
Abstract
Lysosomal storage disorders are strong candidates for the development of specific innovative therapies. The discovery of enzyme deficiencies is an important milestone in understanding the underlying cause of disease. Being able to replace the first missing enzyme in a lysosomal storage required three decades of dedicated research. Successful drug development for lysosomal storage disorders was fostered by the U.S. Orphan Drug Act. Various optimization strategies have the potential to overcome the current limitations of enzyme replacement therapies. In addition, substrate reduction therapies are an alternative approach to treat lysosomal storage disorders, chemical chaperones enhance residual enzyme activity, and small molecules can facilitate substrate transport through subcellular compartments. Bone-marrow derived multipotent stem cells and gene therapies have received FDA orphan drug designation status. The science of small clinical trials played an essential role: non-neurological endpoints, biomarker, and regulatory alignment are key factors in successful drug development for lysosomal storage disorders. Being able to treat brain disease is the next frontier. This review is dedicated to the memory of Roscoe O. Brady, an early pioneer in the research of lysosomal storage diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Lysosomal storage disorders as candidates for the development of specific innovative therapies
Eukaryotic cells are surrounded by a plasma membrane that contains glycosphingolipids. The membrane is constantly remodeled requiring continous degradation of macromolecules and smaller substances. The final degradation products, such as sphingosine, fatty acids, monosaccharides, sialic acids, or sulfate, leave the lysosome via diffusion or transport systems and are subsequently recycled for synthesis of macromolecules or further degraded in order to provide energy. If any of the exohydrolases or an activator protein are deficient, non-degradable substrates will accumulate in the cells and cause a lysosomal storage disorder, such as sphingolipidoses, mucopolysaccharidoses (MPS), glycoprotein, or glycogen storage disease (Sandhoff and Kolter 2003; Sabatini and Adesnik 2005); Kolter and Sandhoff 2005). With an increasing understanding of molecular concepts and mechanisms of lysosomal storage diseases, a variety of therapeutical or potential therapeutical options received consideration: replacement of the missing enzyme (enzyme replacement therapy), reduction of substrate formation (substrate reduction therapy), stabilization of misfolded protein (pharmacological chaperone therapy), replacement of defect cells (stem cell transplantation), replacement of the deficient gene (gene therapy), and correction of gene transcription (stop codon read through).
The pathway toward the understanding of the underlying cause of lysosomal storage disorders led to the discovery of enzyme deficiencies
Clinical case reports of conditions that were later understood to be lysosomal storage disorders were initially published in the last quarter of the nineteenth century, such as Tay-Sachs-disease (Tay 1881), Gaucher disease (Gaucher 1882), and Fabry disease (Anderson 1898; Fabry 1898). In 1934, the identification of accumulated glucocerebroside as the underlying mechanism of Gaucher disease by Aghion set an important milestone for identification of potential therapeutic targets and subsequent drug development (Aghion 1934). In the same year, Klenk found that sphingomyelin accumulated in patients with Niemann–Pick disease (Klenk 1934). In 1962, Svennerholm reported ganglioside GM2 accumulated in Tay–Sachs disease (Svennerholm 1962), and in 1963 ceramidetrihexoside was identified to accumulate in Fabry disease (Sweeley and Klionsky 1963). The concept of a lysosomal storage disorder was proposed after the discovery that the deficiency of the enzyme acid maltase causes Pompe disease (Baudhuin et al 1964). Further underlying enzyme deficiencies were discovered in the 1960s: glucocerebrosidasedeficiency in Gaucher disease (Fig. 1) (Brady et al 1965), sphingomyelinase deficiency in Niemann-Pick disease (Brady et al 1966), ceramidetrihexosidase deficiency in Fabry disease (Brady et al 1967), and the biochemical evidence of inefficient enzymatic degradation of mucopolysaccharides in MPS I and II (Fratantoni et al 1968a).

Crystal structure of glucocerebrosidase (PDBe 2017)
The principle of enzyme replacement therapy is based upon the observation of an “experiment of nature”: in lysosomal storage disorders, such as Fabry disease, residual enzyme activity protects the patient to a certain degree from the full blown, severe classical disease phenotype. For instance, in Fabry disease, patients with residual enzyme activity exhibit a later onset of renal involvement, a decreased prevalence of neuropathic pain, a lower incidence of cornea verticillata, and a lower incidence of hearing loss compared with patients with complete enzyme deficiency (Altarescu et al 2001b; Branton et al 2002; Ries et al 2005, 2006a, b; 2007a, b2007a, b). Conzelmann and Sandhoff proposed the threshold theory of residual enzyme activity to explain the correlation between severity of the disease and residual enzyme activity. While the mutated catabolic enzyme may have residual catalytic ability, mutation specific altered kinetic properties, such as a different Km or Vmax may be present. Only if the velocity of substrate load into the lysosome is larger than the velocity of degradation (Vmax) by the corresponding enzyme, glycosphingolipids will be stored. Milder forms of sphingolipidoses dispose sufficient residual enzyme activity facilitating very slow accumulation of degradation products (Conzelmann and Sandhoff 1983). This model could be verified in vitro in lysosomal storage disorders such as GM2-gangliosidosis, metachromatic leukodystrophy (Leinekugel et al 1992), and Gaucher disease (Schueler et al 2004). Therefore, enzyme replacement therapy aims at substituting the missing enzyme to a level at least as high as the minimally effective level of residual enzyme activity and ideally be able to convert a classical clinical phenotype at least into an attenuated one.
Replacing a missing enzyme: three decades of dedicated research
Based on the observation that there is a lysosomal accumulation of degradation material caused by enzyme deficiency, the strategy to replace the missing enzyme was proposed by Christian de Duve and Roscoe Brady in the late 1960s:
“In our pathogenic speculations and in our therapeutic attempts, it may be well to keep in mind that any substance which is taken up intracellularly in an endocytic process is likely to end up within lysosomes. This obviously opens up many possibilities for interaction, including replacement therapy” (De Duve 1964)
“Since there is good evidence for a specific enzyme deficiency in at least three of the sphingolipidoses a potential approach might be the replacing or supplementing of these deficiencies by exogenous administration of the respective enzyme” (Brady 1966).
The translation of these visionary ideas into clinical practice would require sustained commitment for almost three decades: In 1991, the first enzyme replacement therapy, i.e., imiglucerase for Gaucher disease, received orphan drug approval by the Food and Drug Administration (FDA) (Mechler et al 2015). Mutual cross correction of the respective deficient enzyme by simultaneous cell-culture of fibroblasts from patients with MPS I and MPS II, demonstrated by the group of Elizabeth Neufeld in 1968, constitutes an early preclinical proof of concept (Fratantoni et al 1968b). The production of the therapeutic enzyme—a biological—and long-term supply for patients in sufficient quantities was an early concern for the feasibility of this approach. Pentchev et al suggested the isolation of glucocerebrosidase from human tissue and proposed placentas as a constantly available source of material for clinical trials and long term treatment (Pentchev et al 1973; Furbish et al 1977). The research for enzyme replacement therapy at the National Institutes of Health moved forward in parallel for Gaucher disease and Fabry disease. An important milestone was the demonstration of a pharmacodynamic proof of concept: Ceramidetrihexosidase isolated from human placenta was able to induce a rapid reduction of ceramidetrihexoside in two male patients with Fabry disease (Brady et al 1973). Likewise, the administration of purified glucocerebrosidase in patients having Gaucher disease decreased glucocerebroside concentrations in the liver and that associated with red blood cells in the circulation of two patients with Gaucher disease (Brady et al 1974). In order to improve the therapeutic response, exogenous glucocerebrosidase was biochemically modified and targeted to mannose lectin on macrophages (Brady et al 1994). The next step was to demonstrate a clinical response to therapy. In one patient with type 1 Gaucher disease, hemoglobin and platelets increased and bone manifestations improved with repeated infusions of purified glucocerebrosidase (Barton et al 1990). Subsequently, a sustained therapeutic response to enzyme replacement therapy, i.e., an increase of hemoglobin and platelets, decrease in serum acid phosphatase, decrease of spleen size, and improved skeleton, was shown in a larger group of patients (N = 12) with Gaucher disease. These observations eventually led to the FDA approval of imiglucerase for the treatment of Gaucher disease, which was the first FDA approved orphan drug for a lysosomal storage disorder (Barton et al 1991; Mechler et al 2015). In order to further minimize infection risk and to become independent from external tissue supply, the manufacturing method was later changed from purified placenta derived enzyme to a recombinant source based on Chinese hamster ovary (CHO) cells (Grabowski et al 1995). The development of enzyme replacement therapy with agalsidase alfa was done in a similar way a decade later. First, the reduction of disease inherent storage material, i.e., globotriaosylceramide in liver tissue and shed renal tubular epithelial cells was demonstrated (Schiffmann et al 2000). Clinical efficacy and safety was first tested in a randomized clinical trial in adult patients (Schiffmann et al 2001) and subsequently investigated in children (Ries et al 2006a, b; 2007a, 2007a, b).
Successful drug development for lysosomal storage disorders was fostered by the U.S. Orphan Drug Act
The US Orphan Drug Act of 1983 was instrumental in translating biochemical and genetic discoveries into innovative therapies for patients with lysosomal storage disorders. The Orphan Drug Act intends to stimulate the investment into the development of medicines for rare diseases. Various incentives are proposed such as seven years’ marketing exclusivity, tax credit for 50% of clinical trial costs, protocol assistance, FDA fee waiver, and orphan grants programs (Haffner et al 2008). By 18 January 2017, a total of 590 orphan indications were approved by the FDA (FDA) including therapies for lysosomal storage disorders (Table 1).
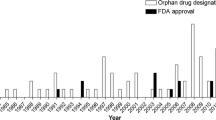
The four key factors that played an essential role in successful orphan drug development or orphan drug designations were prevalence of disease, endpoints in clinical trials, regulatory precedent (a drug approval in the same or a clinically very similar disease such as the MPS group), and finally the pharmacological technology platform (Mechler et al 2015). The first orphan drug designation for a compound intended to treat a lysosomal storage disorder occurred in 1985 for alglucerase which was approved by the FDA in 1991 for the treatment of Gaucher disease and became the first approved orphan drug for a lysosomal storage disorder. Successful development seeded further innovation: in the three decades between 1983 and 2013, 14 drugs for seven conditions received FDA approval (Fig. 2). By today (January 2017), there are ten lysosomal conditions with FDA and/or EMA drug approvals, i.e., Fabry disease, cystinosis, Gaucher disease, lysosomal acid lipase deficiency, Niemann-Pick disease type C, Pompe disease, MPS I, MPS II, MPS IVA, and MPS VI. Most of these FDA approved drugs were enzyme replacement therapies indicating that, until today, enzyme replacement therapy was the most successful technology. Substrate reduction therapies, and small molecules facilitating lysosomal substrate transportation were developed, too (Fig. 3). Of interest, Fabry disease, Gaucher disease and cystinosis had multiple drug approvals by the FDA and/or EMA. Successful FDA approval was significantly associated with higher disease prevalence and clinical development programs that did not require a primary neurological endpoints. As such, drug development in lysosomal storage disorders focused on more common diseases and primarily neurological lysosomal storage disorders were neglected. Small clinical trials with somatic or biomarker endpoints were successful study designs leading to FDA drug approval.

Number of orphan drug designations (open bars) and FDA approvals (full bars) for compounds intended to treat lysosomal storage disorders by year (Mechler et al 2015).

Time to approval of compounds intended to treat lysosomal storage disorders by A) technology platform and B) disease. Lines indicate means (Mechler et al 2015).
Orphan drug status was designated for enzymes, modified enzymes, fusion proteins, chemical chaperones, small molecules leading to substrate reduction, or facilitating subcellular substrate transport, stem cells as well as gene therapies (Fig. 4) (Mechler et al 2015). Therapeutic enzymes are manufactured in a variety of cell types. FDA approved enzyme replacement therapies were extracted from human placenta (Barton et al 1991), or derived from human (Muenzer et al 2007), animal (Chinese hamster ovaries), (Grabowski et al 1995; Eng et al 2001; Harmatz et al 2004; Wraith et al 2004; Kishnani et al 2007), or plant cells (carrot) production systems (Zimran et al 2011). Orphan drug status was also assigned for enzymes made in hen oviduct cells and insect cells (Chen et al 2000; Leavitt et al 2013). Although these different systems deliver enzymes with different biochemical and pharmacological properties which can be tested and compared in cellular and animal models, the true clinical significance of these differences in humans remains to be investigated through appropriately designed and sufficiently powered studies or meta-analyses in human. Such a comparison can be difficult and has recently been made in children and adolescents with diabetes type I, not without eliciting a considerable debate among stake holders involved. Specifically, there were no significant differences for patient-relevant outcomes between various rapidly acting insulin analogues and human insulin (IQWiG 2005).

A) Year of orphan drug designation for compounds intended to treat lysosomal storage disorders by technology platform. B) Year of orphan drug designation for compounds intended to treat lysosomal storage disorders by disease (Mechler et al 2015).
Optimization strategies have the potential to overcome the current limitations of enzyme replacement therapies
The effect of enzyme replacement therapy is mainly compromised by late initiation of treatment, immune reactions against the therapeutic protein and incomplete bioavailability in certain tissues, such as skeletal muscle, bone, and especially brain (Altarescu et al 2001a; Ries et al 2006a, b; Strothotte et al 2010; Ohashi 2012). Dose and frequency of enzyme administration are further important questions (Schiffmann et al 2007, 2015).
Various strategies have been employed to address the issue of the blood brain barrier. High dose intravenous enzyme replacement therapy has been proposed, however, significant effects on neurological outcomes could not be detected in a clinical study in patients with Gaucher disease type 3 (Altarescu et al 2001a; Goker-Alpan et al 2008). More recently, Ou et al reported the feasibility of this approach in MPS I mice and Blanz et al in alpha-mannosidosis mice (Blanz et al 2008; Ou et al 2014). Given the clinical experience in Gaucher disease, further studies in humans will be of particular interest. To overcome the blood brain barrier in an experimental setting, one patient with Gaucher disease type 2 was infused recombinant glucocerebrosidase directly into the brainstem by convection-enhanced delivery (CED) without signs of toxicity (Lonser et al 2007). However, long-term feasibility, safety, and efficacy of this approach require further studies. For intrathecal enzyme administration in MPS II and MPS IIIA, an orphan drug designation was granted. Iduronate-2-sulfatase was successfully administered intrathecally by intracerebroventricular and lumbar routes in healthy dogs and nonhuman primates, as well as in the MPS II mouse model (Calias et al 2012). Monthly intrathecal enzyme administration was studied in cynomolgus monkeys in a six month animal toxicology study (Felice et al 2011). A human clinical trial was conducted between 2009 and 2012 in patients with MPS II ((Muenzer et al 2016). Another clinical trial for a compound with orphan drug designation investigated intrathecal enzyme administration into the cerebrospinal fluid in patients with MPS IIIA through an intrathecal drug delivery device between 2010 and 2012 (Jones et al 2016). Sohn et al proposed continuous intrathecal enzyme infusions with iduronate-2-sulfatase which was tested in the MPS II mouse model instead of administering enzyme at long and therefore non-physiological time intervals (Sohn et al 2013).
Modification of therapeutic enzymes is a strategy to improve targeting properties of approved and designated orphan compounds. Human placental glucocerebrosidase (Gaucher disease) was deglycosylated to yield a macrophage-targeted mannose-terminated preparation allowing an uptake mediated by mannose receptors (Barton et al 1990). The addition of polyethylene glycol to glucocerebrosidase facilitating an increased plasma half-life was proposed and tested in a clinical trial (clinicaltrials.gov identifier NCT00001410). A PEGylated version of alpha-galactosidase A (Fabry disease) is currently under investigation in human (clinicaltrials.gov identifier NCT01678898).
Targeting the cation-independent mannose-6-phosphate receptor, recombinant human alpha glucosidase (Pompe disease) was conjugated with bis-mannose-6-phosphate bearing synthetic glycans in order to improve muscular uptake of the enzyme (Zhou et al 2011).
Orphan drug designations were granted for fusion proteins. In lyososmal storage disorders fusion proteins are enzymes coupled with another protein or part of a protein with high affinity for a certain receptor which enhances the ability for the large molecule to cross barriers between compartments. As such, recombinant human α-N-acetyl-glucosaminidase (MPS IIIB) was modified by fusing the enzyme to the receptor binding motif of insulin-like growth factor-II (IGF-II). In order to enhance the cellular and lysosomal uptake, this fusion protein is targeted at the cation-independent mannose-6-phosphate receptor, which is also the receptor for IGF-II at a different binding site (Kan et al 2014). Human acid α-glucosidase (Pompe disease) was fused to a portion of IGF-II targeting the cation-independent mannose-6-phosphate receptor in order to increase cellular uptake (Maga et al 2013). Human alpha-L-iduronidase (MPS I) and iduronate-2-sulfatase (MPS II), were each coupled with the heavy chain of a chimeric monoclonal antibody to the human insulin receptor. These fusion proteins were designed to cross the blood–brain-barrier facilitated by the endogenous insulin receptor (Boado et al 2008) and are under investigation in clinical trials in MPS I and MPS II (clinicaltrials.gov identifier NCT02371226 and NCT02262338)
Substrate reduction therapies are an alternative approach to treat lysosomal storage disorders
Substrate reduction therapies are based on small molecules that reduce the formation of substrate for a compromised enzyme by inhibiting the respective synthase of the particular non-degradable substrate. The first FDA approved substrate inhibitor was miglustat (Fig. 5) for Gaucher disease (Cox et al 2000). This compound inhibits the ceramide-specific glucosyltransferase which initiates the glycosphingolipid biosynthetic pathway and catalyzes the formation of glucocerebroside (Fig. 6) (Cox et al 2000). The drug approval was based on biomarker and visceral endpoints, i.e., on non-neurological measures. Small molecules are of particular interest for lysosomal storage disorders with neurological manifestations because they can cross the blood brain barrier. However, demonstrating the clinical effect can be challenging as illustrated by the results of a clinical trial with miglustat in Gaucher disease type 3 where effects on neurological endpoints could not be detected (Schiffmann et al 2008). More recently, another oral substrate inhibitor, eligustat (Fig. 7), was approved for the treatment of Gaucher disease (Cox et al 2015; Mistry et al 2015). Eligustat inhibits glucosylceramide synthase. It is different from miglustat as eliglustat resembles the ceramide rather than the glucose moiety and does not cross the blood–brain-barrier (Cox et al 2015).

Chemical structure of miglustat (NCBI 2017c)

Chemical structure of glucocerebroside (NCBI 2017a)

Chemical structure of eliglustat (NCBI 2017b)
Chemical chaperones enhance residual enzyme activity
Chemical chaperones are low molecular weight competitive enzyme inhibitors at high concentrations, but at very low concentrations they stabilize certain misfolded mutant enzymes (Fan et al 1999). Consecutively, these stabilized enzymes are protected from degradation in the endoplasmic reticulum/Golgi system leaving the endoplasmic reticulum and are transported to the lysosome resulting in an augmentation of residual enzyme activity after dissociation of the chaperone from the enzyme (Tropak et al 2004; Clarke et al 2011; Germain et al 2012; Khanna et al 2012; Sun et al 2012; Zimran et al 2013). Chemical chaperones are suitable only for a selected group of disease-causing missense mutations. Pathognomonic nonsense mutations would biochemically not be responsive, because the coded protein is truncated. As chemical chaperones can both inhibit and enhance enzyme activity depending on their concentrations in the local subcellular environment, the dose rationale in patients requires careful consideration. By 2013, six chemical chaperones have received orphan drug designation and clinical trials are ongoing (Mechler et al 2015). In 2016, migalastat was approved for treatment of Fabry disease in the EU, in the US the compound is investigated in further clinical trials (Germain et al 2016; Schiffmann and Ries 2016).
Small molecules facilitate substrate transport through subcellular compartments
Three small molecules, i.e., all derivatives of cysteamine, received orphan drug approval for the treatment of cystinosis, either applied systemically or topically in the eye (Gahl et al 1987; MacDonald et al 1990; Dohil et al 2010). In cystinosis, a cystine transporter gene mutation leads to an intralysosomal accumulation of cystine resulting among others in renal Fanconi syndrome and the formation of corneal crystals associated with photophobia. Cysteamine depletes cells from cystine by forming cysteine and cysteine-cysteamin complexes that can leave the lysosome. The most recent orphan drug approval for cystinosis was an extended release formulation of cysteamine that requires less frequent drug administration (BID instead of QID for the precurosor product) (Dohil et al 2010). In addition two cysteamine derivatives, i.e., phospohocysteamine and cyclodextrin, received orphan drug designation but not approval to date. Phosphocysteamine was intended to be developed as a formulation with higher bioavailability and better tolerance but the orphan drug status was withdrawn after pharmacokinetic studies in healthy volunteers did not confirm this hypothesis (Tenneze et al 1999). Cyclodextrin is a small molecule faciliating the intracellular transport of cholesterol in mouse models Niemann-Pick type C disease, another lysosomal transporter disease (Davidson et al 2009). Orphan drug designation was granted in 2013.
Bone-marrow derived multipotent stem cells and gene therapies have received FDA orphan drug designation status
A stem cell system—based on adult adherent bone marrow-derived multipotent stem cells—has received orphan drug designation for MPS I. These stem cells derived from a donor are subsequently expanded and scaled up in order to yield enough material to be used in a large number of patients. The concept was recently tested in MPS I mice that received bone marrow-derived multipotent progenitor cell cell transplants into the lateral cerebral ventricles (Nan et al 2012). Gene therapies have been granted orphan drug designation status for lysosomal storage disorders. Gene therapy is of particular interest in this context because there is the potential to continuously deliver enzyme into the organism rather than intermittent, thus, less physiological administration than in enzyme replacement therapy. Similar to chemical chaperones, these orphan drug designations were granted only recently (Fig. 4). The two older designations of 1993 and 1997 were based on a retroviral vector system. Designations after 2007 are based on adenovirus or lentivirus vectors (Dunbar et al 1998; Sun et al 2005; Ferla et al 2013; Haurigot et al 2013). One phase I/II clinical trial investigating adenovirus vector based gene therapy with a single intracerebral injection into both cerebral hemispheres in patients with MPS IIIA was conducted between 2011 and 2013 (clinicaltrials.gov identifier NCT 01474343).
The science of small clinical trials in lysosomal storage disorders and the next frontier, being able to treat the brain
In all lysosomal storage disorders pivotal trials leading to FDA or EMA approval were, by nature, small clinical trials (Table 1). In order to better understand inherent differences between orphan drug development and traditional drug development programs for more frequent diseases, the example of the rivaroxaban drug approval package is instructive and illustrative. Pivotal studies in the development program for rivaroxaban, an oral direct acting anticoagulant (factor Xa inhibitor), for the prophylaxis of deep vein thrombosis were conducted in three RCTs with a total of 9011 patients. Overall, the application of rivaroxaban was supported by 44 human clinical studies, eight population pharmacokinetic studies, 13 in vitro studies, and 16 biopharmaceutics studies (FDA 2011). These included human drug-drug-interaction studies, studies in populations with renal and hepatic impairment, a thorough QT interval prolongation study, and a food-effect study which are usually not part of an orphan drug development program. Pediatric studies were not conducted in the rivaroxaban program whereas a substantial proportion of patients in clinical registration trials for lysosomal storage disorders were children or adolescents. Of interest, none of the primary endpoints in the pivotal clinical trials for lysosomal storage disorders was purely neurological, although the majority of these diseases are associated with neurological involvement, such as neurodegeneration. The distance in a 6 minute walk test was a frequently used primary endpoint in registration trials for MPS I, MPS II, MPS VI (12 minute walk test), and Pompe disease (Table 1). The 6 minute walk test was initially developed and validated in patients with heart and lung diseases. Specifically, the test predicts morbidity and mortality in patients with chronic cardiopulmonary diseases such as primary pulmonary hypertension or advanced congestive heart failure (Miyamoto et al 2000; Shah et al 2001). Other genetic diseases with neuromuscular or pulmonary involvement, such as Duchenne muscular dystrophy or cystic fibrosis were also functionally assessed and quantified with the 6 minute walk test (Gulmans et al 1996; McDonald et al 2010).
Orphan drugs for lysosomal storage disorders were designated or developed either for mainly non-neurological and relatively more frequent conditions, or for diseases with a similar regulatory approach toward drug registration. Main pathophysiological biochemical pathways were known in all conditions with approved orphan drugs which renders lysosomal storage disorders interesting from a drug development perspective, because as exemplified in oncology, the understanding of molecular mechanisms of diseases is directly connected to the search for novel therapies (Stockklausner et al 2016). On the other hand, the precise mechanism of disease in lysosomal storage disorder is not fully understood, therefore, further research may provide insights into new therapeutic approaches. In addition, it would be desirable to have robust and unfragmented long-term outcome data for treatment in order to assess the potential for further innovation more thoroughly (Hollak et al 2011).
Currently available therapies show incomplete therapeutic response on advanced cardiac and skeletal manifestations and being able to treat brain disease is the next frontier. Drug development in neuronopathic lysosomal storage disorders may be facilitated through the availability of instruments assessing neurological and behavioral functions in a standardized way (Gershon et al 2010). The journey may be long and challenging—first, approved orphan drugs are costly (Crow 2016). Second, as it took 30 years to develop drugs for seven lysosomal storage disorders and assuming that progress occurs at the same speed, by facing the obstacle of CNS bioavailabilty and smaller patient populations, it may still take many years of ambition and dedicated research to have one approved drug, for each of the 50 known lysosomal storage disorders today. Third, the availability of effective treatment may allow more and more LSDs to be added to newborn screening programs, and early treatment may subsequently improve outcome further.
References
Aghion H (1934) La maladie de Gaucher dans l’enfance. In Editor ed.^eds. Book La maladie de Gaucher dans l’enfance Paris
Altarescu G, Hill S, Wiggs E et al (2001a) The efficacy of enzyme replacement therapy in patients with chronic neuronopathic Gaucher’s disease. J Pediatr 138:539–547
Altarescu GM, Goldfarb LG, Park KY et al (2001b) Identification of fifteen novel mutations and genotype-phenotype relationship in Fabry disease. Clin Genet 60:46–51
Anderson W (1898). A case of “angeiokeratoma”. Br J Dermatol 10:113–17
Barton NW, Furbish FS, Murray GJ, Garfield M, Brady RO (1990) Therapeutic response to intravenous infusions of glucocerebrosidase in a patient with Gaucher disease. Proc Natl Acad Sci U S A 87:1913–1916
Barton NW, Brady RO, Dambrosia JM et al (1991) Replacement therapy for inherited enzyme deficiency--macrophage-targeted glucocerebrosidase for Gaucher’s disease. N Engl J Med 324:1464–1470
Baudhuin P, Hers HG, Loeb H (1964) An electron microscopic and biochemical study of type II glycogenosis. Lab Invest 13:1139–1152
Blanz J, Stroobants S, Lullmann-Rauch R et al (2008) Reversal of peripheral and central neural storage and ataxia after recombinant enzyme replacement therapy in alpha-mannosidosis mice. Hum Mol Genet 17:3437–3445
Boado RJ, Zhang Y, Zhang Y, Xia CF, Wang Y, Pardridge WM (2008) Genetic engineering of a lysosomal enzyme fusion protein for targeted delivery across the human blood–brain barrier. Biotechnol Bioeng 99:475–484
Brady RO (1966) The sphingolipidoses. N Engl J Med 275:312–318
Brady RO, Kanfer JN, Shapiro D (1965) Metabolism of glucocerebrosides. II. Evidence of an enzymatic deficiency in Gaucher’s Disease. Biochem Biophys Res Commun 18:221–225
Brady RO, Kanfer JN, Mock MB, Fredrickson DS (1966) The metabolism of sphingomyelin. II. evidence of an enzymatic deficiency in Niemann-Pick diseae. Proc Natl Acad Sci U S A 55:366–369
Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L (1967) Enzymatic defect in Fabry’s disease—ceramidetrihexosidase deficiency. N Engl J Med 276:1163–1167
Brady RO, Tallman JF, Johnson WG et al (1973) Replacement therapy for inherited enzyme deficiency. use of purified ceramidetrihexosidase in Fabry’s disease. N Engl J Med 289:9–14
Brady RO, Pentchev PG, Gal AE, Hibbert SR, Dekaban AS (1974) Replacement therapy for inherited enzyme deficiency. use of purified glucocerebrosidase in Gaucher’s disease. N Engl J Med 291:989–993
Brady RO, Murray GJ, Barton NW (1994) Modifying exogenous glucocerebrosidase for effective replacement therapy in Gaucher disease. J Inherit Metab Dis 17:510–519
Branton MH, Schiffmann R, Sabnis SG et al (2002) Natural history of Fabry renal disease: influence of alpha-galactosidase A activity and genetic mutations on clinical course. Medicine 81:122–138
Calias P, Papisov M, Pan J et al (2012) CNS penetration of intrathecal-lumbar idursulfase in the monkey, dog and mouse: implications for neurological outcomes of lysosomal storage disorder. PLoS One 7:e30341
Chen Y, Jin M, Goodrich L, Smith G, Coppola G, Calhoun DH (2000) Purification and characterization of human alpha-galactosidase A expressed in insect cells using a baculovirus vector. Protein Expr Purif 20:228–236
Clarke JT, Mahuran DJ, Sathe S et al (2011) An open-label phase I/II clinical trial of pyrimethamine for the treatment of patients affected with chronic GM2 gangliosidosis (Tay-Sachs or Sandhoff variants). Mol Genet Metab 102:6–12
Conzelmann E, Sandhoff K (1983) Partial enzyme deficiencies: residual activities and the development of neurological disorders. Dev Neurosci 6:58–71
Cox T, Lachmann R, Hollak C et al (2000) Novel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet (London, England) 355:1481–1485
Cox TM, Drelichman G, Cravo R et al (2015) Eliglustat compared with imiglucerase in patients with Gaucher’s disease type 1 stabilised on enzyme replacement therapy: a phase 3, randomised, open-label, non-inferiority trial. Lancet (London, England) 385:2355–2362
Crow JM (2016) Advocacy: strong foundations. Nature 537:S152–S154
Davidson CD, Ali NF, Micsenyi MC et al (2009) Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS One 4:e6951
De Duve C (1964) From cytases to lysosomes. Fed Proc 23:1045–1049
Dohil R, Fidler M, Gangoiti JA, Kaskel F, Schneider JA, Barshop BA (2010) Twice-daily cysteamine bitartrate therapy for children with cystinosis. J Pediatr 156(71–75):e71–e73
Dunbar CE, Kohn DB, Schiffmann R et al (1998) Retroviral transfer of the glucocerebrosidase gene into CD34+ cells from patients with Gaucher disease: in vivo detection of transduced cells without myeloablation. Hum Gene Ther 9:2629–2640
Eng CM, Guffon N, Wilcox WR et al (2001) Safety and efficacy of recombinant human alpha-galactosidase A--replacement therapy in Fabry’s disease. N Engl J Med 345:9–16
Fabry J (1898) Ein Beitrag zur Kenntniss der Purpura haemorrhagica nodularis (Purpura papulosa haemorrhagica Hebrae). Arch Dermatol Syph 43:187–200
Fan JQ, Ishii S, Asano N, Suzuki Y (1999) Accelerated transport and maturation of lysosomal alpha-galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat Med 5:112–115
FDA (2011) “Drug Approval Package Xarelto (Rivaroxaban) 10 mg immediate release tablets.” Retrieved 3 Sept 2015, from http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/022406Orig1s000TOC.cfm
FDA “Search Orphan Drug Designations and Approvals’ Retrieved 20 Dec 2013, from http://www.accessdata.fda.gov/scripts/opdlisting/oopd/index.cfm
Felice BR, Wright TL, Boyd RB et al (2011) Safety evaluation of chronic intrathecal administration of idursulfase-IT in cynomolgus monkeys. Toxicol Pathol 39:879–892
Ferla R, O’Malley T, Calcedo R et al (2013) Gene therapy for mucopolysaccharidosis type VI is effective in cats without pre-existing immunity to AAV8. Hum Gene Ther 24:163–169
Fratantoni JC, Hall CW, Neufeld EF (1968a) The defect in Hurler’s and Hunter’s syndromes: faulty degradation of mucopolysaccharide. Proc Natl Acad Sci U S A 60:699–706
Fratantoni JC, Hall CW, Neufeld EF (1968b) Hurler and Hunter syndromes: mutual correction of the defect in cultured fibroblasts. Science 162:570–572
Furbish FS, Blair HE, Shiloach J, Pentchev PG, Brady RO (1977) Enzyme replacement therapy in Gaucher’s disease: large-scale purification of glucocerebrosidase suitable for human administration. Proc Natl Acad Sci U S A 74:3560–3563
Gahl WA, Reed GF, Thoene JG et al (1987) Cysteamine therapy for children with nephropathic cystinosis. N Engl J Med 316:971–977
Gaucher PCE (1882) De l’épithélioma primitif de la rate. Hypertrophie idiopathique de la rate sans leucémie. In Editor ed.^eds. Book De l’épithélioma primitif de la rate. Hypertrophie idiopathique de la rate sans leucémie: Paries
Germain DP, Giugliani R, Hughes DA et al (2012) Safety and pharmacodynamic effects of a pharmacological chaperone on alpha-galactosidase A activity and globotriaosylceramide clearance in Fabry disease: report from two phase 2 clinical studies. Orphanet J Dis 7:91
Germain DP, Hughes DA, Nicholls K et al (2016) Treatment of Fabry’s disease with the pharmacologic chaperone migalastat. N Engl J Med 375:545–555
Gershon RC, Cella D, Fox NA, Havlik RJ, Hendrie HC, Wagster MV (2010) Assessment of neurological and behavioural function: the NIH toolbox. Lancet Neurol 9:138–139
Goker-Alpan O, Wiggs EA, Eblan MJ et al (2008) Cognitive outcome in treated patients with chronic neuronopathic Gaucher disease. J Pediatr 153:89–94
Grabowski GA, Barton NW, Pastores G et al (1995) Enzyme therapy in type 1 Gaucher disease: comparative efficacy of mannose-terminated glucocerebrosidase from natural and recombinant sources. Ann Intern Med 122:33–39
Gulmans VA, van Veldhoven NH, de Meer K, Helders PJ (1996) The six-minute walking test in children with cystic fibrosis: reliability and validity. Pediatr Pulmonol 22:85–89
Haffner ME, Torrent-Farnell J, Maher PD (2008) Does orphan drug legislation really answer the needs of patients? Lancet (London, England) 371:2041–2044
Harmatz P, Whitley CB, Waber L et al (2004) Enzyme replacement therapy in mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). J Pediatr 144:574–580
Haurigot V, Marco S, Ribera A, et al (2013) Whole body correction of mucopolysaccharidosis IIIA by intracerebrospinal fluid gene therapy. J Clin Investig 123:3254–3271
Hollak CE, Aerts JM, Ayme S, Manuel J (2011) Limitations of drug registries to evaluate orphan medicinal products for the treatment of lysosomal storage disorders. Orphanet J Rare Dis 6:16
IQWiG (2005) Rapid-acting insulin analogues in children and adolescents with diabetes mellitus type 1 - follow-up commission. Executive summary of final report A08-01, Version 1.0. In Institute for quality and efficiency in health care: Executive Summaries Cologne, Germany
Jones SA, Breen C, Heap F et al (2016) A phase 1/2 study of intrathecal heparan-N-sulfatase in patients with mucopolysaccharidosis IIIA. Mol Genet Metab 118:198–205
Kan SH, Troitskaya LA, Sinow CS et al (2014) Insulin-like growth factor II peptide fusion enables uptake and lysosomal delivery of alpha-N-acetylglucosaminidase to mucopolysaccharidosis type IIIB fibroblasts. Biochem J 458:281–289
Khanna R, Flanagan JJ, Feng J et al (2012) The pharmacological chaperone AT2220 increases recombinant human acid alpha-glucosidase uptake and glycogen reduction in a mouse model of Pompe disease. PLoS One 7:e40776
Kishnani PS, Corzo D, Nicolino M et al (2007) Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology 68:99–109
Klenk E (1934) Über die Natur der Phosphatide der Milz bei Niemann-Pickscher Krankheit. Z Physiol Chem 229
Kolter T, Sandhoff K (2005) Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu Rev Cell Dev Biol 21:81–103
Leavitt M, Rutkowski JV, Xu H, Canty D, Quinn AG (2013) Biochemical evidence of the effects of SBC-103, a recombinant human alpha-N-acetylglucosaminidase in a mucopolysaccharidosis IIIB mouse model using an improved analytical method for substrate quantification. Mol Genet Metab 108:S58–S59
Leinekugel P, Michel S, Conzelmann E, Sandhoff K (1992) Quantitative correlation between the residual activity of beta-hexosaminidase A and arylsulfatase A and the severity of the resulting lysosomal storage disease. Hum Genet 88:513–523
Lonser RR, Warren KE, Butman JA et al (2007) Real-time image-guided direct convective perfusion of intrinsic brainstem lesions. technical note. J Neurosurg 107:190–197
MacDonald IM, Noel LP, Mintsioulis G, Clarke WN (1990) The effect of topical cysteamine drops on reducing crystal formation within the cornea of patients affected by nephropathic cystinosis. J Pediatr Ophthalmol Strabismus 27:272–274
Maga JA, Zhou J, Kambampati R et al (2013) Glycosylation-independent lysosomal targeting of acid alpha-glucosidase enhances muscle glycogen clearance in pompe mice. J Biol Chem 288:1428–1438
McDonald CM, Henricson EK, Han JJ et al (2010) The 6-minute walk test as a new outcome measure in Duchenne muscular dystrophy. Muscle Nerve 41:500–510
Mechler K, Mountford WK, Hoffmann GF, Ries M (2015) Pressure for drug development in lysosomal storage disorders—a quantitative analysis thirty years beyond the US orphan drug act. Orphanet J Rare Dis 10:46
Mistry PK, Lukina E, Ben Turkia H et al (2015) Effect of oral eliglustat on splenomegaly in patients with Gaucher disease type 1: the ENGAGE randomized clinical trial. Jama 313:695–706
Miyamoto S, Nagaya N, Satoh T et al (2000) Clinical correlates and prognostic significance of six-minute walk test in patients with primary pulmonary hypertension. Comparison with cardiopulmonary exercise testing. Am J Respir Crit Care Med 161:487–492
Muenzer J, Gucsavas-Calikoglu M, McCandless SE, Schuetz TJ, Kimura A (2007) A phase I/II clinical trial of enzyme replacement therapy in mucopolysaccharidosis II (Hunter syndrome). Mol Genet Metab 90:329–337
Muenzer J, Hendriksz CJ, Fan Z et al (2016) A phase I/II study of intrathecal idursulfase-IT in children with severe mucopolysaccharidosis II. Genet Med: Off J Am Coll Med Genet 18:73–81
Nan Z, Shekels L, Ryabinin O et al (2012) Intracerebroventricular transplantation of human bone marrow-derived multipotent progenitor cells in an immunodeficient mouse model of mucopolysaccharidosis type I (MPS-I). Cell Transplant 21:1577–1593
NCBI (2017a) National Center for Biotechnology Information. PubChem Compound Database; CID=20057354, https://pubchem.ncbi.nlm.nih.gov/compound/20057354. Accessed 20 Jan 2017
NCBI (2017b) National Center for Biotechnology Information. PubChem Compound Database; CID=23652731, https://pubchem.ncbi.nlm.nih.gov/compound/23652731. Accessed 20 Jan 2017
NCBI (2017c) National Center for Biotechnology Information. PubChem Compound Database; CID=51634, https://pubchem.ncbi.nlm.nih.gov/compound/51634. Accessed 20 Jan 2017
Ohashi T (2012) Enzyme replacement therapy for lysosomal storage diseases. Pediatr Endocrinol Rev 10(Suppl 1):26–34
Ou L, Herzog T, Koniar BL, Gunther R, Whitley CB (2014) High-dose enzyme replacement therapy in murine Hurler syndrome. Mol Genet Metab 111:116–122
PDBe (2017) Protein Data Bank in Europe 3gxi, author: Lieberman, RL. Crystal structure of acid-beta-glucosidase at pH 5.5, http://www.ebi.ac.uk/pdbe/entry/pdb/3gxi. Accessed 20 Jan 2017
Pentchev PG, Brady RO, Hibbert SR, Gal AE, Shapiro D (1973) Isolation and characterization of glucocerebrosidase from human placental tissue. J Biol Chem 248:5256–5261
Ries M, Gupta S, Moore DF et al (2005) Pediatric Fabry disease. Pediatrics 115:e344–e355
Ries M, Clarke JT, Whybra C et al (2006a) Enzyme-replacement therapy with agalsidase alfa in children with Fabry disease. Pediatrics 118:924–932
Ries M, Moore DF, Robinson CJ et al (2006b) Quantitative dysmorphology assessment in Fabry disease. Genet Med: Off J Am Coll Med Genet 8:96–101
Ries M, Clarke JT, Whybra C et al (2007a) Enzyme replacement in Fabry disease: pharmacokinetics and pharmacodynamics of agalsidase alpha in children and adolescents. J Clin Pharmacol 47:1222–1230
Ries M, Kim HJ, Zalewski CK et al (2007b) Neuropathic and cerebrovascular correlates of hearing loss in Fabry disease. Brain: J Neurol 130:143–150
Sabatini DD, Adesnik MB (2005) The biogenesis of membranes and organelles, chap 16. McGraw-Hill, New York
Sandhoff K, Kolter T (2003) Biosynthesis and degradation of mammalian glycosphingolipids. Philos Trans R Soc Lond B Biol Sci 358:847–861
Schiffmann R, Ries M (2016) Fabry disease: a disorder of childhood onset. Pediatr Neurol 64:10–20
Schiffmann R, Murray GJ, Treco D et al (2000) Infusion of alpha-galactosidase A reduces tissue globotriaosylceramide storage in patients with Fabry disease. Proc Natl Acad Sci U S A 97:365–370
Schiffmann R, Kopp JB, Austin HA 3rd et al (2001) Enzyme replacement therapy in Fabry disease: a randomized controlled trial. Jama 285:2743–2749
Schiffmann R, Askari H, Timmons M et al (2007) Weekly enzyme replacement therapy may slow decline of renal function in patients with Fabry disease who are on long-term biweekly dosing. J Am Soc Nephrol: JASN 18:1576–1583
Schiffmann R, Fitzgibbon EJ, Harris C et al (2008) Randomized, controlled trial of miglustat in Gaucher’s disease type 3. Ann Neurol 64:514-522
Schiffmann R, Swift C, Wang X, Blankenship D, Ries M (2015) A prospective 10-year study of individualized, intensified enzyme replacement therapy in advanced Fabry disease. J Inherit Metab Dis 38:1129-36
Schueler UH, Kolter T, Kaneski CR, Zirzow GC, Sandhoff K, Brady RO (2004) Correlation between enzyme activity and substrate storage in a cell culture model system for Gaucher disease. J Inherit Metab Dis 27:649–658
Shah MR, Hasselblad V, Gheorghiade M et al (2001) Prognostic usefulness of the six-minute walk in patients with advanced congestive heart failure secondary to ischemic or nonischemic cardiomyopathy. Am J Cardiol 88:987–993
Sohn YB, Lee J, Cho SY et al (2013) Improvement of CNS defects via continuous intrathecal enzyme replacement by osmotic pump in mucopolysaccharidosis type II mice. Am J Med Genet A 161A:1036–1043
Stockklausner C, Lampert A, Hoffmann GF, Ries M (2016) Novel treatments for rare cancers: The U.S. orphan drug act is delivering-a cross-sectional analysis. Oncologist 21:487–493
Strothotte S, Strigl-Pill N, Grunert B et al (2010) Enzyme replacement therapy with alglucosidase alfa in 44 patients with late-onset glycogen storage disease type 2: 12-month results of an observational clinical trial. J Neurol 257:91–97
Sun B, Zhang H, Franco LM et al (2005) Efficacy of an adeno-associated virus 8-pseudotyped vector in glycogen storage disease type II. Mol Therapy: J Am Soc Gene Therapy 11:57–65
Sun Y, Liou B, Xu YH et al (2012) Ex vivo and in vivo effects of isofagomine on acid beta-glucosidase variants and substrate levels in Gaucher disease. J Biol Chem 287:4275–4287
Svennerholm L (1962) The chemical structure of normal human brain and Tay-Sachs gangliosides. Biochem Biophys Res Commun 9:436–441
Sweeley CC, Klionsky B (1963) Fabry’s disease: classification as a sphingolipidosis and partial characterization of a novel glycolipid. J Biol Chem 238:3148–3150
Tay W (1881) Symmetrical changes in the region of the yellow spot in each eye of an infant. Trans Ophthalmol Soc 1:55–57
Tenneze L, Daurat V, Tibi A, Chaumet-Riffaud P, Funck-Brentano C (1999) A study of the relative bioavailability of cysteamine hydrochloride, cysteamine bitartrate and phosphocysteamine in healthy adult male volunteers. Br J Clin Pharmacol 47:49–52
Tropak MB, Reid SP, Guiral M, Withers SG, Mahuran D (2004) Pharmacological enhancement of beta-hexosaminidase activity in fibroblasts from adult Tay-Sachs and Sandhoff Patients. J Biol Chem 279:13478–13487
Wraith JE, Clarke LA, Beck M et al (2004) Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human alpha-L-iduronidase (laronidase). J Pediatr 144:581–588
Zhou Q, Stefano JE, Harrahy J et al (2011) Strategies for Neoglycan conjugation to human acid alpha-glucosidase. Bioconjug Chem 22:741–751
Zimran A, Brill-Almon E, Chertkoff R et al (2011) Pivotal trial with plant cell-expressed recombinant glucocerebrosidase, taliglucerase alfa, a novel enzyme replacement therapy for Gaucher disease. Blood 118:5767–5773
Zimran A, Altarescu G, Elstein D (2013) Pilot study using ambroxol as a pharmacological chaperone in type 1 Gaucher disease. Blood Cells Mol Dis 50:134–137
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Marc Patterson
Rights and permissions
About this article

Cite this article
Ries, M. Enzyme replacement therapy and beyond—in memoriam Roscoe O. Brady, M.D. (1923–2016). J Inherit Metab Dis 40, 343–356 (2017). https://doi.org/10.1007/s10545-017-0032-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-017-0032-8