
Abstract
Mucopolysaccharidosis (MPS) type IIIA, or Sanfilippo syndrome, is a neurodegenerative lysosomal storage disorder caused by a deficiency of the lysosomal enzyme N-sulfoglucosamine sulfohydrolase (SGSH), involved in the catabolism of heparan sulfate. The clinical spectrum is broad and the age of symptom onset and the degree of preservation of cognitive and motor functions appears greatly influenced by genotype. To explore this further, we generated a conditional knockout (Sgsh KO) mouse model with ubiquitous Sgsh deletion, and compared the clinical and pathological phenotype with that of the spontaneous Sgsh D31N MPS-IIIA mouse model. Phenotypic deficits were noted in Sgsh KO mice prior to Sgsh D31N mice, however these outcomes did not correlate with any shift in the time of appearance nor rate of accumulation of primary (heparan sulfate) or secondary substrates (GM2/GM3 gangliosides). Other disease lesions (elevations in lysosomal integral membrane protein-II expression, reactive astrocytosis and appearance of ubiquitin-positive inclusions) were also comparable between affected mouse strains. This suggests that gross substrate storage and these neuropathological markers are neither primary determinants, nor good biomarkers/indicators of symptom generation, confirming similar observations made recently in MPS-IIIA patients. The Sgsh KO mouse will be a useful tool for elucidation of the neurological basis of disease and assessment of the clinical efficacy of new treatments for Sanfilippo syndrome.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mucopolysaccharidosis (MPS)-IIIA (OMIM252900) is caused by mutations in the SGSH gene resulting in insufficient N-sulfoglucosamine sulfohydrolase (SGSH; EC3.10.1.1) activity and consequent accumulation of heparan sulfate (HS) oligosaccharides. Patients typically present in toddlerhood with delays in reaching developmental milestones, particularly speech (Valstar et al 2008, 2010). This is generally followed by progressive neurological deterioration featuring hyperactivity, aggression, sleep disturbance and regression in cognitive and motor function. Unsteady gait and seizures occur and there is regression to a vegetative state. No treatment is available and patients generally die in their teens. However, the clinical spectrum is broad and the age of symptom onset and the degree of preservation of cognitive and motor skills appears greatly influenced by genotype. For example, MPS-IIIA has been diagnosed in a 45-year-old who presented with hypertension in the absence of neurologic impairment (Van Hove et al 2003). Likewise, a patient with homozygous p.S298P mutations was able to speak some words and walk independently shortly before his death at age 51-years (Valstar et al 2010).
Pre-symptomatic neonates with MPS-IIIA are expected to be identified in trials of newborn screening (Matern et al 2015), therefore understanding what determines the age of symptom onset and the rate of symptom progression is both an ethical imperative and essential for the cogent application of treatments. To identify determinants of disease expression, animal models that faithfully recapitulate the cardinal features of the disease are of considerable use. Several MPS III animal models exist including the spontaneously-occurring D31N MPS-IIIA mouse model (Sgsh D31N) which has approximately 3–4% SGSH activity (Bhaumik et al 1999; Crawley et al 2006). Here, we generated a novel conditional Sgsh KO MPS-IIIA mouse model with ubiquitous deletion of Sgsh and compared the behavioral and pathological phenotype to the Sgsh D31N model. We hypothesized that genotype influences phenotype, such that Sgsh KO mice exhibit accelerated disease progression, c.f. Sgsh D31N mice.
Materials and methods
Additional details can be found in the Supplementary information.
Mouse line generation and husbandry
The pDTA-SGSH-CKO targeting plasmid (Fig. 1a–ii) was electroporated into Bruce4 embryonic stem cells. Clonal cell lines with the targeting cassette in intron 2 of the murine Sgsh gene (Fig. 1a–iii) were injected into BALB/c blastocysts and chimeric male progeny were mated with C57BL/6 dams. Mice containing the targeted allele were bred with B6.Cg-Tg(ACTFLPe)9205Dym/J or B6.C-Tg(CMV-Cre)1Cgn/J mice to generate Sgsh flox mice (Sgsh tm1Ldru; Fig. 1a–iv) or Sgsh KO MPS-IIIA mice (Sgsh tm1.1Ldru; Fig. 1a-v), respectively. Congenic C57BL/6 Sgsh +/+ or Sgsh +/− (collectively referred to as “wild-type”) and D31N MPS-IIIA (B6.Cg-Sgsh mps3a or Sgsh D31N) mice were obtained from a breeding colony at the Women’s and Children’s Health Network Animal Care Facility (Crawley et al 2006). Genotyping was conducted using the conditions described in Suppl. Table 1 or in Lau et al (2013). Sequencing, alloenzyme electrophoresis and single nucleotide variations/polymorphisms (SNP) analysis were performed by commercial laboratories. All institutional and national guidelines for the care and use of laboratory animals were followed.

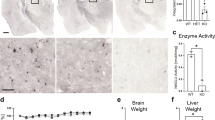
Conditional Sgsh gene deletion and effect on mRNA and protein expression. (a) (i) Wild-type murine Sgsh gene with open boxes representing exons 1 to 8 (Sgsh +). (ii) The pDTA-SGSH-CKO targeting vector. The arrows represent chloramphenicol resistance (Ch), neomycin resistance (Neo) or diphtheria toxin A (DTA) genes. Triangles and crescents represent loxP or FRT sites, respectively. (iii) The targeted allele in mice following recombination. (iv) Sgsh flox mice were generated by breeding with ACTB-FLPe mice and were deleted of the Neo gene. (v) Conditional Sgsh KO mice (Sgsh KO) were generated by breeding with CMV-Cre mice causing the excision of exon 2 of Sgsh which is predicted to result in a frameshift mutation causing a predicted premature stop codon in exon 3. Shaded rectangles indicate exons that are frame shifted. (b) Genotyping of wild-type (Sgsh +/+), Sgsh KO MPS-IIIA or Sgsh flox mice by multiplex PCR amplification of genomic DNA. Expected bands of sizes 516, 475, and 387 bp are expected for Sgsh flox, Sgsh KO, and Sgsh +/+ alleles. Amplification of a control gene (IL2) is observed at 324 bp. (c) Sgsh expression in 20-week-old brain homogenates was determined using semi-quantitative PCR. Full length and truncated forms of Sgsh were detected at 276 and 115 bp, respectively (n = 3 mice/genotype). (d) SGSH activity measured with 4MU-conjugated substrate in brain or liver homogenates from 20- to 21-week-old mice, or in mouse fibroblast cell extracts (n = 3–4 mice/group). Inset, SGSH activity measured with radio-labeled natural substrate in brain homogenates (n = 3 mice/group). n.d. not detected. +/+, Sgsh +/+ or Sgsh flox; +/−, heterozygote; −/−, Sgsh D31N or Sgsh KO MPS-IIIA. Error bars ±1 SEM. * p < 0.05
mRNA and protein expression
Total RNA was extracted with Trizol. cDNA was generated using 1-μg of DNase I-treated RNA using a Superscript III first strand synthesis system kit and oligo(dT)20 primer (ThermoFisher Scientific). Semi-quantitative PCR was conducted using 5′-CCTGCTGCACAATTCTGTTGG-3′ and 5′-TGAAGTGATGCACATCCTGGT-3′ primers.
SGSH activity was assessed in 15-μg of total protein equivalent of PBS-perfused brain or liver homogenates or in mouse fibroblasts cell extracts after dialysis overnight in 0.2 M sodium acetate, pH 6.5, using a 4MU-conjugated αGlcNS substrate (Carbosynth Limited, Berkshire, UK; EM06602) according to Whyte et al (2015).
Behavior
All male mice were separated into single cages at ∼10-weeks of age due to increased aggression in Sgsh D31N and Sgsh KO males. Testing was performed in a randomized order without knowledge of genotype/colony. Open field activity was evaluated using HVS Image Field2020 software (Lau et al 2008). Cognition was examined in the Morris water maze using HVS Image Water2020 software using a modified protocol of Hemsley et al (2009). Motor function was tested using pole and neuromuscular grip strength tests (Hemsley and Hopwood 2005; Griffey et al 2006).
Mass spectrometry
Brain HS and gangliosides were determined using established methods (Marshall et al 2015; Trim et al 2015). Samples were analyzed on an API4000 QTrap hybrid triple quadrupole/linear ion trap mass spectrometer and peak areas integrated with Analyst 1.4.2 software (ABSciex; Concord, Ontario, Canada).
Immunohistochemistry
Mice were fixation-perfused and the brains embedded in paraffin wax. Lysosomal integral membrane protein (LIMP)-II, glial fibrillary acidic protein (GFAP), and ubiquitin staining was performed (Hemsley et al 2009).
Statistics
Kruskal-Wallis, log-linear analysis for a three-way contingency table, repeated measures ANOVA or ANOVA were used to compare the main effects of genotype or colony. Post-hoc testing was conducted using the Bonferroni test. p ≤ 0.05 was considered statistically significant.
Results
Generation and breeding outcomes of Sgsh flox and Sgsh KO mouse lines
Gene targeting was used to insert loxP and FRT sites flanking exon 2 of murine Sgsh and the neo gene, respectively (Fig. 1a). Sgsh flox/+; FLPe+/− mice were generated by breeding mice containing the targeted allele with the ACTB-FLPe mice. The neo gene was excised when FLP recombinase was expressed, leaving the loxP sites and introduced restriction enzyme sites in the intronic DNA. Also, mice containing the targeted allele were paired with the CMV-Cre mice to generate Sgsh KO/+; Cre+/−progeny. Cre-mediated excision of exon 2 is predicted to result in a frameshift and the introduction of a nonsense mutation in exon 3 (Fig. 1 ; Suppl. Fig. 1). Specific PCR amplification confirmed recombinase sequence deletion and allowed discrimination of Sgsh + , Sgsh flox, and Sgsh KO alleles for genotyping (Fig. 1b).
One Sgsh KO; Cre −/− and Sgsh flox; FLPe −/− foundation pair subsequently produced Sgsh flox/KO progeny which were inter-crossed to give Sgsh flox , Sgsh flox/KO Sgsh KO mice on an identical C57BL6 genetic background. Sequencing confirmed deletion of the neomycin and/or Sgsh exon 2 in Sgsh flox and Sgsh KO mice, respectively (data not shown). Allozyme electrophoresis profiles from both colonies were like the C57BL/6 reference. However, subsequent SNP analysis showed 7% variation compared to the C57BL6/J reference (41/588 SNP; Suppl. Table 2). The SNP polymorphisms were not linked to a specific chromosome. Two SNPS (0.3%) were divergent in Sgsh +/D31N mice.
Sgsh KO mice were fertile until at least ∼19-weeks of age. However, pairing with at least one Sgsh KO parent reduced the number of successful pregnancies compared to Sgsh flox/?xSgsh flox/? breeding pairs (Suppl. Table 3). Reduced fertility was also observed in Sgsh D31N pairings. No differences were measured in the pairing-to-birth interval. Litter sizes were smaller for Sgsh D31N pairs compared to the other combinations in that colony. In contrast, litter sizes were equivalent for all mating combinations in the Sgsh flox/KO colony. Between 9 and 17% of pups born were lost by weaning in the Sgsh flox/KO colony, mainly due to cannibalism or maternal neglect.
Sgsh mRNA and protein expression
Stable Sgsh expression was observed in wild-type, Sgsh +/D31N, and Sgsh D31N mice (Fig. 1c). Sgsh was also expressed in Sgsh flox and Sgsh flox/KO brains. In Sgsh flox/KO and Sgsh KO mice, a stable, truncated Sgsh transcript was detected corresponding with exon 2 deletion. SGSH activity was readily detectable in wild-type and Sgsh flox brain but was not detected in either MPS-IIIA line using fluorogenic or natural substrates (Fig. 1d) (Hopwood and Elliott 1982). In livers and fibroblast extracts, no significant differences in activity were measured between Sgsh D31N and Sgsh KO.
Clinical phenotype
Sgsh flox and Sgsh KO mice were phenotypically indistinguishable at birth. Chronic urinary retention was observed in Sgsh D31N and Sgsh KO males by 20 weeks of age. Sgsh D31N and Sgsh KO mice were heavier than age-matched wild-type and Sgsh flox counterparts, respectively (Fig. 2a). Sgsh flox mice were smaller than age-matched wild-type mice, as were Sgsh KO mice compared to Sgsh D31N mice (p < 0.05). Liver enlargement was observed in both MPS-IIIA models, with a 37% and 40% increase recorded for Sgsh D31N and Sgsh KO mice, respectively (Fig. 2b). Both MPS-IIIA cohorts also exhibited heavier spleens (120% and 114% of control for Sgsh D31N and Sgsh KO, respectively; Fig. 2c).

Body/organ weights and cognitive and motor abilities. (a) Average total body weight of male Sgsh D31N and Sgsh KO and control mice up to 20 weeks of age (n = 19–23/group). (b, c) Average liver and spleen weight from 21-week-old male mice (n = 15–16/group). (d-f) Thirteen-week-old mice were tested in their ability to find a submerged platform in opaque water using distal cues over a five-day training period (Acquisition Phase). The (d) time and (e) distance to find the hidden platform and the (f) percentage of time swimming in a thigmotactic pattern (<12 cm from the pool wall) were recorded. (g-i) On day 6, the platform was removed from the pool and the mice were tested in their ability to remember where the platform was previously located (Probe Phase). The percentage of (g) time and (h) path length spent in the target quadrant compared to the average of the non-target quadrants and the (i) average swim speed of the mice were measured. (J, K) At 15-weeks of age, mice were tested in a motor test battery. (j) In the pole test, the time taken for mice to turn around and climb down a vertical pole was timed for up to 120 s. Mice who fell from the pole received a maximum score of 120 s. (k) Neuromuscular grip strength was measured up to a maximum of 60 s (n = 15–16/group). Error bars ±1 SEM. * p < 0.05 wild-type vs Sgsh D31N ; ^ Sgsh flox vs Sgsh KO ; # Sgsh D31N vs Sgsh KO
There were no genotype-dependent changes in the exploratory activity at 3 weeks of age in either colony (Suppl. Fig. 2). Although significant colony effects were found for both path length and cell entries (p < 0.05), pairwise comparisons by genotype or colony did not show significant differences.
All mice exhibited the capacity to use distal cues to find a hidden platform with training in the Morris Water Maze (Fig. 2d,e). However, Sgsh KO mice were slower than Sgsh flox mice to find the platform on days 2 to 5 of training. No impairment was observed in the Sgsh D31N mice. Likewise, Sgsh KO mice swam further than the other groups to find the platform (Fig. 2e). Sgsh KO (but not Sgsh D31N) mice also showed a greater tendency for thigmotaxis compared to controls (Fig. 2f). In the probe phase, all mice spent the largest amount of time and path length swimming in the quadrant where the platform had been located, c.f. other quadrants, suggesting that they had learnt the position of the hidden platform (Fig. 2g–h). Swim speed differences were noted in Sgsh KO mice (Fig. 2i).
In the pole test, wild-type and Sgsh flox mice rarely fell from the pole (0% and 6.3%, respectively). In contrast, Sgsh D31N and Sgsh KO mice fell more frequently than their respective control groups (18.8% and 31.2%). The time taken to climb down the pole was greater in Sgsh KO animals compared to Sgsh flox mice (Fig. 2j). However, despite significant genotype differences in the two-way ANOVA analysis (p = 0.03), pairwise comparisons revealed no differences between wild-type and Sgsh D31N mice, or Sgsh flox and Sgsh KO mice. Neuromuscular grip strength was reduced in both diseased groups compared to controls (Fig. 2k). However, post-hoc testing revealed significant differences only between Sgsh flox and Sgsh KO mice (p < 0.05).
Substrate accumulation
Low levels of HS primary substrate were detected in the brain of wild-type and Sgsh flox mice at all ages assessed (Fig. 3a). At one-day of age, both MPS-IIIA models showed significant elevations in HS, with four- and five-fold control levels measured in Sgsh D31N and Sgsh KO mouse brains, respectively. HS steadily accumulated with age (16- to 18-fold control at 20-weeks).

Primary and secondary substrate accumulation. (a) HS accumulation in brain with age. (b) HS in the brain and peripheral organs from the same cohorts of mice. (c-f) Secondarily-stored gangliosides GM2 and GM3 (expressed as pmol ganglioside per mg of protein; n = 3–5 mice/group). Error bars ±1 SEM. * p < 0.05 wild-type vs Sgsh D31N ; ^ Sgsh flox vs Sgsh KO ; # Sgsh D31N vs Sgsh KO
HS was also elevated in a panel of organs from both MPS-IIIA models with liver and kidney homogenates showing the greatest abundance (Fig. 3b). The degree of HS accumulation varied with tissue type, with 138-, 28-, 18-, 18-, 10-, and six-fold control levels measured in liver, spleen, brain, kidney, heart, and lung, respectively, from Sgsh KO mice. Spleen homogenates from Sgsh KO mice yielded 29% more HS than Sgsh D31N mice (p < 0.01). Similar levels were observed from affected mice from both colonies in all other organs.
Secondarily-stored GM2 ganglioside steadily accumulated with age in diseased mice compared to controls (Fig. 3c,d). At one day of age, both GM2 species were two- to three-fold higher in affected mice, reaching four- to six-fold control at 20-weeks of age. GM3 ganglioside accumulated in a similar manner (Fig. 3e,f). By 20-weeks of age, elevated GM3 36:1 (eight- and seven-fold control for Sgsh D31N and Sgsh KO mice, respectively) and GM3 38:1 (12-fold for both models) were measured in MPS-IIIA brain. There was no difference in GM2 or GM3 ganglioside concentrations between the two MPS-IIIA models. GM1 and GT1 gangliosides showed no consistent genotype/colony differences (Suppl. Fig. 3).
Histology
Brain sections were assessed immunohistochemically with a panel of disease markers (Fig. 4). LIMP-II was used as a marker of endosomal/lysosomal membrane content and diffuse, cytoplasmic staining was observed in wild-type and Sgsh flox mice at all ages. Sgsh D31N and Sgsh KO mice showed strong, punctate staining in most regions. LIMP-II progressively accumulated with age in affected mice, becoming significantly different from controls in the hippocampus and rostral cortex from one- and three-weeks of age, respectively.

Time course of appearance of brain lesions in Sgsh D31N and Sgsh KO. (a) Representative images of immunohistochemical staining in 20-week old brain from each of the four genotypes. Lysosomal integral membrane protein (LIMP)-II and reactive glial fibrillary acidic protein (GFAP) staining in the rostral cerebral cortex (secondary motor cortex) is depicted. Images from the intermediate reticular nucleus of the brainstem of ubiquitin-stained sections are also shown, with a hematoxylin counterstain. Arrows indicate the type of inclusion that was included in the analysis if the diameter of the narrowest region of the lesion area exceed 5 μm. Scale bar, 50 μm. (b) The percentage of stained LIMP-II area was examined in wild-type, Sgsh D31N, Sgsh flox, and Sgsh KO mice at different ages ranging from 1 to 20-weeks of age (n = 3–4 mice/group, except for the hippocampal CA1/stratum radiatum region of the 1-week-old Sgsh flox group where n = 2 mice). The percentage of stained GFAP and the number of ubiquitin-positive inclusions were measured in this same cohort of mice (n = 3–4 mice/group). The central nucleus of the inferior colliculus was examined for LIMP-II and ubiquitin-stained sections. Error bars ±1 SEM. * p < 0.05 wild-type vs Sgsh D31N ; ^ Sgsh flox vs Sgsh KO
GFAP staining identifies reactive astrocytes. Pale GFAP-positive staining was observed throughout the brain of wild-type and Sgsh flox mice, and significant changes with age were not detected. In contrast, intense cytoplasmic staining of reactive astrocytes was observed in most brain regions in both MPS-IIIA models from three-weeks of age. In the cortex and inferior colliculus, astrogliosis was first detected in Sgsh D31N mice from three- and six-weeks of age, respectively, whilst significant changes were seen from three-weeks of age in these brain regions in Sgsh KO mice (p < 0.05).
Ubiquitinylated inclusions were not observed in wild-type or Sgsh flox brains. Inclusions were not evident in the two areas studied in Sgsh D31N or Sgsh KO mice until after six-weeks of age. By 10-weeks, affected mice exhibited intra-cytoplasmic, ubiquitin-positive inclusions that were variable in size and staining intensity. The number of ubiquitin-positive inclusions increased progressively with age in both MPS-IIIA models. However, no differences were observed between MPS-IIIA models for any of these stains.
Discussion
Genotype/phenotype correlations have previously been made in MPS-IIIA patients, where those with p.R245H, p.Q380R, p.S66W, or c.1080delC homozygous or compound heterozygous mutations display earlier symptom onset than p.S298P homozygotes (NCBI reference sequence identifiers NM000199.3 and NP_000190.1; Valstar et al 2010). To explore this relationship further, we generated a conditional Sgsh knockout mouse model and compared it to the well-characterized Sgsh D31N strain. Phenotypic differences were noted between the diseased mouse lines, with Sgsh KO mice taking longer and swimming a greater distance to find the hidden platform in the Morris Water Maze, a test of spatial memory, c.f. Sgsh D31N MPS-IIIA mice. Whilst Sgsh D31N mice did not exhibit cognitive dysfunction at this age, they eventually develop impairments at approximately 20-weeks of age (e.g., Fraldi et al 2007). Interpretation of this outcome is hampered however, by the observation that the Sgsh KO swam slower than the three other experimental groups, therefore motor deficits present at this age (as highlighted by the impaired grip strength noted in the Sgsh KO mice) may have contributed to the significant increase in latency to find the platform. Differences in activity were not found in either colony and this may relate to the young age selected for testing (3 weeks), as hypoactivity is not evident in Sgsh D31N mice until ∼10 weeks of age (Lau et al 2008). Whilst these cognitive and motor tests display face validity for the human condition, it should be noted that they are not as refined as the developmental assessments that can be conducted in patients, e.g., Bayley Scales of Infant Development II or the Vineland Adaptive or the Behavior Scales II (Valstar et al 2011; Shapiro et al 2016).
We were surprised to observe no obvious difference in the age of appearance and rate of primary and secondary substrate storage between the two MPS IIIA strains. Brain HS was significantly elevated at birth, and accumulated at a similar rate to 20-weeks of age in affected mice of both strains. Whilst the time-course of heparan sulfate accumulation observed here is different to that seen in previous studies of Sgsh D31N mice (Crawley et al 2006), those differences can be attributed to the detection methods employed and the disaccharide selected for analysis (a pathogenic HS disaccharide absent in normal/heterozygote mouse brain was quantified in earlier studies, whereas total HS was quantified using a representative disaccharide in the present study). Further, there was no difference in the time of appearance or rate of accumulation of GM2/GM3 gangliosides. This apparent ‘uncoupling’ of the expression of phenotypic disease and the amount of primary and secondary substrate in brain at the level of the organ is analogous to reports of a lack of correlation between the appearance of HS glycosaminoglycans in cerebrospinal fluid from MPS-IIIA patients and the rate of disease progression (Shapiro et al 2016). These findings have significant implications for the utility (or lack thereof) of HS/gangliosides as markers of disease progression or ‘severity’ in patients. Whilst they are excellent indicators of disease presence, they are unable to stratify early- and late-onset disease-expressing patients, or Sgsh KO and Sgsh D31N mice. However, using these methods, we cannot exclude that there are differences in the rate of accumulation or storage amount in specific cell types or brain regions. Therefore, more subtle markers of neurodegeneration are required.
Several disease-related lesions that have previously been reported in congenic Sgsh D31N MPS-IIIA mice (Savas et al 2004; Hemsley et al 2008) were also examined. No consistent differences were noted in either the age of onset or rate of lesion development between the two MPS IIIA models. Astrogliosis was similar in Sgsh D31N and Sgsh KO brain and can be induced by cytokines such as interleukin-1 (Giulian et al 1988). Gene expression profiling has shown that inflammatory genes are highly up-regulated in the Sgsh D31N brains, including the cytokine MIP-1α, interleukin-1β, and interleukin-10 (Arfi et al 2011). Ubiquitin-positive axonal spheroids developed similarly in affected mice, and may interfere with axonal transport and trafficking of synaptic vesicles, proteins and other cellular organelles. This may ultimately impair neurotransmission, as evidenced by reduced catecholamine secretion in Sgsh D31N chromaffin cells (Keating et al 2012). In summary, these lesion types with respect to their age of onset and rate of progression, do not appear to underlie the earlier onset of symptoms in the Sgsh KO mice. Shapiro et al (2016) reported that developmental quotients and gray matter volume estimates in MPS IIIA patients correlated with the rate of disease progression (as determined by the age of diagnosis in combination with genotype). The latter is a gross measurement of cortical pathology, which may be useful to explore further in these mouse cohorts.
Whilst cortical thickness does not change with disease progression in Sgsh D31N or MPS IIIB mice and cell counts have failed to find differences in cortical neuron numbers or cell death (Villani et al 2007; Vitry et al 2009; Wilkinson et al 2012), this does not preclude more refined degenerative changes occurring within the cortical architecture. Reduced numbers of synaptophysin-positive puncta, but not synaptobrevin-positive puncta, in the rostral cerebral cortex in MPS IIIB mouse brain by 10 days of age was interpreted as indicating an alteration in the ratio of synapse components, rather than synapse loss per se (Vitry et al 2009). Future studies will determine whether subtle changes in neuronal connectivity and communication might underpin the earlier onset of cognitive impairment in Sgsh KO mice and provide potential biomarkers of clinical disease progression.
We did not observe significant differences in SGSH activity between Sgsh D31N and Sgsh KO brain, liver or skin fibroblasts using the 4MU fluorimetric assay. The mixed and C57BL/6 strain Sgsh D31N mice were originally described as having 3–4% residual SGSH activity in brain, kidney or liver homogenates, using a natural, heparin-derived substrate (Bhaumik et al 1999; Crawley et al 2006). Using this method, SGSH activity in untreated or mock-treated Sgsh D31N brain was either not detected or measured at very low levels (Lau et al 2010, 2013). Using the fluorigenic substrate, we have previously been unable to measure SGSH activity in Sgsh D31N brain (Whyte et al 2015) though other laboratories have observed ∼2–10% wild-type activity depending on the brain area analyzed (Haurigot et al 2013; Sergijenko et al 2013). Reports quantifying the activity of SGSH in Sgsh D31N mouse liver are also inconsistent, with either undetectable (Haurigot et al 2013) or low level SGSH activity observed (Sergijenko et al 2013; Sorrentino et al 2013). Due to the limit of sensitivity in the activity assays at the low end of the spectrum, we are unable to conclusively determine whether the MPS IIIA mouse models show no or low levels of SGSH activity. Sgsh KO mice are predicted to be null, and genomic DNA sequencing confirmed exon deletion. Other knockout models such as the two Hgsnat KO models of MPS IIIC also show small amounts of HGSNAT activity when assessed with 4MU-conjugated substrate (Martins et al 2015; Marco et al 2016).
One caveat to our observations regarding the phenotypic differences between the two MPS-IIIA mouse models is that despite our efforts to generate mice on a genetically pure C57BL/6 background, SNP analysis showed divergence between the Sgsh flox/KO and Sgsh +/D31N colonies. In contrast, allozyme electrophoresis suggested that both colonies were C57BL/6. These differences are likely attributed to the number of loci assessed (15 versus 588 loci). The Sgsh flox/KO colony was generated using Bruce4 C57BL/6 embryonic stem cells and mice obtained from Monash University, which were then inter-crossed with C57BL/6 J mouse Cre/FLP-deleter lines from Jackson Laboratories. In contrast, the congenic Sgsh D31N mouse colony has been back-crossed every five- to seven-generations with wild-type C57BL/6NHsd mice originally sourced from Harlan Laboratories. These SNP polymorphisms may have contributed to the weight differences between the colonies.
Our study also resulted in the generation of the Sgsh tm1Ldru or Sgsh flox mouse line that contains a conditional Sgsh allele that, when bred with mice that express Cre recombinase under the control of a cell-specific promotor, can be used to generate targeted ablation of Sgsh for discrimination of cell autonomous from non-cell autonomous traits. An MPS-I mouse line with conditional Idua expression has been generated (Russell 2003), but its characterization has not been reported. This approach has however been harnessed in mice with Npc1-deletion in mature cerebellar Purkinje neurons (Elrick et al 2010). Cell-autonomous Purkinje cell loss and subsequent impairment of motor function was evident, however, the weight loss and early death that is characteristic of global Npc1-deleted mice was not evident, indicating that Purkinje neuron degeneration is not responsible for those phenotypes (Elrick et al 2010). These genetic tools will be important for dissecting the molecular mechanisms that underlie disease etiology in MPS-IIIA patients.
In summary, we have shown that the MPS-IIIA conditional Sgsh KO mouse model faithfully reflects the human condition. However, behavioral defects were uncoupled to the age of onset and rate of accumulation of substrates, suggesting that they are not the primary determinants of symptom generation. Future studies will examine fine structural and functional deficits as the basis for the different rates of disease progression, with the hope of identifying prognostic biomarkers with human relevance.
GFAP, glial fibrillary acidic protein; HS, heparan sulfate; KO, knockout; LIMP, lysosomal integral membrane protein; MPS, Mucopolysaccharidosis; SGSH, N-sulfoglucosamine sulfohydrolase.
References
Arfi A, Richard M, Gandolphe C, Bonnefont-Rousselot D, Therond P, Scherman D (2011) Neuroinflammatory and oxidative stress phenomena in MPS IIIA mouse model: the positive effect of long-term aspirin treatment. Mol Genet Metab 103:18–25
Bhaumik M, Muller VJ, Rozaklis T et al (1999) A mouse model for mucopolysaccharidosis type III a (Sanfilippo syndrome). Glycobiology 9:1389–1396
Crawley AC, Gliddon BL, Auclair D et al (2006) Characterization of a C57BL/6 congenic mouse strain of mucopolysaccharidosis type IIIA. Brain Res 1104:1–17
Elrick MJ, Pacheco CD, Yu T et al (2010) Conditional Niemann-Pick C mice demonstrate cell autonomous Purkinje cell neurodegeneration. Hum Mol Genet 19:837–847
Fraldi A, Hemsley K, Crawley A et al (2007) Functional correction of CNS lesions in a MPS-IIIA mouse model by intracerebral AAV-mediated delivery of sulfamidase and SUMF1 genes. Hum Mol Genet 16:2693–2702
Giulian D, Woodward J, Young DG, Krebs JF, Lachman LB (1988) Interleukin-1 injected into mammalian brain stimulates astrogliosis and neovascularization. J Neurosci 8:2485–2490
Griffey MA, Wozniak D, Wong M et al (2006) CNS-directed AAV2-mediated gene therapy ameliorates functional deficits in a murine model of infantile neuronal ceroid lipofuscinosis. Mol Ther 13:538–547
Haurigot V, Marco S, Ribera A et al (2013) Whole body correction of mucopolysaccharidosis IIIA by intracerebrospinal fluid gene therapy. J Clin Invest 123:3254–3271
Hemsley KM, Beard H, King BM, Hopwood JJ (2008) Effect of high dose, repeated intra-CSF injection of sulphamidase on neuropathology in MPS IIIA mice. Genes Brain Behav 7:740–753
Hemsley KM, Hopwood JJ (2005) Development of motor deficits in a murine model of mucopolysaccharidosis type IIIA (MPS-IIIA). Behav Brain Res 158:191–199
Hemsley KM, Luck AJ, Crawley AC et al (2009) Examination of intravenous and intra-CSF protein delivery for treatment of neurological disease. Eur J Neurosci 29:1197–1214
Hopwood JJ, Elliott H (1982) Diagnosis of Sanfilippo a syndrome by estimation of sulphamidase activity using a radiolabelled tetrasaccharide substrate. Clin Chim Acta 123:241–250
Keating DJ, Winter MA, Hemsley KM et al (2012) Exocytosis is impaired in mucopolysaccharidosis IIIA mouse chromaffin cells. Neuroscience 227:110–118
Lau AA, Crawley AC, Hopwood JJ, Hemsley KM (2008) Open field locomotor activity and anxiety-related behaviors in mucopolysaccharidosis type IIIA mice. Behav Brain Res 91:130–136
Lau AA, Hopwood JJ, Kremer EJ, Hemsley KM (2010) SGSH gene transfer in mucopolysaccharidosis type IIIA mice using canine adenovirus vectors. Mol Genet Metab 100:168–175
Lau AA, Shamsani NJ, Winner LK et al (2013) Neonatal bone marrow transplantation in MPS IIIA mice. JIMD reports 8:121–132
Marco S, Pujol A, Roca C, et al (2016) Progressive neurologic and somatic disease in a novel model of human mucopolysaccharidosis type IIIC. Dis Model Mech doi:10.1242/dmm.025171
Marshall NR, Hassiotis S, King B et al (2015) Delivery of therapeutic protein for prevention of neurodegenerative changes: comparison of different CSF-delivery methods. Exp Neurol 263:79–90
Martins C, Hulkova H, Dridi L et al (2015) Neuroinflammation, mitochondrial defects and neurodegeneration in mucopolysaccharidosis III type C mouse model. Brain 138:336–355
Matern D, Gavrilov D, Oglesbee D, Raymond K, Rinaldo P, Tortorelli S (2015) Newborn screening for lysosomal storage disorders. Semin Perinatol 39:206–216
Russell C (2003) Transgenic expression of human alpha-L-iduronidase in mouse and characterization of the long term pathophysiology of murine alpha-L-iduronidase deficiency. In: Transgenic expression of human alpha-L-iduronidase in mouse and characterization of the long term pathophysiology of murine alpha-L-iduronidase deficiency. University of British Colombia, p 177
Savas PS, Hemsley KM, Hopwood JJ (2004) Intracerebral injection of sulfamidase delays neuropathology in murine MPS-IIIA. Mol Genet Metab 82:273–285
Sergijenko A, Langford-Smith A, Liao AY et al (2013) Myeloid/microglial driven autologous hematopoietic stem cell gene therapy corrects a neuronopathic lysosomal disease. Mol Ther 21:1938–1949
Shapiro EG, Nestrasil I, Delaney KA et al (2016) A prospective natural history study of mucopolysaccharidosis type IIIA. J Pediatr 170(278–287):e274
Sorrentino NC, D’Orsi L, Sambri I et al (2013) A highly secreted sulphamidase engineered to cross the blood-brain barrier corrects brain lesions of mice with mucopolysaccharidoses type IIIA. EMBO molecular medicine 5:675–690
Trim PJ, Hopwood JJ, Snel MF (2015) Butanolysis derivatization: improved sensitivity in LC-MS/MS quantitation of heparan sulfate in urine from Mucopolysaccharidosis patients. Anal Chem 87:9243–9250
Valstar MJ, Marchal JP, Grootenhuis M, Colland V, Wijburg FA (2011) Cognitive development in patients with mucopolysaccharidosis type III (Sanfilippo syndrome). Orphanet journal of rare diseases 6:43
Valstar MJ, Neijs S, Bruggenwirth HT et al (2010) Mucopolysaccharidosis type IIIA: clinical spectrum and genotype-phenotype correlations. Ann Neurol 68:876–887
Valstar MJ, Ruijter GJ, van Diggelen OP, Poorthuis BJ, Wijburg FA (2008) Sanfilippo syndrome: a mini-review. J Inherit Metab Dis 31:240–252
Van Hove JL, Wevers RA, Van Cleemput J et al (2003) Late-onset visceral presentation with cardiomyopathy and without neurological symptoms of adult Sanfilippo a syndrome. Am J Med Genet A 118:382–387
Villani GR, Gargiulo N, Faraonio R, Castaldo S, Gonzalez YRE, Di Natale P (2007) Cytokines, neurotrophins, and oxidative stress in brain disease from mucopolysaccharidosis IIIB. J Neurosci Res 85:612–622
Vitry S, Ausseil J, Hocquemiller M, Bigou S, Dos Santos CR, Heard JM (2009) Enhanced degradation of synaptophysin by the proteasome in mucopolysaccharidosis type IIIB. Mol Cell Neurosci 41:8–18
Whyte L, Hopwood J, Hemsley K, Lau A (2015) Variables influencing fluorimetric N-sulfoglucosamine sulfohydrolase (SGSH) activity measurement in brain homogenates. Molecular Genetics and Metabolism Reports 5:60–62
Wilkinson FL, Holley RJ, Langford-Smith KJ et al (2012) Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS One 7:e35787
Acknowledgements
This work was supported by the Lysosomal Diseases Research Unit (SAHMRI, Australia). The Monash Gene Targeting Facility was contracted to generate the targeting construct and mice containing the targeted allele. We thank Meghan Setford and Andrew Shoubridge for assistance in genotyping and mouse husbandry; Lynn Garrard and staff at the Women’s and Children’s Health Network Animal Care Facility for care of the mice; Dr. Mark Corbett (Adelaide Neurogenetics Research Group; The University of Adelaide) for providing the CMV-Cre mice, Dr. Mark Adams (Evolutionary Biology Unit, South Australian Museum) for the allozyme electrophoresis and the Australian Genome Research Facility Ltd. for SNP genotyping.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest statement
None.
Animal rights
All institutional and national guidelines for the care and use of laboratory animals were followed.
Informed consent
This article does not contain any studies with human subjects performed by any of the authors.
Funding details
This work was supported by the Lysosomal Diseases Research Unit (SAHMRI, Australia).
Additional information
Communicated by: Carla E. Hollak
Electronic supplementary material
ESM 1
(DOCX 608 kb)
Rights and permissions
About this article

Cite this article
Lau, A.A., King, B.M., Thorsen, C.L. et al. A novel conditional Sgsh knockout mouse model recapitulates phenotypic and neuropathic deficits of Sanfilippo syndrome. J Inherit Metab Dis 40, 715–724 (2017). https://doi.org/10.1007/s10545-017-0044-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-017-0044-4