
Abstract
Alkaptonuria (AKU) is an ultra-rare inborn error of metabolism developed from the lack of homogentisic acid oxidase activity, causing homogentisic acid (HGA) accumulation that produces an HGA-melanin ochronotic pigment, of hitherto unknown composition. Besides the accumulation of HGA, the potential role and presence of unidentified proteins has been hypothesized as additional causal factors involved in ochronotic pigment deposition. Evidence has been provided on the presence of serum amyloid A (SAA) in several AKU tissues, which allowed classifying AKU as a novel secondary amyloidosis. In this paper, we will briefly review all direct and indirect lines of evidence related to the presence of amyloidosis in AKU. We also report the first data on abnormal SAA serum levels in a cohort of AKU patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alkaptonuria (AKU, OMIM: 203500) is an ultra-rare (one in 250,000–1,000,000) dismetabolic condition due to a deficiency of homogentisate 1,2-dioxygenase (HGD, E.C.1.13.11.5), the enzyme converting homogentisic acid (HGA) to maleylacetoacetic acid in the degradation pathway of tyrosine and phenylalanine (Ranganath et al 2014). Mutations in HGD (Zatkova 2011) impair HGD catalytic activity and the resulting HGA increase leads to ochronosis, that is the “hallmark” of AKU, a dark brown-black discoloration prominent in connective tissues. Ochronosis causes dramatic tissue degeneration due to still unknown mechanisms. AKU is asymptomatic at the beginning, but starting from the third decade problems occur at several body districts, mainly joints and heart, but also kidney, eyes, prostate, gall bladder, etc. For this reason, AKU is now considered a multisystemic disease (Helliwell et al 2008).
Besides the accumulation of HGA, the potential role and presence of unidentified proteins has been hypothesized as additional causal factors involved in both intra- and extra-cellular ochronotic pigment deposition (Taylor et al 2010).
Evidence has been provided on the presence of serum amyloid A (SAA) and serum amyloid P (SAP) in AKU, which allowed classifying AKU as a secondary (AA) amyloidosis (Spencer et al 2004; Millucci et al 2012). Amyloidosis is a pathological process centred on the conversion of normally soluble proteins into insoluble protein fibrillar aggregates, called amyloid, that deposit either systemically or in a specific organ and may be associated with a diverse range of disorders. Amyloidoses are progressive diseases characterized by a lag phase, analogously to what happens in AKU whose nature is also progressive similarly to other rheumatic joint diseases where secondary amyloidosis is well ascertained (Obici et al 2009; Nakamura 2011; Bely and Apathy 2012).
In this paper, we will briefly review all direct and indirect evidences of amyloidosis in AKU.
Materials and methods
Samples
Sera were obtained from three different cohorts of AKU patients. Two cohorts were represented by AKU patients from United Kingdom and Slovakia who had been enrolled in a clinical trial for the use of nitisinone for AKU treatment: Suitability Of Nitisinone In Alkaptonuria 1 (SONIA 1), an international, multicentre, randomized, open-label, no-treatment controlled, parallel-group, dose-response study (Ranganath et al 2014). The third cohort was represented by Italian AKU patients under control in the AKU reference rheumatologic clinic of Siena University Hospital (Prof. M. Galeazzi). Blood samples were collected in fasting conditions in non-gel serum tubes; serum samples were then kept frozen at -80 °C until analysis.
Alkaptonuric specimens were obtained from the Italian cohort of AKU patients. Healthy human articular cartilage and synovia were obtained from patients without any history of rheumatic diseases who underwent surgical knee joint sampling. Tissue was removed only from healthy, glossy and intact articular cartilage surface.
Amyloid staining
Amyloid fibrils were revealed by Congo red, as previously described (Millucci et al 2012).
Transmission electron microscopy (TEM)
AKU cartilage, aortic valve, salivary gland and knee fat specimens were fixed in 2.5 % glutaraldehyde in 0.1 M cacodylate buffer (CB) pH 7.2 for 3 h at 4 °C, post-fixed in 1 % osmium tetroxide in CB for 2 h at 4 C, dehydrated in a graded series of ethanol and embedded in a mixture of Epon–Araldite resins. Thin sections, obtained with a Reichert ultramicrotome, were stained with uranyl acetate and lead citrate and observed with a TEM FeiTecnai G2 spirit at 80 Kv.
Histology
AKU synovial specimens were placed in 5 % formaldehyde. Samples were dehydrated with increasing concentrations of alcohol and embedded in paraffin. Sections were then stained with hematoxylin-eosin (H/E).
Determination of SAA serum levels
SAA serum levels were determined by means of a commercial ELISA assay (kit code KHA0012, Invitrogen-Life Technologies, Carlsbad, CA, USA) according to manufacturer’s instruction after the recommended dilution (DF = 200). Plates were read on a VersaMax microplate reader (Molecular Devices, Sunnyvale, CA, USA) using Ascent software (Thermo Scientific, Waltham, MA, USA). SAA quantification was obtained against a second order polynomial standard curve (R2 = 0.999) generated with SAA standards.
Amyloid evidence observation
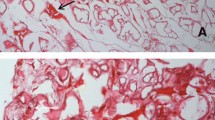
Amyloid has specific structural and staining properties and the gold standard for the diagnosis of amyloidosis is the histological identification of amyloid by the birefringence of Congo Red (CR) stain under polarizing light. Such a typical CR birefringence has been reported to be visible in cartilage (Millucci et al 2012; Spencer et al 2004) and synovia (Millucci et al 2012) from AKU patients (Fig. 1). Remarkably, the amyloid presence was mainly found in correspondence of ochronotic areas and shards, thus suggesting a strict intimate structural correlation between pigment and amyloid (Millucci et al 2012; Spencer et al 2004). Amyloid fibrils appeared as twisted rods composed of cross-beta sheet structures that selectively bound the dye CR and Thioflavin-T, thus this latter fluorescent dye confirmed the presence of amyloid aggregates in cartilage and synovial tissue from AKU patients (Millucci et al 2012). The presence of amyloid deposit was observable also in chondrocytes isolated from ochronotic cartilage of AKU patients (Millucci et al 2012), in keeping with evidence that synoviocytes and chondrocytes may be important producers of amyloid (Momohara et al 2008). Moreover, both synoviocytes and chondrocytes express HGD, thus contributing to the local production of ochronotic pigment and amyloid (Laschi et al 2012).

Congo red staining of amyloid in AKU cartilage (left) and synovia (right). Birefringent amyloid deposits in strongly degenerated AKU articular cartilage (left) were detected, particularly localized in pigmented areas although amyloid infiltrates were evident all throughout the tissue. AKU synovia (right) also showed massive and extensive amyloid infiltration, predominantly in the superficial layer of the synovial membrane, and especially in concomitance with pigmented areas and with ochronotic shards. Local inflammation and tissue damage were evidenced by the presence, in both AKU cartilage and synovia samples, of inflammatory infiltrates, consisting mainly of monocytes and macrophages
Moreover, amyloid was detected also in heart (Millucci et al 2014a, c), prostatic stones, periumbelical abdominal (Millucci et al 2012), and articular fat and labial salivary gland (Millucci et al 2014d) of tissues from ochronotic patients (Fig. 2), thus suggesting the systemic nature of AKU-related amyloidosis and the case of a secondary amyloidosis. In fact, SAA deposition was revealed in all AKU amyloid specimens (Millucci et al 2012, 2014a, b, c, d), clearly indicating that AA amyloidosis is related to ochronosis. Immunofluorescence also allowed highlighting the co-localization of amyloid with the melanin-like ochronotic pigment that has intrinsic fluorescence properties (Millucci et al 2012, 2014a, c, d; Spencer et al 2004; Spreafico et al 2013), and such superimposition was particularly remarkable in correspondence of synovial ochronotic shards and in AKU aortic valve (Millucci et al 2012, 2014a, c). Localized amyloid was noted in ochronotic cartilage in all cases (Millucci et al 2012, 2014b; Spencer et al 2004), and calcium pyrophosphate crystals deposition was also seen in knee and elbow joints (Spencer et al 2004). Immunohistochemistry confirmed the presence of SAP component in all AKU tested specimens, while no positivity was observed for the presence of immunoglobulin light chains, pre-albumin, beta-2 microglobulin, α-synuclein, and Pmel17 (Millucci et al 2012; Spencer et al 2004). Transmission electron microscopy (TEM) observations indicated that the amyloid fibrils detected in AKU tissues ranged 6-13 nm in diameter and were always in close proximity of ochronotic pigmented areas (Millucci et al 2012, 2014a, c; Millucci et al 2014d) (Fig. 2). Apart from visible signs of cell death and autophagy, dense layers of amyloid and scattered amyloid aggregates and fibrils among collagen fibres were always evident. These latter appeared usually strongly loaded by drop-like pigment elements as also previously reported (Taylor et al 2010).

TEM observation of amyloid in AKU tissues. Articular AKU cartilage showed the presence of aggregates of amyloid fibrils between recognizable broken collagen fibrils, finally sprinkled with pigment drop-like granules (bar 2.5 μm). AKU aortic valve revealed the presence of intracellular and extracellular deposits of ochronotic pigment. A massive infiltration of fine amyloid fibrils in proximity of a large pigment patch is shown (bar 100 nm). AKU salivary gland is the tissue where amyloid deposits were observed more abundantly. Ultrastructural observation showed a region of the interstitial glandular stroma that contained fine amyloid fibrils, interspersed with bundles of collagen fibrils. Pigment deposits were present on broken collagen fibres scattered between amyloid fibrils (bar 2.5 μm). AKU knee fat showed the presence of numerous and pervading long, straight and parallel bundles of amyloid fibrils (bar 5 μm)
Supporting evidences of amyloidosis
Cellular events may be associated with AA amyloidogenesis that can be observable in AKU cell and tissues.
It is known that AA amyloidosis is related to the expression/presence of SAA in tissues, but a connection with an oxidized and inflammatory status is also very often present. Data suggest that AA is a secondary complication of AKU due to a chronic inflammatory status derived from HGA-benzoquinone acetic acid (BQA)-melanin-induced oxidative stress.
Such indirect observations further support amyloidosis in AKU.
Cell and tissue degeneration in AKU
Emerging evidence has demonstrated that a large fraction of SAA amyloid is generated in intracellular compartments rather than as systemic deposition (Magy et al 2007). Similarly, in AKU, intracellular SAA may be sequestered by autophagy-lysosomal machinery along with damaged organelles. AKU cells often appear irregular with dense cytoplasm filled with multiple transcytotic vesicles (Millucci et al 2012, 2014b, c, d). Ultrastructural observation of AKU chondrocytes showed typical signs of a primary autophagy-lysosomal injury that may sit at the top of a pathogenic hierarchy, eventually developing into the plethora of heterogeneous pathologic signs including structural tissues defects, extracellular diffuse amyloid deposition, amyloid plaques, intracellular tangles, etc. (Millucci et al 2014b). In AKU, AA amyloid fibril accumulation was associated with membrane lesions in basal, cytoplasmic organelle (endoplasmic reticulum, mitochondria), and nuclear membranes. It is plausible that some of lysosomal vesicles may secret their stored monomeric or oligomeric SAA peptides into extracellular spaces. Highly concentrated SAA aggregates stored in enlarged autophagy-lysosomal vesicles may contribute to the development of amyloid plaques. Thus the cellular autophagy-lysosomal pathway appears to play a central role in amyloid deposition associated also in AKU. The vesicle damage and subsequent leakage is a primary causative event that results in secondary pathogenic lesions as evident by extensive membrane disruption occurring in cytoplasmic, nuclear and other organelle membranes (Millucci et al 2014b). In AKU cells and tissues, disrupted membranes are discontinuous with large gaps or exhibit irregularly multilamellar or indistinct cloud-like morphology (Millucci et al 2014b, c), suggesting that membrane disruption results from structural destabilization likely due to an altered intracellular microenvironment rather than direct interaction between amyloid and lipid bilayers. Moreover, mitochondrial deficits appear as a prominent pathogenic lesion in AKU tissues (Millucci et al 2012, 2014b, c, d). Morphological changes (decreased size, abnormal cristae) and accumulation of osmiophilic materials were often observable. Mitochondria provide the energy to support cellular activities but also produce free radicals and other oxidative molecules. Indeed, metabolic defects, energy deficiency and increased oxidative stress are common pathogenic lesions found in AKU. In addition to being the major source of intracellular reactive oxygen species, mitochondria are also particularly vulnerable to oxidative damage. Oxidative stress may thus result in a self-amplifying pathogenic lesion.
Inflammation
Chronic inflammation has long been recognized as central to the induction of AA amyloidosis, although its role has not yet been fully investigated. Chronic inflammation, superimposed by amyloid fibril deposition, is believed to trigger the cascade of oxidative stress response in the affected organs and tissues (Kamalvand and Ali-Khan 2004). These markers were co-localized bound to AA fibrils infiltrating tissues.
HGA, if injected into joints produces disabling damage, ochronosis, necrosis and inflammatory reactions (Martin and Batkoff 1987). Indeed, an inflammatory status can be observed in AKU tissues (Selvi et al 2000; Taylor et al 2010), where macrophages may surround the pigmented areas (Taylor et al 2010), while HGA-treated chondrocytes and synoviocytes show phagocytic features (Castillo and Kourí 2004). Local inflammation signs have been previously reported in AKU (Selvi et al 2000). Lymphocyte aggregates and infiltrating macrophages were found in the synovial (Fig. 3) and aortic valve tissues (Millucci et al 2014a, c), especially in the proximity of ochronotic shards. In AKU aortic valves large deposits of ochronotic pigment were found in the vicinity of areas of valvular calcification, tissue fibres adjacent to calcium deposits interconnecting bundles of collagen were demonstrated and EDS spectra of pigmented areas of AKU aortic valve proved the presence of indicate hydroxyapatite concretions (Millucci et al 2014a, c). Such a valvular calcification, typical of stenosis, is always in strong correlation with an inflammatory local condition (Dweck et al 2012).

Haematoxylin and eosin histology of AKU synovia indicates local inflammation. Representative haematoxylin and eosin micrographs of synovium biopsies from an AKU total knee (left) and shoulder (right) arthroplasty (both magnification 40×). Synovial membrane showed hyperplasia of the lining layer with a region of predominantly lymphocytic inflammation in the sublining layer with plasma cells and macrophages. Ochronotic cartilage shards embedded in severely degraded synovium were evident. Dense cellularity with inflammatory cells, containing many or large lymphoid aggregates were also present. Lymphocytes, macrophages and even clumps of plasma cells infiltrating the sublining region, mainly in correspondence of large ochronotic shards were visible. The synovial intimal lining, here not appreciable due to the extremely decayed conditions of the tissue, was ever hyperplastic with an increased number of macrophages and fibroblasts
Oxidized lipids and their components have strong pro-inflammatory properties. Lipid peroxidation (LPO) is a hallmark of amyloidosis. Co-localization of LPO and amyloid has always been reported in all types of amyloidosis, LPO being also a contributing factor to the generation of amyloid A fibrils. In AKU, LPO positively correlated with pigmented areas, suggesting the association of intraleaflet ochronosis and oxidative stress (Millucci et al 2014a, c). AKU valve contained macrophages in the subendothelial layer of the fibrosa, in the vicinity of ochronotic deposits. LPO are pro-inflammatory and attract inflammatory cells, the predominant cell type in aortic valve lesions, like T lymphocytes and macrophages. Possibly, products generated by HGA/BQA-induced LPO are involved in the inflammatory process present in the AKU valves. Myelin figures, well-known indicators of LPO and membrane degeneration, were also detected in AKU (Millucci et al 2014a, c).
Local inflammation and oxidation in AKU joints have also been indicated by the characterization of AKU chondrocytes, obtained from the ochronotic cartilage of AKU patients. AKU cells, once cultured in vitro, release nitric oxide (Braconi et al 2011), a well-known compound related to inflammation and oxidative stress, and pro-inflammatory cytokines. AKU chondrocytes also release high levels of pro-inflammatory cytokines, like IL-1beta, IL-6, IL-8, IL-10 and TNF-alpha, related to cartilage degeneration (Braconi et al 2012, 2013). The treatment of normal cells with HGA (Tinti et al 2010, 2011a, b) induces the formation of amyloid, the synthesis and the release of SAA, pro-inflammatory cytokines, and cell membrane LPO (Spreafico et al 2013), strongly suggesting that HGA may be the primary cause of local inflammation, oxidation and amyloidosis. It has been proven that HGA produces melanin (Hegedus 2000) that is known cause of inflammation. In AKU, continuing accumulation of HGA and its oxidized derivatives may promote inflammatory responses mediating tissue damage. The chronic presence of HGA-melanin may cause a further inflammatory stimulus resulting in SAA overproduction finally causing the formation of amyloid deposits.
Oxidative stress
Oxidative stress has been indicated as a contributing factor to the generation of AA fibrils and an integral component of amyloidotic tissues. LPO is implicated in protein modification and amyloid fibril formation.
Symptoms in AKU patients may be the consequence of repeated oxidative insults to target tissues initiated by HGA spontaneous oxidation.
In human serum model, HGA proved to induce, concomitantly to ochronotic pigment production, oxidative stress through LPO, decreased activity of glutathione peroxidase and a massive depletion of thiol groups, thus compromizing the so-called ‘plasma antioxidant capacity’ (Braconi et al 2011). HGA induces oxidative stress, which is mediated by thiol depletion (Giustarini et al 2012) and oxidation of protein thiols (which also contribute to plasma redox balance), LPO, and protein post-translational modifications like carbonylation, quinolation and aggregation (Braconi et al 2013). Irreversible structural/functional HGA-induced modification of specific proteins may be relevant for impairment of an appropriate defence against oxidative stress and protein aggregation. BQA, the oxidative metabolite of HGA, and melanin-like ochronotic pigment can both promote and further propagate such an oxidative stress (Braconi et al 2013). Likely, AKU patients experience significant oxidative stress due to the circulating levels of HGA and BQA.
The first clue of AKU-related LPO was shown in an in vitro HGA-treated serum model where a significant increase of TBARS has been revealed (Braconi et al 2011). 4-hydroxy-2-nonenal (HNE) is one of the major aldehydic reactive products generated by LPO and its presence has been generally localized in primary and secondary amyloidosis (Butterfield and Lauderback 2002). HNE, by means of Michael adducts with protein thiol and amino groups, causes cross-linked aggregation, with protein inactivation/functional modification, and renders such aggregates highly resistant to proteolysis. LPO was visible in cartilage and chondrocytes obtained from AKU patients and HNE present in AKU chondroptotic cartilage resulted a key mediator of oxidative stress-induced pathophysiological effects (Millucci et al 2014b). Analogously, ochronotic intraleaflet pigmentation of AKU stenotic valve positively correlated with 4-HNE staining (Millucci et al 2014a, c).
Proteomics
Proteomic analysis of human cells from AKU patients revealed the aberrant expression of several proteins involved in the control of folding/unfolding and amyloidogenic processes (Braconi et al 2012). Cathepsin D under-expression in cells from AKU patients could be relevant since this protein plays a major key physiological role in SAA catabolism, completing its degradation process and thus preventing SAA from accumulating and serving as a precursor of AA amyloid fibrils (Millucci et al 2012; Braconi et al 2012). Insufficient degradation systems for aggregated proteins, as suggested in the case of other secondary amyloidoses (Westermark and Westermark 2009), may be on the origin of AKU amyloidosis. Moreover, proteomics of AKU chondrocytes (Braconi et al 2012) and HGA-treated normal human cells (Braconi et al 2010a, b) indicated relevant alterations in the expression of proteins involved in cell defence, protein folding and cell organization and that these cells experience significant protein oxidation and aggregation.
Ochronosis and amyloidosis
AKU amyloid has been reported to strongly co-localize with HGA-melanin ochronotic pigment. This evidence suggests that HGA polymer may be involved in amyloid deposition or that these two types of deposits (pigment and amyloid, both detergent-, acid-, alkali- and protease-resistant) may be structurally related. The striking co-localization of HGA-melanin and amyloid suggests the participation of oxidized HGA pigment in the formation of amyloid aggregates and a link between HGA oxidation and amyloid deposition.
Mechanical stress, one of the typical functional features of heart valve and also typically present in joint function, may be a linker between amyloid and ochronotic pigment co-presence, since it has been reported to be responsible for both amyloid and ochronotic pigment deposition in AKU. Mechanical stress in heart valves has been positively related to lipoproteins oxidation inducing calcification in cardiac pathologies (Dweck et al 2012). LPO was found to be more intense in AKU cartilage zones where the ochronotic pigment could be detected (Millucci et al 2014b), while superficial not pigmented zones did not present significant traces of HNE. Myelin figures have also been reported to overlap pigmented tissues, suggesting a strict correlation between the cytotoxic action of ochronotic pigment and LPO (Millucci et al 2014c).
Clinical aspects
Epidemiological data are still lacking since a systematic early diagnosis of AA-amyloidosis in AKU cases has not been undertaken so far.
AA-amyloidosis is a complication of chronic diseases in which plasma SAA levels are constantly above 5 mg/L (Westermark and Westermark 2009). The only indication about the prevalence of high SAA serum levels in AKU patients is reported in the present paper. SAA serum levels above normal values were found in 93 % of patients from the UK cohort, in 60 % from the Slovak cohort and 78 % from the Italian cohort (Table 1). These data suggest that SAA serum levels may be useful indicators of inflammation in AKU.
In AA amyloidosis, proteinuria, renal dysfunction and gastrointestinal symptoms are diagnostically informative, but it is important not to overlook these symptoms, and to confirm the presence of amyloidosis by organ biopsy. Although interferences of HGA on some diagnostic assays have been reported (Selvi et al 2010; Curtis et al 2014), actually no proteinuria has been detected in AKU patients. The lack of proteinuria is not unusual (Nakamura 2011), also because it is typical of the final fatal phases in AA amyloidosis. No data have been published so far on renal and gastrointestinal involvement in AKU.
An early diagnosis of amyloidosis is crucial and it has been suggested that histological evaluation of amyloid with CR would be preferable in labial salivary gland biopsies instead of periumbelical fat aspirate that could lead to false negatives in AKU (Millucci et al 2014d).
The best approach to treatment of amyloidosis is to prevent progression by controlling the serum level of SAA, whose suppression below 10 mg/L halts is associated with prolonged survival, amyloid deposition reversal and organ function recovery (Lachmann et al 2007). Anti-oxidants and methotrexate proved to possess a significant capacity of inhibiting both HGA-induced ochronosis and amyloidosis as well as SAA release in in vitro AKU cell models (Tinti et al 2010; Spreafico et al 2013; Millucci et al 2012).
Amyloid and melanin-like pigment
A thorough systematic analysis of amyloid deposits in AKU has not been undertaken so far; however, also in light of the most recent findings discussed here (Millucci et al 2012, 2014a, b, c, d; Spreafico et al 2013), this would definitely deserve further investigations. Besides providing novel insights on AKU pathological mechanisms, such an analysis will contribute to get a better awareness of AKU and related co-morbidities within the medical community. Furthermore, thanks to the structural and functional correlations that can be found between the ochronotic pigment and amyloid (Millucci et al 2012), a deeper characterization of the amyloid deposits found in AKU might help the clarification of the composition (so far largely unknown) of the ochronotic pigment. In fact, though it is without doubt that HGA is the causative agent of the pathological manifestations in AKU, mediated by BQA as the intermediary agent in a so far undefined polymerisation process (Braconi et al 2015), the final structure and composition of the ochronotic pigment deposited in connective tissues of AKU patients are still quite obscure. The co-localization of SAA amyloid with melanin-like ochronotic pigment suggests a structural relationship between these two types of deposits, a unique feature of AKU (Fig. 4). Amyloid production is physiologically related to melanin synthesis in human melanocytes (Watt et al 2013; Hu et al 2011), introducing the concept of “functional amyloid” playing a key role in preventing the cytotoxic effect of melanin (Jiang and Lee 2014; Rochin et al 2013; Theos et al 2013; Leonhardt et al 2013; McGlinchey et al 2011; Delevoye et al 2011). AKU amyloidosis is an intriguing case of amyloid associated with melanin outside the melanosome compartment under pathological conditions. Moreover, AKU is the third pathology after Parkinson's disease (PD) and Alzheimer's disease (AD) where amyloid is associated with a melanin-based pigmentation (Fowler et al 2006; Aquaron et al 1995). Indeed, associations of PD/AD and AKU have been reported and HGD expression in brain have been reported (Bernardini et al 2015).

Hypothesis of ochronotic pigment and amyloid formation in AKU HGA and its auto-oxidation product BQA are progenitors of the melanin-like ochronotic pigment. Both the process of production and melanin itself are causes of induction of oxidative imbalance and consequent cytotoxicity. Such a repetitive oxidative insult also causes a chronic inflammatory status accompanied by inadequate defense responses and aberrant production of amyloidogenic proteins finally resulting in secondary amyloid deposition. Amyloid and its intermediate forms are also cytotoxic and pro-oxidant. On the other hand, analogously to what occurs physiologically in melanogenesis, amyloid may also be produce to attempt to mitigate melanin’s cytotoxicity and/or function as a scaffold for its synthesis
In virtue of this, determining the identity of macromolecules composing the ochronotic pigment and their reciprocal interactions is the basic step to find possible drugs to inhibit its formation or to disrupt/disaggregate it. Any information about pigment composition and reciprocal interactions with amyloid, can be decisive in the achievement of this goal. Altogether, future investigations might help the design of therapeutic approaches for AKU and ochronosis, in line with the aims of the ‘International Rare Disease Research Consortium’, a joint effort by the US NIH and the European Commission for the develop of treatments of all the known rare diseases by the year 2020 (Abbott 2011).
Conclusions
Alkaptonuria is a complicating inflammatory multisystemic disease, involving many different organs where any body district expressing HGD may be affected by ochronosis and related AA amyloidosis.
Amyloid, ochronosis, LPO, inflammation, tissue calcification, cell death and tissue degeneration co-localize in AKU.
These findings are in agreement with high serum SAA levels in AKU patients that may hence be related to the HGA-induced oxidative stress.
HGA/BQA-induced melanin-like ochronotic pigment promotes inflammation, tissue degeneration and self-amplifying oxidative stress. A chronic inflammatory status paralleled by inadequate antioxidant defences may thus promote the synthesis of amyloidogenic proteins ultimately leading to secondary amyloid deposition, which is in turn cytotoxic, pro-inflammatory and pro-oxidant, further complicating AKU pathogenic lesions.
References
Abbott A (2011) Rare-disease project has global ambitions. Nature 472:17
Aquaron R, Fayet G, Barthet C, Désiré S, Viallet F (1995) Parkinson disease and alkaptonuria: fortuitous association or striatonigral ochronosis? Rev Neurol (Paris) 151(1):63–66
Bely M, Apathy A (2012) Amyloid A deposition in rheumatoid arthritis: a retrospective clinicopathologic study of 161 autopsy patients. Amyloid 19:212–213
Bernardini G, Laschi M, Geminiani M, Braconi D, Vannuccini E, Lupetti P, Manetti F, Millucci L, Santucci A (2015) Homogentisate 1,2 dioxygenase is expressed in brain: implications in alkaptonuria. J Inherit Metab Dis – in press
Braconi D, Laschi M, Taylor AM et al (2010a) Proteomic and redox-proteomic evaluation of homogentisic acid and ascorbic acid effects on human articular chondrocytes. J Cell Biochem 111:922–932
Braconi D, Laschi M, Amato L et al (2010b) Evaluation of anti-oxidant treatments in an in vitro model of alkaptonuric ochronosis. Rheumatology (Oxford) 49:1975–1983
Braconi D, Bianchini C, Bernardini G et al (2011) Redox-proteomics of the effects of homogentisic acid in an in vitro human serum model of alkaptonuric ochronosis. J Inherit Metab Dis 34:1163–1176
Braconi D, Bernardini G, Bianchini C et al (2012) Biochemical and proteomic characterization of alkaptonuric chondrocytes. J Cell Physiol 227:3333–3343
Braconi D, Millucci L, Ghezzi L, Santucci A (2013) Redox proteomics gives insights into the role of oxidative stress in alkaptonuria. Expert Rev Proteomics 10:521–535
Braconi D, Millucci L, Bernardini G, Santucci A (2015) Oxidative stress and mechanisms of ochronosis in alkaptonuria. Free Radic Biol Med - in press
Butterfield DA, Lauderback CM (2002) Lipid peroxidation and protein oxidation in Alzheimer's disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med 32:1050–1060
Castillo ECG, Kourí JB (2004) A new role for chondrocytes as non-professional phagocytes. An in vitro study. Microsc Res Tech 64:269–278
Curtis SL, Roberts NB, Ranganath LR (2014) Interferences of homogentisic acid (HGA) on routine clinical chemistry assays in serum and urine and the implications for biochemical monitoring of patients with alkaptonuria. Clin Biochem 47:640–647
Delevoye C, Giordano F, van Niel G, Raposo G (2011) Biogenesis of melanosomes - the chessboard of pigmentation. Med Sci (Paris) 27(2):153–162
Dweck MR, Chow MW, Joshi NV et al (2012) Coronary arterial 18 F-sodium fluoride uptake: a novel marker of plaque biology. J Am Coll Cardiol 59:1539–1548
Fowler DM, Koulov AV, Alory-Jost C, Marks MS, Balch WE, Kelly JW (2006) Functional amyloid formation within mammalian tissue. PLoS Biol 4(1):e6
Giustarini D, Dalle-Donne I, Lorenzini S et al (2012) Protein thiolation index (PTI) as a biomarker of oxidative stress. Free Radical Biol Med 53:907–915
Hegedus ZL (2000) The probable involvement of soluble and deposited melanins, their intermediates and the reactive oxygen side-products in human diseases and aging. Toxicology 145:85–101
Helliwell TR, Gallagher JA, Ranganath L (2008) Alkaptonuria–a review of surgical and autopsy pathology. Histopathology 53:503–512
Hu KN, McGlinchey RP, Wickner RB, Tycko R (2011) Segmental polymorphism in a functional amyloid. Biophys J 101(9):2242–2250
Jiang Z, Lee JC (2014) Lysophospholipid-containing membranes modulate the fibril formation of the repeat domain of a human functional amyloid, pmel17. J Mol Biol 426(24):4074–4086
Kamalvand G, Ali-Khan Z (2004) Immunolocalization of lipid peroxidation/advanced glycation end products in amyloid A amyloidosis. Free Radic Biol Med 36:657–664
Lachmann HJ, Goodman HJ, Gilbertson JA et al (2007) Natural history and outcome in systemic AA amyloidosis. N Engl J Med 356:2361–2371
Laschi M, Tinti L, Braconi D et al (2012) Homogentisate 1,2 dioxygenase is expressed in human osteoarticular cells: implications in alkaptonuria. J Cell Physiol 227:3254–3257
Leonhardt RM, Vigneron N, Hee JS, Graham M, Cresswell P (2013) Critical residues in the PMEL/Pmel17 N-terminus direct the hierarchical assembly of melanosomal fibrils. Mol Biol Cell 24(7):964–981
Magy N, Benson MD, Liepnieks JJ, Kluve-Beckerman B (2007) Cellular events associated with the initial phase of AA amyloidogenesis: insights from a human monocyte model. Amyloid 14:51–63
Martin JP Jr, Batkoff B (1987) Homogentisic acid autoxidation and oxygen radical generation: implications for the etiology of alkaptonuric arthritis. Free Radic Biol Med 3:241–250
McGlinchey RP, Yap TL, Lee JC (2011) The yin and yang of amyloid: insights from α-synuclein and repeat domain of Pmel17. Phys Chem Chem Phys 13(45):20066–20075
Millucci L, Spreafico A, Tinti L et al (2012) Alkaptonuria is a novel human secondary amyloidogenic disease. Biochim Biophys Acta 1822:1682–1691
Millucci L, Ghezzi L, Braconi D et al (2014a) Secondary amyloidosis in an alkaptonuric aortic valve. Int J Cardiol 172:e121–e123
Millucci L, Giorgetti G, Viti C et al (2014b) Chondroptosis in alkaptonuric cartilage. J Cell Physiol 230(5):1148-57
Millucci L, Ghezzi L, Paccagnini E et al (2014c) Amyloidosis, inflammation, and oxidative stress in the heart of an alkaptonuric patient. Mediat Inflamm 2014:258471
Millucci L, Ghezzi L, Bernardini G et al (2014d) Diagnosis of secondary amyloidosis in alkaptonuria. Diagn Pathol 9:185
Momohara S, Okamoto H, Yamanaka H (2008) Chondrocyte of rheumatoid arthritis serve as a source of intra-articular acute-phase serum amyloid A protein. Clin Chim Acta 398:155–156
Nakamura T (2011) Amyloid A amyloidosis secondary to rheumatoid arthritis: pathophysiology and treatments. Clin Exp Rheumatol 29:850–857
Obici L, Raimondi S, Lavatelli F, Bellotti V, Merlini G (2009) Susceptibility to AA amyloidosis in rheumatic diseases: a critical overview. Arthritis Rheum 61:1435–1440
Ranganath LR, Milan A, Hughes AT, et al (2014) Suitability of nitisinone in alkaptonuria 1 (SONIA 1): an international, multicenter, randomized, open-label, no-treatment controlled, parallel-group, dose-response study to investigate the effect of once daily nitisinone on 24-hour urinary homogentisic acid excretion in patients with alkaptonuria after 4 weeks of treatment. Annals of the Rheumatic Diseases. doi:10.1136/annrheumdis-2014-206033
Rochin L, Hurbain I, Serneels L, Fort C, Watt B, Leblanc P, Marks MS, De Strooper B, Raposo G, van Niel G (2013) BACE2 processes PMEL to form the melanosome amyloid matrix in pigment cells. Proc Natl Acad Sci U S A 25:110(26)
Selvi E, Manganelli S, Mannoni A, Benucci M, Minacci C, Marcolongo R (2000) Chronic ochronotic arthritis: clinical, arthroscopic, and pathologic findings. J Rheumatol 27:2272–2274
Selvi E, Garcia-Gonzalez E, Maggio R, Lorenzini S, Santucci A, Galeazzi M (2010) False proteinuria in patients with alkaptonuria. Clin Exp Rheumatol 28:591
Spencer JM, Gibbons CL, Sharp RJ, Carr AJ, Athanasou NA (2004) Arthroplasty for ochronotic arthritis: no failure of 11 replacements in 3 patients followed 6-12 years. Acta Orthop Scand 75:355–358
Spreafico A, Millucci L, Ghezzi L et al (2013) Antioxidants inhibit SAA formation and pro-inflammatory cytokine release in a human cell model of alkaptonuria. Rheumatology (Oxford) 52:1667–1673
Taylor AM, Wlodarski B, Prior IA et al (2010) Ultrastructural examination of tissue in a patient with alkaptonuric arthropathy reveals a distinct pattern of binding of ochronotic pigment. Rheumatology (Oxford) 49:1412–1414
Theos AC, Watt B, Harper DC, Janczura KJ, Theos SC, Herman KE, Marks MS (2013) The PKD domain distinguishes the trafficking and amyloidogenic properties of the pigment cell protein PMEL and its homologue GPNMB. Pigment Cell Melanoma Res 26(4):470–486
Tinti L, Spreafico A, Braconi D et al (2010) Evaluation of antioxidant drugs for the treatment of ochronotic alkaptonuria in an in vitro human cell model. J Cell Physiol 225:84–91
Tinti L, Spreafico A, Chellini F, Galeazzi M, Santucci A (2011a) A novel ex vivo organotypic culture model of alkaptonuria-ochronosis. Clin Exp Rheumatol 29:693–696
Tinti L, Taylor AM, Santucci A et al (2011b) Development of an in vitro model to investigate joint ochronosis in alkaptonuria. Rheumatology (Oxford) 50:271–277
Watt B, van Niel G, Raposo G (2013) Marks MS (2013) PMEL: a pigment cell-specific model for functional amyloid formation. Pigment Cell Melanoma Res 26(3):300–315
Westermark GT, Westermark P (2009) Serum amyloid A and protein AA: molecular mechanisms of a transmissible amyloidosis. FEBS Lett 583:2685–2690
Zatkova A (2011) An update on molecular genetics of Alkaptonuria (AKU). J Inherit Metab Dis 34:1127–1136
Acknowledgments
This work has been supported by Telethon Italy grant GGP10058 and FP7 Research & Innovation Grant 304985-2 – DevelopAKUre. The authors also thank Toscana Life Sciences Orphan_1 project, Fondazione Monte dei Paschi di Siena, and aimAKU – Associazione Italiana Malati di Alcaptonuria (ORPHA263402).
Compliance with ethics guidelines
ᅟ
Conflict of interest
None.
Informed consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients being included in the study.
Animal rights
All institutional and national guidelines for the care and use of laboratory animals were followed.
All experiments were carried out according to the ECC guidelines for animal care (DL 116/92, application of the European Communities Council Directive 86/609/ EEC) and all efforts were made to minimize animal suffering.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Francois Feillet
Rights and permissions
About this article

Cite this article
Millucci, L., Braconi, D., Bernardini, G. et al. Amyloidosis in alkaptonuria. J Inherit Metab Dis 38, 797–805 (2015). https://doi.org/10.1007/s10545-015-9842-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-015-9842-8