
Abstract
Carnosinase (CN1) is a dipeptidase, encoded by the CNDP1 gene, that degrades histidine-containing dipeptides, such as carnosine, anserine and homocarnosine. Loss of CN1 function (also called carnosinase deficiency or aminoacyl-histidine dipeptidase deficiency) has been reported in a small number of patients with highly elevated blood carnosine concentrations, denoted carnosinaemia; it is unclear whether the variety of clinical symptoms in these individuals is causally related to carnosinase deficiency. Reduced CN1 function should increase serum carnosine concentrations but the genetic basis of carnosinaemia has not been formally confirmed to be due to CNDP1 mutations. A CNDP1 polymorphism associated with low CN1 activity correlates with significantly reduced risk for diabetic nephropathy, especially in women with type 2 diabetes, and may slow progression of chronic kidney disease in children with glomerulonephritis. Studies in rodents demonstrate antiproteinuric and vasculoprotective effects of carnosine, the precise molecular mechanisms, however, are still incompletely understood. Thus, carnosinemia due to CN1 deficiency may be a non-disease; in contrast, carnosine may potentially protect against long-term sequelae of reactive metabolites accumulating, e.g. in diabetes and chronic renal failure.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
In 1900, Vladimir Gulewitsch identified carnosine (Gulewitsch and Amiradžibi 1900; Gulewitsch 1905). Carnosine (ß-alanyl-L-histidine), anserine (ß-alanyl-3-methyl-L-histidine) and homocarnosine (γ-aminobutyric acid-L-histidine) belong to the group of histidine-containing dipeptides and meanwhile, at least in experimental settings, it has been shown that these dipeptides exert a variety of protective functions (Boldyrev et al. 2013). Carnosine inhibits glycation (Alhamdani et al. 2007) and acts as ACE inhibitor (Hou et al. 2003; Nakagawa et al. 2006). Its function as antioxidant (Decker et al. 2000; Mozdan et al. 2005; Velez et al. 2008; Hipkiss 2011; Babizhayev et al. 2013) and its capacity to scavenge carbonyls (Negre-Salvayre et al. 2008; Barski et al. 2013; Brings et al. 2017), however, is debated. In vitro, anserine also scavenges carbonyls, may act as an antioxidant (Kohen et al. 1988; Aldini et al. 2005), and in rodents reduces renal sympathetic nerve activity and blood pressure (Tanida et al. 2010). Homocarnosine is a brain-specific dipeptide (Bauer 2005) and has been suggested as a precursor for the neurotransmitter γ-aminobutyric acid (GABA) and also acts as an antioxidant, free radical scavenger, and metal-chelating agent especially for copper(II) and zinc(II) (Grasso et al. 2014). The formation of carnosine and homocarnosine is catalyzed by carnosine synthase (CS; EC 6.3.2.11). CS belongs to the ATP-grasp family of ligases and the gene (ATPGD1) is mainly present in skeletal and heart muscle and certain brain regions (Drozak et al. 2010), but also in kidney (Peters et al. 2015a). The olfactory neurons display very high CS expression, which is in agreement with its very high carnosine content (Margolis et al. 1987). In other brain regions, however, homocarnosine rather than carnosine is the main dipeptide. Recombinant mouse and human CS catalyzes the ATP-dependent synthesis of carnosine and, with a lower affinity, homocarnosine and other related dipeptides (Drozak et al. 2010). The enzyme is localized in the cytosol (Ng and Marshall 1978; Harding and O’Fallon 1979). Little is known about the regulation of CS expression and activity; a number of highly conserved cysteine residues indicates redox regulation (Drozak et al. 2010). The formation of anserine is more likely achieved through N-methylation of carnosine rather than enzymatic condensation of ß-alanine with N-methylhistidine (Bauer and Schulz 1994; Drozak et al. 2010). CS activity in diabetes and potential benefits of increased CS activity, e.g. under diabetic conditions, have not yet been studied.
Carnosinases
The carnosine-degrading enzyme carnosinase was first described and partially purified from porcine kidney (Hanson and Smith 1949) and in 1973, two electrophoretic forms of carnosinase in normal tissue extracts were identified, only one of which was lacking in a patient with carnosinaemia (Murphey et al. 1973). Lenny et al. confirmed two different metal-dependent porcine carnosinases, denoted homocarnosinase and carnosinase, with distinct differences in their substrate specificity (Lenney 1976; Lenney et al. 1977). In 1982, human serum carnosinase (EC 3.4.13.20) was isolated from human plasma (Lenney et al. 1983) and in 2003, two human carnosinase genes denoted CN1 (now CNDP1) and CN2 (now CNDP2) were characterized (Teufel et al. 2003). The term “carnosinase” is predominantly used for the enzyme also known as “serum carnosinase” or CN1 encoded by CNDP1. CN1 has a narrow substrate spectrum for histidine-containing dipeptides, such as carnosine, anserine and homocarnosine. In contrast, CN2 is a cytosolic nonspecific dipeptidase (EC 3.4.13.18) with a broader specificity of substrates, previously named prolinase (Lenney 1990). Both enzymes are members of the M20 family of metalloproteases (Teufel et al. 2003) and show 53% sequence identity in humans. CNDP1 and CNDP2 are located immediately adjacent on human chromosome 18q22.3 in a head-to-tail position. Although structurally related, CN1 and CN2 have quite different properties.
CN1
CN1 (EC 3.4.13.20) is the only dipeptidase with substrate specificity for carnosine (Fig. 1), anserine and homocarnosine. The pH activity curve of CN1 enzyme shows a broad maximum between pH 7.5 and 8.5. In humans, the CN1 gene CNDP1 is expressed in the central nervous system, the liver (Teufel et al. 2003) and kidney (Peters et al. 2015a). Rat and mouse orthologues of human carnosinase are found in the kidney but are not expressed in the CNS, supporting previous results describing a homocarnosine-splitting enzyme activity in the kidney of these animals (Margolis et al. 1983; Teufel et al. 2003). In human serum and cerebrospinal fluid (CSF) CN1 activity increases with age (Lenney et al. 1982) and varies greatly between individuals (Peters et al. 2011), with higher activities in females compared to males (Bando et al. 1984). Lower CN1 activity in children is not due to lower CN1 protein concentrations but different allosteric conformations of CN1 in children and adults (Peters et al. 2010; Adelmann et al. 2012). The human enzyme is present as a monomer or dimer (Pavlin et al. 2016). Carnosine is the best substrate for CN1; hydrolysis rates for homocarnosine and anserine in human serum are 50-fold and 200-fold lower than for carnosine (Peters et al. 2011). Homocarnosine and anserine effectively inhibit carnosine degradation by CN1, whereas related compounds such as carcinine, (β-alanylhistamine), do not (Peters et al. 2011). Secretion of CN1 is influenced by a common leucine repeat polymorphism in the signal peptide region of CNDP1 (exon 2) (Riedl et al. 2007) and by N-glycosylation (Riedl et al. 2010). CN1 has three N-glycosylation sites and its activity increases when all three sites are N-glycosylated (Riedl et al. 2010). Further, CN1 is a metal ion-dependent dipeptidase and its activity can be activated by addition of cadmium at low concentrations (0.1–3 μM) (Teufel et al. 2003).

Carnosine, anserine and homocarnosine pathway. CN1 degrades histidine-containing dipeptides, such as carnosine, anserine and homocarnosine. The formation of anserine by methylation is more likely than the enzymatic condensation of ß-alanine with N-methylhistidine by carnosine-synthase
Carnosinemia
Carnosinaemia as an inherited metabolic disease was first described in 1967 and confirmed to be due to serum carnosinase deficiency in 1968 (Perry et al. 1967; Perry et al. 1968; Jakobs et al. 1993; Scriver 2001). Since then several patients with this biochemical phenotype and a range of clinical symptoms have been reported (Fig. 2). In addition, reduced CN1 activity in serum may also cause a condition called homocarnosinosis with elevated concentrations of homocarnosine in cerebral spinal fluid (Gjessing and Sjaastad 1974; Sjaastad et al. 1976). Clinical features associated with serum carnosinase deficiency and carnosinaemia/homocarnosinaemia are highly variable (Bando et al. 1984, 1986; Duane and Peters 1988; Schoen et al. 2003; Balion et al. 2007), and a review of 23 patients in 1985 found no correlation between type and severity of neurological symptoms and residual serum CN1 activity (Cohen et al. 1985). This suggested that carnosinaemia may be an incidental finding of metabolic studies in children with symptoms with a different, independent cause (Cohen et al. 1985). The conclusion is indirectly supported by the report of a CNDP1 frameshift null mutation c.48_49insTGCTG (p.Leu17fs*20, rs532358622) with a carrier frequency of 1–1.4% in Europeans (Zschocke et al. 2006). Homozygosity for this mutation, and consequently complete absence of CN1 function, is expected to have a prevalence of 1:20.000–1:40.000, and if harmful should have been found in a much higher number of individuals.

Carnosine, anserine and homocarnosine have cytoprotective properties, i.e. scavenging carbonyls, such as methylglyoxal (MG), and act as antioxidants by scavenging reactive oxygen species (ROS). Increased CN1 activity under diabetic conditions may lead to organ damage by reactive metabolites due to decreased carnosine, anserine and homocarnosine tissue levels. Whether inborn deficiency of CN1 and subsequent carnosinaemia causes clinical symptoms is uncertain; experimental and clinical association studies suggest protection from reactive metabolites and diabetic nephropathy
Dietary intake of carnosine usually results in rapid degradation upon absorption, yet this was assumed to be less pronounced in subjects with low CN1 activity (Everaert et al. 2012). However, the half-life of carnosine in the human circulation is minutes only, even in subjects with low CN1 activity/content (Baguet et al. 2014). In patients with liver cirrhosis, serum CN1 abundance and activity was found to be more than 10-fold lower than in healthy controls (Peters et al. 2011), most likely caused by reduced hepatic expression of CN1, but plasma carnosine levels were not increased in these patients (Peters et al. 2011).
CNDP1 and the risk of diabetic nephropathy
Susceptibility to diabetic nephropathy is strongly associated with a leucine repeat polymorphism in the signal peptide region of CNDP1. Homozygosity for five leucine repeats, the shortest allelic variant (denoted “Mannheim allele”) is more common in patients with type 2 diabetes without nephropathy and associated with lower CN1 activities (Janssen et al. 2005). Mooyaart et al. showed that the effect may be gender-specific, with a stronger protective effect of the homozygous 5–5 genotype in women than in men (Mooyaart et al. 2010). This finding was recently reconfirmed in patients with biopsy-proven diabetic nephropathy (Albrecht et al. 2017). The association was also reported in European Americans (Freedman et al. 2007), South Asian Surinamese (Mooyaart et al. 2009) and in North Indians (Yadav et al. 2016) but not in African-Americans (Freedman et al. 2007; McDonough et al. 2009), Japanese (Kurashige et al. 2013) and in a small number of Scandinavian patients (Ahluwalia et al. 2011). In Japanese women with type 2 diabetes, a deep intronic SNP in CNDP1 was associated with overt proteinuria but not with end-stage renal disease (Kurashige et al. 2013). In the Scandinavian cohort of type 2 diabetic patients, a haplotype comprising three SNPs in the 3′ untranslated region of CNDP2 and the promotor region of CNDP1 was associated with DN (Ahluwalia et al. 2011). In type 1 diabetes, a possible role of the CNDP1 leucine repeat polymorphism is controversial (Bakker et al. 2008; Wanic et al. 2008; Craig et al. 2009; Alkhalaf et al. 2010). In non-diabetic children, the Mannheim allele is associated with slower progression of chronic kidney disease caused by glomerulonephritis but not by tubulointerstitial nephritis (Kiliś-Pstrusińska et al. 2010; Peters et al. 2016). On the other hand, women with the Mannheim allele (5 L–5 L genotype) were found to have a higher cardiovascular mortality risk (Alkhalaf et al. 2015).
Taken together, several association studies suggest renoprotection from lower CN1 activity in patients with diabetes mellitus and chronic glomerulonephritis. The molecular mechanisms underlying the association between CNDP1 variants and renoprotection, however, remain to be clarified. The Mannheim allele is not associated with increased plasma carnosine levels (Peters et al. 2011). In diabetic mice, renal CN1 activity is increased due to post-translational modifications (Peters et al. 2015b), and histidine-containing dipeptide concentrations are decreased (Peters et al. 2012). We speculate that the CNDP1 Mannheim allele may cause reduced CN1 activity and increased carnosine levels within the renal tissue; further work is required, e.g. in renal CN1 knock out models and respective human tissue.
In rodents, no serum CN1 is present and in diabetic mice, carnosine supplementation mitigates DN, reduces renal vasculopathy, normalizes vascular permeability (Peters et al. 2012) and improves wound healing (Ansurudeen et al. 2012) (Fig. 3). In diabetic rats, carnosine treatment prevents apoptosis of glomerular cells (Riedl et al. 2011; Peters et al. 2014), podocyte loss and vascular damage (Pfister et al. 2011). In humans, dietary supply of carnosine does not increase systemic histidine dipeptide concentrations due to rapid degradation by the serum CN1. An alternative approach to increase tissue carnosine concentrations is pharmacological inhibition of CN1 activity. We recently showed that cysteine-compounds inhibit CN1 activity by allosteric interactions through S-cysteinylation (Peters et al. 2017). The homocarnosine concentration in rodent renal tissue is below detection in health and diabetes, thus a role in protection from nephropathy is unlikely.

Feeding of diabetic mice (db/db) with carnosine over 4 weeks (a) normalized increased renal CN1 activity, (b) improved wound healing (Ansurudeen et al. 2012, adapted with permission from publisher) and (c) improved albuminuria, normalized vascular permeability (measured by Evans blue injection intravenously into the tail veins of mice) and reduced vasculopathy (by morphological and stereological evaluation) (Peters et al. 2012). *** = p < 0.01
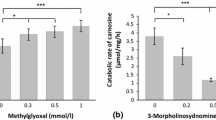
Under diabetic conditions, renal CN1 activity is increased (Fig. 2) due to post-translational modifications in diabetic mice and humans (Peters et al. 2015b). Reactive metabolites such as methylglyoxal (MG), reactive oxygen species (ROS) and nitrogen oxide (NO), increase CN1 activity by carbonylation and S-nitrosylation. Substitution of the two cysteine residues of CN1 by serine revealed the importance of cysteine at position 102 for enzyme activity, whereas cysteine at position 229 is irrelevant for CN1 activity. S-Nitrosylation of cysteine residue at position 102, but not at position 229, reduces CN1 activity (Peters et al. 2015b).
CN2
Whereas CN1 is well characterized, little is known about the regulation and function of CN2. The enzyme is a cytosolic nonspecific dipeptidase (EC 3.4.13.18), and as such not limited to histidine-containing dipeptides. It hydrolyzes carnosine only at alkaline pH with an optimum at pH 9.5 and does not degrade homocarnosine. CN2 RNA and protein are expressed in central and peripheral human tissues and in peripheral blood leukocytes (Lenney et al. 1985; Teufel et al. 2003). A recent crystallographic study of mouse CN2 revealed that each subunit consists of two domains, an A domain with catalytic and metal binding activity, and a B domain for dimerization (Unno et al. 2008). In contrast to CN1, CN2 can be inhibited by bestatin, a compound known to specifically inhibit various amino- and dipeptidases (Suda et al. 1976). Bound to domain A, bestatin interacts with several residues of domain B of the other subunit. These interactions are likely to be essential for enzyme activity (Unno et al. 2008). Furthermore, immunohistochemical staining of the rat hypothalamus with anti-CN2 antibody demonstrated high CN2 expression in histaminergic neurons of the tuberomammillary nucleus, suggesting that it may supply histidine to these neurons for histamine biosynthesis (Otani et al. 2008). Whether CN2 plays a role in carnosine metabolism remains unknown. Jansen et al. (2015) recently reported a different function of CN2, the formation of N-lactoyl-amino acids. N-lactoyl-aminoacids are rapidly formed by reverse proteolysis. The plasma levels of these metabolites strongly correlate with plasma levels of lactate and amino acids. In patients with phenylketonuria (PKU), N-lactoyl-phenylalanine (N-lac-Phe) plasma levels showed a positive correlation with plasma phenylalanine levels (Jansen et al. 2015).
Additional dipeptidases involved in carnosine metabolism
A cytosolic β-alanyl-lysine dipeptidase (PM20D2) activity was described in rodent muscle. This enzyme is an M20 metalloprotease which belongs to the same family as CN1 and CN2. It cannot degrade carnosine or homocarnosine but hydrolyzes β-alanyl-lysine, β-alanyl-ornithine, γ-aminobutyryl-lysine, and γ-aminobutyryl-ornithine. It assists carnosine synthase by degrading abnormal biosynthetic dipeptides and thus protects the physiologically relevant carnosine and homocarnosine from the accumulation of these compounds (Veiga-da-Cunha et al. 2014). In humans, PM20D2 is also present but less active. Another dipeptidase called anserinase is found in fish (EC 3.4.13.5); it also belongs to the M20 metalloprotease family and shows broad substrate specificity which includes carnosine, anserine and homocarnosine. In bacteria, two further carnosine-cleaving enzymes denoted peptidase V and peptidase D (EC 3.4.13.3) are known, for details see (Bellia et al. 2014).
Carnosine-methyltransferase
Anserine can be formed by N-methylation of carnosine in skeletal muscle (McManus 1962). Drozak et al. identified a carnosine N-methyltransferase in chicken named HNMT-like protein (CMT, EC 2.1.1.22), catalyzing the transfer of methylgroup from S-adenosyltransferase (SAM) onto carnosine (Drozak et al. 2013). Amphibian and mammalian genomes do not contain orthologues of the CMT gene, indicating another anserine-producing enzyme in mammals.
Carnosine transporter
Cellular uptake of carnosine and anserine occurs by proton-coupled oligopeptide transporters (H+)(POTS). These are membrane proteins that translocate various small peptides and peptide-like drugs across the biological membrane via an inwardly-directed proton gradient and negative membrane potential (Daniel and Kottra 2004). At present, four members of the POT family — PEPT1, PEPT2, PHT1 and PHT2 — have been identified in mammals. The impact of carnosine transporter expression and carnosine transport activity on tissue and cellular carnosine homeostasis is largely unknown as are the potential clinical consequences. PEPT1 mediates intestinal absorption of luminal di/tripeptides from dietary protein digestion, while PEPT2 mainly allows for renal tubular reabsorption of di/tripeptides from the glomerular ultrafiltration. PHT2 and PHT1 possibly interact with di/tripeptides and histidine in certain immune cells (Verri et al. 2017). In rats, PHT1 is widely expressed and transports carnosine and histidine in brain and retina of rats (Yamashita et al. 1997). PEPT2 is responsible for the cellular uptake of exogenous carnosine in mice (Kamal et al. 2009) and mediates >90% of cellular carnosine uptake in the choroid plexus (Teuscher et al. 2004).
Conclusion
The biological relevance of carnosine and dipeptide metabolism is only partially understood. Carnosinaemia due to serum carnosinase deficiency has been suggested as an inherited neurometabolic disorder, but there is also evidence to suggest that this condition is clinically irrelevant. In contrast, low CN1 serum activity due to a functional leucine repeat polymorphisms in CDNP1 is associated with a lower incidence of diabetic nephropathy and possibly a slower progression of chronic renal failure in children with non-diabetic glomerulopathies.
In diabetic mice, increased renal CN1 activity causes decreased histidine-containing dipeptide concentrations. Exogenous carnosine supply in rodents exerts a range of nephroprotective effects such as reductions of proteinuria, renal vasculopathy, and podocyte loss. Carnosine therefore may be a promising therapeutic target in patients with diabetes mellitus. While potential benefits of oral carnosine supplementation in humans are counteracted by a high serum CN1 activity, inhibition of CN1 or upregulation of CS activity might protect diabetic patients from damage exerted by hyperglycemia and accumulated reactive metabolites.
References
Adelmann K, Frey D, Riedl E et al. (2012) Different conformational forms of serum carnosinase detected by a newly developed sandwich ELISA for the measurements of carnosinase concentrations. Amino Acids 43:143–151
Ahluwalia TS, Lindholm E, Groop LC (2011) Common variants in CNDP1 and CNDP2, and risk of nephropathy in type 2 diabetes. Diabetologia 54:2295–2302
Albrecht T, Zhang S, Braun JD et al. (2017) The CNDP1 (CTG)5 polymorphism is associated with biopsy-proven diabetic nephropathy, time on hemodialysis, and diabetes duration. J Diabetes Res 2017:9506730
Aldini G, Facino RM, Beretta G, Carini M (2005) Carnosine and related dipeptides as quenchers of reactive carbonyl species: from structural studies to therapeutic perspectives. BioFactors 24:77–87
Alhamdani M, Al-Azzawie HF, Abbas FK (2007) Decreased formation of advanced glycation end-products in peritoneal fluid by carnosine and related peptides. Perit Dial Int 27:86–89
Alkhalaf A, Bakker SJ, Bilo HJ et al. (2010) A polymorphism in the gene encoding carnosinase (CNDP1) as a predictor of mortality and progression from nephropathy to end-stage renal disease in type 1 diabetes mellitus. Diabetologia 53:2562–2568
Alkhalaf A, Landman GW, van Hateren KJ et al. (2015) Sex specific association between carnosinase gene CNDP1 and cardiovascular mortality in patients with type 2 diabetes (ZODIAC-22). J Nephrol 28:201–207
Ansurudeen I, Sunkari VG, Grunler J et al. (2012) Carnosine enhances diabetic wound healing in the db/db mouse model of type 2 diabetes. Amino Acids 43:127–134
Babizhayev MA, Lankin VZ, Savel’Yeva EL, Deyev AI, Yegorov YE (2013) Diabetes mellitus: novel insights, analysis and interpretation of pathophysiology and complications management with imidazole-containing peptidomimetic antioxidants. Recent Pat Drug Deliv Formul 7:216–256
Baguet A, Everaert I, Yard B et al. (2014) Does low serum carnosinase activity favor high-intensity exercise capacity? J Appl Physiol (1985) 116:553–559
Bakker SJ, Alkhalaf A, Tarnow L, Navis G (2008) Re: Exclusion of polymorphisms in carnosinase genes (CNDP1 and CNDP2) as a cause of diabetic nephropathy in type 1 diabetes: results of large case-control and follow-up studies. Diabetes 57:e16 author reply e17
Balion CM, Benson C, Raina PS, Papaioannou A, Patterson C, Ismaila AS (2007) Brain type carnosinase in dementia: a pilot study. BMC Neurol 7:38
Bando K, Shimotsuji T, Toyoshima H, Hayashi C, Miyai K (1984) Fluorometric assay of human serum carnosinase activity in normal children, adults and patients with myopathy. Ann Clin Biochem 21(Pt 6):510–514
Bando K, Ichihara K, Shimotsuji T et al. (1986) Reduced serum carnosinase activity in hypothyroidism. Ann Clin Biochem 23(Pt 2):190–194
Barski OA, Xie Z, Baba SP et al. (2013) Dietary carnosine prevents early atherosclerotic lesion formation in apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol 33:1162–1170
Bauer K (2005) Carnosine and homocarnosine, the forgotten, enigmatic peptides of the brain. Neurochem Res 30:1339–1345
Bauer K, Schulz M (1994) Biosynthesis of carnosine and related peptides by skeletal muscle cells in primary culture. Eur J Biochem 219:43–47
Bellia F, Vecchio G, Rizzarelli E (2014) Carnosinases, their substrates and diseases. Molecules 19:2299–2329
Boldyrev AA, Aldini G, Derave W (2013) Physiology and pathophysiology of carnosine. Physiol Rev 93:1803–1845
Brings S, Fleming T, De Buhr S et al. (2017) A scavenger peptide prevents methylglyoxal induced pain in mice. Biochim Biophys Acta 1863:654–662
Cohen M, Hartlage PL, Krawiecki N, Roesel RA, Carter AL, Hommes FA (1985) Serum carnosinase deficiency: a non-disabling phenotype? J Ment Defic Res 29(Pt 4):383–389
Craig DW, Millis MP, DiStefano JK (2009) Genome-wide SNP genotyping study using pooled DNA to identify candidate markers mediating susceptibility to end-stage renal disease attributed to type 1 diabetes. Diabet Med 26:1090–1098
Daniel H, Kottra G (2004) The proton oligopeptide cotransporter family SLC15 in physiology and pharmacology. Pflugers Archiv 447:610–618
Decker E, Livisay SA, Zhou S (2000) A re-evaluation of the antioxidant activity of purified carnosine. Biochemistry 65:766–770
Drozak J, Veiga-da-Cunha M, Vertommen D, Stoobant V, Van Schaftingen E (2010) Molecular identification of carnosine synthase as ATP-grasp domain-containing protein 1 (ATPGD1). J Biol Chem 2685:9346–9356
Drozak J, Chrobok L, Poleszak O, Jagielski AK, Derlacz R (2013) Molecular identification of carnosine N-methyltransferase as chicken histamine N-methyltransferase-like protein (hnmt-like). PLoS One 8:e64805
Duane P, Peters TJ (1988) Serum carnosinase activities in patients with alcoholic chronic skeletal muscle myopathy. Clin Sci 75:185–190
Everaert I, Taes Y, De Heer E et al. (2012) Low plasma carnosinase activity promotes carnosinemia after carnosine ingestion in humans. Am J Physiol Renal Physiol 302:F1537–F1544
Freedman BI, Hicks PJ, Sale MM et al. (2007) A leucine repeat in the carnosinase gene CNDP1 is associated with diabetic end-stage renal disease in European Americans. Nephrol Dial Transplant 22:1131–1135
Gjessing LR, Sjaastad O (1974) Letter: Homocarnosinosis: a new metabolic disorder associated with spasticity and mental retardation. Lancet 2:1028
Grasso GI, Arena G, Bellia F, Rizzarelli E, Vecchio G (2014) Copper(II)-chelating homocarnosine glycoconjugate as a new multifunctional compound. J Inorg Biochem 131:56–63
Gulewitsch W (1905) On the nature of substances extracted from muscle: a communication about carnitine. Zeitschrift fur Physiologische Chemie 45:326–330
Gulewitsch W, Amiradžibi S (1900) Ueber das carnosin, eine neue organische Base des Fleischextractes. Eur J Inorg Chem 33:1902–1903
Hanson HT, Smith EL (1949) Carnosinase; an enzyme of swine kidney. J Biol Chem 179:789–801
Harding JW, O’Fallon JV (1979) The subcellular distribution of carnosine, carnosine synthetase, and carnosinase in mouse olfactory tissues. Brain Res 173:99–109
Hipkiss AR (2011) Energy metabolism, proteotoxic stress and age-related dysfunction - protection by carnosine. Mol Asp Med 32:267–278
Hou W, Chen HJ, Lin YH (2003) Antioxidant peptides with Angiotensin converting enzyme inhibitory activities and applications for Angiotensin converting enzyme purification. J Agric Food Chem 51:1706–17093
Jakobs C, Jaeken J, Gibson KM (1993) Inherited disorders of GABA metabolism. J Inherit Metab Dis 16:704–715
Jansen RS, Addie R, Merkx R et al. (2015) N-lactoyl-amino acids are ubiquitous metabolites that originate from CNDP2-mediated reverse proteolysis of lactate and amino acids. Proc Natl Acad Sci U S A 112:6601–6606
Janssen B, Hohenadel D, Brinkkoetter P et al. (2005) Carnosine as a protective factor in diabetic nephropathy: association with a leucine repeat of the carnosinase gene CNDP1. Diabetes 54:2320–2327
Kamal MA, Jiang H, Hu Y, Keep RF, Smith DE (2009) Influence of genetic knockout of Pept2 on the in vivo disposition of endogenous and exogenous carnosine in wild-type and Pept2 null mice. Am J Physiol Regul Integr Comp Physiol Physiol 296:R986–R991
Kiliś-Pstrusińska K, Zwolińska D, Grzeszczak W, Study Group (2010) Is carnosinase 1 gene (CNDP1) polymorphism associated with chronic kidney disease progression in children and young adults? Results of a family-based study. Arch Med Res 41:356–362
Kohen R, Yamamoto Y, Cundy KC, Ames BN (1988) Antioxidant activity of carnosine, homocarnosine, and anserine present in muscle and brain. Proc Natl Acad Sci U S A 85:3175–3179
Kurashige M, Imamura M, Araki S et al. (2013) The influence of a single nucleotide polymorphism within CNDP1 on susceptibility to diabetic nephropathy in Japanese women with type 2 diabetes. PLoS One 8:e54064
Lenney JF (1976) Specificity and distribution of mammalian carnosinase. Biochim Biophys Acta 429:214–219
Lenney JF (1990) Human cytosolic carnosinase: evidence of identity with prolinase, a non-specific dipeptidase. Biol Chem Hoppe Seyler 371:167–171
Lenney JF, Kan SC, Siu K, Sugiyama GH (1977) Homocarnosinase: a hog kidney dipeptidase with a broader specificity than carnosinase. Arch Biochem Biophys 184:257–266
Lenney J, Georg RP, Weiss AM, Kucera CM, Chan PW, Rinzler GS (1982) Human serum carnosinase: characterization, distinction from cellular carnosinase, and activation by cadmium. Clin Chim Acta 123:221–231
Lenney JF, Peppers SC, Kucera CM, Sjaastad O (1983) Homocarnosinosis: lack of serum carnosinase is the defect probably responsible for elevated brain and CSF homocarnosine. Clin Chim Acta 132:157–165
Lenney JF, Peppers SC, Kucera-Orallo CM, George RP (1985) Characterization of human tissue carnosinase. Biochem J 228:653–660
Margolis FL, Grillo M, Grannot-Reisfeld N, Farbman AI (1983) Purification, characterization and immunocytochemical localization of mouse kidney carnosinase. Biochim Biophys Acta 744:237–248
Margolis FL, Grillo M, Hempstead J, Morgan JI (1987) Monoclonal antibodies to mammalian carnosine synthetase. J Neurochem 48:593–600
McDonough C, Hicks PJ, Lu L, Langefeld CD, Freedman BI, Bowden DW (2009) The influence of carnosinase gene polymorphisms on diabetic nephropathy risk in African-Americans. Hum Genet 126(2):265–275
McManus IR (1962) Enzymatic synthesis of anserine in skeletal muscle by N-methylation of carnosine. J Biol Chem 237:1207–1211
Mooyaart A, van Valkengoed IG, Shaw PK, Peters V, Baelde HJ, Rabelink TJ, Bruijn JA, Stronks K, de Heer E (2009) Lower frequency of the 5/5 homozygous CNDP1 genotype in South Asian Surinamese. Diabetes Res Clin Pract 85:272–278
Mooyaart AL, Zutinic A, Bakker SJ et al. (2010) Association between CNDP1 genotype and diabetic nephropathy is sex specific. Diabetes 59:1555–1559
Mozdan M, Szemraj J, Rysz J, Nowak D (2005) Antioxidant properties of carnosine re-evaluated with oxidizing systems involving iron and copper ions. Basic Clin Pharmacol Toxicol 96:352–360
Murphey WH, Lindmark DG, Patchen LI, Housler ME, Harrod EK, Mosovich L (1973) Serum carnosinase deficiency concomitant with mental retardation. Pediatr Res 7:601–606
Nakagawa K, Ueno A, Nishikawa Y (2006) Interactions between carnosine and captopril on free radical scavenging activity and angiotensin-converting enzyme activity in vitro. Yakugaku Zasshi 126:37–42
Negre-Salvayre A, Coatrieux C, Ingueneau C, Salvayre R (2008) Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. Br J Pharmacol 153:6–20
Ng RH, Marshall FD (1978) Regional and subcellular distribution of homocarnosine-carnosine synthetase in the central nervous system of rats. J Neurochem 30:I87–I90
Otani H, Okumura A, Nagai K, Okumura N (2008) Colocalization of a carnosine-splitting enzyme, tissue carnosinase (CN2)/cytosolic non-specific dipeptidase 2 (CNDP2), with histidine decarboxylase in the tuberomammillary nucleus of the hypothalamus. Neurosci Lett 445:166–169
Pavlin M, Rossetti G, De Vivo M, Carloni P (2016) Carnosine and Homocarnosine Degradation Mechanisms by the Human Carnosinase Enzyme CN1: Insights from Multiscale Simulations. Biochemistry 55:2772–2784
Perry TL, Hansen S, Tischler B, Bunting R, Berry K (1967) Carnosinemia. A new metabolic disorder associated with neurologic disease and mental defect. N Engl J Med 277:1219–1227
Perry TL, Hansen S, Love DL (1968) Serum-carnosinase deficiency in carnosinaemia. Lancet 1:1229–1230
Peters V, Kebbewar M, Jansen EW et al. (2010) Relevance of allosteric conformations and homocarnosine concentration on carnosinase activity. Amino Acids 38:1607–1615
Peters V, Jansen EE, Jakobs C et al. (2011) Anserine inhibits carnosine degradation but in human serum carnosinase (CN1) is not correlated with histidine dipeptide concentration. Clin Chim Acta 412:263–267
Peters V, Schmitt CP, Zschocke J, Gross ML, Brismar K, Forsberg E (2012) Carnosine treatment largely prevents alterations of renal carnosine metabolism in diabetic mice. Amino Acids 42:2411–2416
Peters V, Riedl E, Braunagel M et al. (2014) Carnosine treatment in combination with ACE inhibition in diabetic rats. Regul Pept 194-195:36–40
Peters V, Klessens CQ, Baelde HJ et al. (2015a) Intrinsic carnosine metabolism in the human kidney. Amino Acids 47(12):2541–2550
Peters V, Lanthaler B, Amberger A et al. (2015b) Carnosine metabolism in diabetes is altered by reactive metabolites. Amino Acids 47:2367–2376
Peters V, Kebbewar M, Janssen B et al. (2016) CNDP1 genotype and renal survival in pediatric nephropathies. J Pediatr Endocrinol Metab 29(7):827–833
Peters V, Schmitt CP, Weigand T et al. (2017) Allosteric inhibition of carnosinase (CN1) by inducing a conformational shift. J Enzyme Inhib Med Chem 32:1102–1110
Pfister F, Riedl E, Wang Q et al. (2011) Oral carnosine supplementation prevents vascular damage in experimental diabetic retinopathy. Cell Physiol Biochem 28:125–136
Riedl E, Koeppel H, Brinkkoetter P et al. (2007) A CTG polymorphism in the CNDP1 gene determines the secretion of serum carnosinase in Cos-7 transfected cells. Diabetes 56:2410–2413
Riedl E, Koeppel H, Pfister F et al. (2010) N-glycosylation of carnosinase influences protein secretion and enzyme activity: implications for hyperglycemia. Diabetes 59:1984–1990
Riedl E, Pfister F, Braunagel M et al. (2011) Carnosine prevents apoptosis of glomerular cells and podocyte loss in STZ diabetic rats. Cell Physiol Biochem 28:279–288
Schoen P, Everts H, de Boer T, van Oeveren W (2003) Serum carnosinase activity in plasma and serum: validation of a method and values in cardiopulmonary bypass surgery. Clin Chem 49:1930–1932
Scriver CR (2001) The metabolic & molecular bases of inherited disease. McGraw-Hill, New York
Sjaastad O, Berstad J, Gjesdahl P, Gjessing L (1976) Homocarnosinosis. 2. A familial metabolic disorder associated with spastic paraplegia, progressive mental deficiency, and retinal pigmentation. Acta Neurol Scand 53:275–290
Suda H, Aoyagi T, Takeuchi T, Umezawa H (1976) Inhibition of aminopeptidase B and leucine aminopeptidase by bestatin and its stereoisomer. Arch Biochem Biophys 177:196–200
Tanida M, Shen J, Kubomura D, Nagai K (2010) Effects of anserine on the renal sympathetic nerve activity and blood pressure in urethane-anesthetized rats. Physiol Res 59:177–185
Teufel M, Saudek V, Ledig JP, Bernhardt A, Boularand S, Carreau A, Cairns NJ, Carter C, Cowley DJ, Duverger D, Ganzhorn AJ, Guenet C, Heintzelmann B, Laucher V, Sauvage C, Smirnova T (2003) Sequence identification and characterization of human carnosinase and a closely related non-specific dipeptidase. J Biol Chem 278:6251–6531
Teuscher NS, Shen H, Shu C, Xiang J, Keep RF, Smith DE (2004) Carnosine uptake in rat choroid plexus primary cell cultures and choroid plexus whole tissue from PEPT2 null mice. J Neurochem 89:375–382
Unno H, Yamashita T, Ujita S et al. (2008) Structural basis for substrate recognition and hydrolysis by mouse carnosinase CN2. J Biol Chem 283:27289–27299
Veiga-da-Cunha M, Chevalier N, Stroobant V, Vertommen D, Van Schaftingen E (2014) Metabolite proofreading in carnosine and homocarnosine synthesis: molecular identification of PM20D2 as beta-alanyl-lysine dipeptidase. J Biol Chem 289:19726–19736
Velez S, Nair NG, Reddy VP (2008) Transition metal ion binding studies of carnosine and histidine: biologically relevant antioxidants. Colloids Surf B: Biointerfaces 66:291–294
Verri T, Barca A, Pisani P, Piccinni B, Storelli C, Romano A (2017) Di- and tripeptide transport in vertebrates: the contribution of teleost fish models. J Comp Physiol B 187:395–462
Wanic K, Placha G, Dunn J, Smiles A, Warram JH, Krolewski AS (2008) Exclusion of polymorphisms in carnosinase genes (CNDP1 and CNDP2) as a cause of diabetic nephropathy in type 1 diabetes: results of large case-control and follow-up studies. Diabetes 57:2547–2551
Yadav AK, Sinha N, Kumar V, Bhansali A, Dutta P, Jha V (2016) Association of CTG repeat polymorphism in carnosine dipeptidase 1 (CNDP1) gene with diabetic nephropathy in north Indians. Indian J Med Res 144:32–37
Yamashita T, Shimada S, Guo W et al. (1997) Cloning and functional expression of a brain peptide/histidine transporter. J Biol Chem 272:10205–10211
Zschocke J, Nebel A, Wicks K et al. (2006) Allelic variation in the CNDP1 gene and its lack of association with longevity and coronary heart disease. Mech Ageing Dev 127:817–820
Acknowledgements
Part of this work was supported by the Deutsche Forschungsgemeinschaft (DFG; SFB 1118).
Author information
Authors and Affiliations
Contributions
All authors contributed equally and each were involved in planning this article, interpretation of literature, and in drafting and critically revising the manuscript. All authors reviewed the final manuscript and gave approval for submission.
Corresponding author
Ethics declarations
This article is a review of previously published work and does not present any new previously unpublished studies with human or animal subjects performed by the any of the authors.
Animal rights
The article does not contain animal subjects.
Conflict of interest
V. Peters, J. Zschocke and C. P. Schmitt declare that they have no conflict of interest.
Additional information
Communicated by: Eva Morava
Rights and permissions
About this article

Cite this article
Peters, V., Zschocke, J. & Schmitt, C.P. Carnosinase, diabetes mellitus and the potential relevance of carnosinase deficiency. J Inherit Metab Dis 41, 39–47 (2018). https://doi.org/10.1007/s10545-017-0099-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-017-0099-2