholamine metabolism in the human brain. Brain Res 47:469–476
Moller HE, Ullrich K, Weglage J (2000) In vivo proton magnetic resonance spectroscopy in phenylketonuria. Eur J Pediatr 159 Suppl 2:S121–S125
Ogawa S, Ichinose H (2006) Effect of metals and phenylalanine on the activity of human tryptophan hydroxylase-2: comparison with that on tyrosine hydroxylase activity. Neurosci Lett 401:261–265
Oh HJ, Lee H, Park JW et al (2005) Reversal of gene expression profile in the phenylketonuria mouse model after adeno-associated virus vector-mediated gene therapy. Mol Genet Metab 86 Suppl 1:S124–S132
Park JW, Park ES, Choi EN, Park HY, Jung SC (2009) Altered brain gene expression profiles associated with the pathogenesis of phenylketonuria in a mouse model. Clin Chim Acta 401:90–99
Pascucci T, Andolina D, Mela IL et al (2009) 5-Hydroxytryptophan rescues serotonin response to stress in prefrontal cortex of hyperphenylalaninaemic mice. Int J Neuropsychopharmacol 12:1067–1079
Pascucci T, Giacovazzo G, Andolina D et al (2012) In vivo catecholaminergic metabolism in the medial prefrontal cortex of ENU2 mice: an investigation of the cortical dopamine deficit in phenylketonuria. J Inherit Metab Dis 35:1001–1009
Pascucci T, Ventura R, Puglisi-Allegra S, Cabib S (2002) Deficits in brain serotonin synthesis in a genetic mouse model of phenylketonuria. Neuroreport 13:2561–2564
Pey AL, Stricher F, Serrano L, Martinez A (2007) Predicted effects of missense mutations on native-state stability account for phenotypic outcome in phenylketonuria, a paradigm of misfolding diseases. Am J Hum Genet 81:1006–1024
Pietz J, Kreis R, Rupp A et al (1999) Large neutral amino acids block phenylalanine transport into brain tissue in patients with phenylketonuria. J Clin Invest 103:1169–1178
Puglisi-Allegra S, Cabib S, Pascucci T, Ventura R, Cali F, Romano V (2000) Dramatic brain aminergic deficit in a genetic mouse model of phenylketonuria. Neuroreport 11:1361–1364
Rebuffat A, Harding CO, Ding Z, Thony B (2010) Comparison of adeno-associated virus pseudotype 1, 2, and 8 vectors administered by intramuscular injection in the treatment of murine phenylketonuria. Hum Gene Ther 21:463–477
Reinhard JF Jr, Smith GK, Nichol CA (1986) A rapid and sensitive assay for tyrosine-3-monooxygenase based upon the release of 3H2O and adsorption of [3H]-tyrosine by charcoal. Life Sci 39:2185–2189
Sarkissian CN, Boulais DM, McDonald JD, Scriver CR (2000) A heteroallelic mutant mouse model: a new orthologue for human hyperphenylalaninemia. Mol Genet Metab 69:188–194
Sarkissian CN, Ying M, Scherer T, Thöny B, Martinez A (2012) The mechanism of BH4 -responsive hyperphenylalaninemia--as it occurs in the ENU1/2 genetic mouse model. Hum Mutat 33:1464–1473
Shen N, Heintz C, Thiel C, Okun JG, Hoffmann GF, Blau N (2016) Co-expression of phenylalanine hydroxylase variants and effects of interallelic complementation on in vitro enzyme activity and genotype-phenotype correlation. Mol Genet Metab 117:328–335
Smith CB, Kang J (2000) Cerebral protein synthesis in a genetic mouse model of phenylketonuria. Proc Natl Acad Sci U S A 97:11014–11019
Thony B, Calvo AC, Scherer T et al (2008) Tetrahydrobiopterin shows chaperone activity for tyrosine hydroxylase. J Neurochem 106:672–681
van Spronsen FJ, Hoeksma M, Reijngoud DJ (2009) Brain dysfunction in phenylketonuria: is phenylalanine toxicity the only possible cause? J Inherit Metab Dis 32:46–51
Werner ER, Blau N, Thony B (2011) Tetrahydrobiopterin: biochemistry and pathophysiology. Biochem J 438:397–414
Wettstein S, Underhaug J, Perez B et al (2015) Linking genotypes database with locus-specific database and genotype-phenotype correlation in phenylketonuria. Eur J Hum Gen 23:302–309
Winn SR, Scherer T, Thöny B et al (2017) Blood phenylalanine reduction corrects CNS dopamine and serotonin deficiencies and partially improves behavioral performance in adult phenylketonuric mice. Mol Genet Metabol. https://doi.org/10.1016/j.ymgme.2017.10.009
Acknowledgements
We thank Dr. Ernst Werner for helpful discussions and the Institute for Animal Science at the University of Zürich for cooperation. Financial support was given by the Stiftung für wissenschaftliche Forschung University of Zürich (Baumgarten Stiftung to B.T.), the Swiss National Science Foundation (to B.T.) SNF 310030-162547, the NIH R01NS080866 to C.O.H., the Novartis “Stiftung für medizinisch-biologische Forschung” (to B.T.), The Research Council of Norway (Programmes FORNY and FRIMEDBIO to A.M.), and the K. G. Jebsen Centre for Neuropsychiatric Disorders (to A.M.).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
T.Scherer, G.Allegri, C. Sarkissian, M. Ying, H.M. Grisch-Chan, A. Rassi, S. Winn, C. Harding, A. Martinez and B. Thöny declare that they have no conflict of interest.
Informed consent
This article does not contain any studies with human subjects performed by the any of the authors.
Animal rights
All institutional and national guidelines for the care and use of laboratory animals were followed (see also chapter “Materials, methods, and animal husbandry”).
Additional information
Communicated by: Viktor Kožich
Electronic supplementary material
Suppl. Fig. S1
Inhibition of GPTCH activity by 2,4,-diamino-6-hydroxypyrimidine (DAHP) in crude brain extracts from either wild-type or ENU2/2 (PKU) mice. Activity was assayed as the quantity of 7,8,-dihydroneopterin triphosphate produced by 80 mg of brain extract from either wild-type or ENU1/2 mice in the presence of increasing amounts of added DAHP. Assays were performed in triplicates. Two tailed Student‘s t-test, ***p < 0.005, **p < 0.05, *p < 0.1; n = 3 (wild-type and PKU) (GIF 10 kb)
Rights and permissions
About this article

Cite this article
Scherer, T., Allegri, G., Sarkissian, C.N. et al. Tetrahydrobiopterin treatment reduces brain L-Phe but only partially improves serotonin in hyperphenylalaninemic ENU1/2 mice. J Inherit Metab Dis 41, 709–718 (2018). https://doi.org/10.1007/s10545-018-0150-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-018-0150-y