
Abstract
Background
In glutaric aciduria type 1 (GA1) the neurotoxic metabolites glutaric acid (GA) and 3-hydroxyglutaric acid (3-OH-GA) accumulate within the brain. Due to limited efflux across the blood–brain-barrier biochemical monitoring of intracerebrally accumulating toxic metabolites is as yet not possible.
Aims
To investigate brain metabolic patterns in glutaric aciduria type 1 using 1H magnetic resonance spectroscopy (1H-MRS) with focus on detecting the disease-related neurotoxic metabolites GA and 3-OH-GA.
Patients and methods
Short echo time 1H-MRS was performed in 13 treated metabolically stable patients. Twenty-one white matter and 16 basal ganglia spectra from 12 patients (age range 7 months - 22 years) were included. Subgroups based on age, biochemical phenotype and/or associated MRI changes were compared with control spectra.
Results
GA was elevated in white matter of patients. 3-OH-GA was elevated in white matter of older patients with associated signal changes on MRI, which was structurally characterized by decreased creatine and phosphocreatine (tCr) and elevated choline (Cho). Metabolite changes differed with biochemical phenotype and disease duration: Low excretors with up to 30 % residual enzyme activity had only mildly, non-significantly elevated GA and mildly subnormal N-acetylaspartate (tNAA). High excretors with complete lack of enzyme activity had significantly increased GA, tNAA was mildly subnormal in younger and decreased in older high excretors.
Conclusions
GA and 3-OH-GA are detectable by in vivo 1H-MRS, which might finally allow biochemical follow-up monitoring of intracerebrally accumulating neurotoxic metabolites in GA1. A high excreting phenotype appears to be a risk factor for cerebral GA accumulation and progressive neuroaxonal compromise despite a similar clinical course in younger high and low excreting patients. This might have consequences for long-term outcome.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Glutaric aciduria type 1 (GA1; OMIM#231670) is an autosomal-recessively inherited neurometabolic disorder due to deficiency of glutaryl-CoA dehydrogenase (GCDH, EC1.3.99.7) resulting in accumulation of the putatively neurotoxic dicarboxylic acids glutaric acid (GA) and 3-hydroxy-glutaric acid (3-OH-GA), and the non-toxic glutarylcarnitine. Without treatment most patients develop an acute encephalopathic crisis during the first 6 years of life, resulting in acute striatal injury and, subsequently, dystonia (Kölker et al 2006). Treatment according to evidence-based guideline recommendations with low lysine diet, carnitine supplementation and emergency treatment in catabolic state has dramatically improved the neurological outcome in GA1 patients (Heringer et al 2010; Kölker et al 2011). Two biochemical phenotypes, called low and high excretors, have been defined based on excretion of GA in urine (Baric et al 1999). While there is a correlation between genotype and biochemical phenotype (Christensen et al 2004), none is as yet known between biochemical phenotype and neurological outcome (Christensen et al 2004; Kölker et al 2006).
1H-MRS allows in vivo assessment of cerebral metabolite changes in neurometabolic disorders, namely (1) changes related to structural alterations, e.g., changes of myelin, glial and neuronal cells, (2) elevated lactate indicating impaired oxidative phosphorylation, and (3) disease-specific changes due to the enzymatic defect. Previous case reports of 1H-MRS in GA1 include normal spectra (Bähr et al 2002; Elster 2004; Santos and Roach 2005), metabolite alterations related to assumed structural changes (Bodamer et al 2004; Kurul et al 2004; Oguz et al 2005; Sijens et al 2006; Sonmez et al 2007; Pérez-Duenas et al 2009; Cakmakci et al 2010), and impaired oxidative phosphorylation (Oguz et al 2005). Detection of disease-specific metabolites has not yet been described. We performed short echo time 1H-MRS in a cohort of 13 treated, metabolically stable patients with focus on detecting the disease-related dicarboxylic metabolites GA and 3-OH-GA. Due to limited efflux capacity of the blood–brain-barrier for dicarboxylic acids and consequent intracerebral accumulation of GA and 3-OH-GA, their serum and urine values do not reflect the concentrations in the brain compartment (Funk et al 2005; Külkens et al 2005). The purpose of this study was to detect GA and 3-OH-GA by 1H-MRS, which might finally provide a diagnostic tool for monitoring treatment efficacy, and to investigate patterns of metabolite changes in relation to the patients’ age, biochemical phenotype, and coexistent white matter changes.
Patients and methods
A total of 22 white matter and 19 basal ganglia spectra were acquired in 13 patients as part of a prospective follow-up study on GA1. Written informed consent was obtained from patients and/or parents according to the declaration of Helsinki. The study was approved by the Institutional Ethics Committee of Heidelberg University (S-049/2010). One spectrum was excluded due to artefact and three other spectra due to poor quality (SNR 4-6, full width at half maximum 0.084–0.107 ppm, as estimated by LCModel). The remaining 21 white matter and 16 basal ganglia spectra from 12 patients (age range 7 months - 22 years, mean 9.2 years, median 10.1 years) were compared with control spectra of basal ganglia (n = 13) and white matter of the centrum semiovale (n = 16) from 17 patients (age range 9 months - 17 years, mean 8.9 years, median 11.6 years) with normal MRI findings who had undergone MRI for headache (n = 7), syncope (n = 2), isolated facial nerve paresis, visual disturbances, sinusitis, vertigo, seizure, suspected orbital haemangioma, staging of pelvic rhabdomyosarcoma, and cephalic dysplastic skin changes (each n = 1).
Patient characteristics are summarized in Table 1. All patients received treatment according to guidelines (Kölker et al 2007a, 2011). No patient had suffered from acute onset of striatal injury. Conventional MRI (T2, T1, FLAIR) of the same session was assessed for atrophy and/or T2-hyperintensity of pallidum and/or putamen, delayed myelination, and the presence of abnormal white matter signal not corresponding to the normal pattern of myelinated and unmyelinated structures of a younger child, more prominent than the signal of unmyelinated white matter, and not restricted to parieto-occipital white matter (Harting et al 2009).
Single voxel short echo time 1H-MRS (PRESS, TE 30 ms, TR 2000 ms, 96 averages, 3.92 ml) were acquired at 1.5 T (Siemens Avanto, Germany). Spectra were processed with LCModel using the standard basis set of 17 metabolites (Provencher 1993) as well the standard basis set with added GA, 3-OH-GA, 2-hydroxy-glutaric acid (2-OH-GA), 3-hydroxy-butyric acid (3-OH-But), or L-carnitine. Metabolites were measured at our scanner with the exception of 2-OH-GA and 3-OH-GA, which were simulated (VeSPA) using reported data (HMDB; Bal and Gryff-Keller 2002). Metabolite concentrations were expressed as concentration ratios with respect to tCr. tCr was also expressed relative to the signal of unsuppressed water in order to detect changes due to tissue rarefaction and/or increased water signal. In addition summed spectra were generated by LCModel analysis of the added phase corrected and referenced individual spectra.
To account for metabolite changes during normal maturation (Kreis et al 1993; Pouwels et al 1999) we separately analyzed younger (≤36 months, n = 5 patients/7 MRS) and older patients (>36 months, n = 7 patients/14 MRS). Moreover, patient subgroups based on biochemical phenotype and/or associated MRI changes were compared. Two of the five younger patients were examined during their first and second year of life (pat. 2, 3). Three of the seven older patients were re-examined within 4–5 months with differently strict diet (pat. 7, 8, 10), pat. 6 had routine follow-up after 28 months and pat. 9 after 11, 12, and 25 months. While the multiple measurements are a potential bias, averaging reduces discrimination between already small groups. Therefore metabolite values obtained with and without averaging were compared. If group mean values obtained with and without averaging were clearly different, bias due to multiple measurements was assumed. For metabolites with similar mean group values but lower discrimination between groups compared, we considered loss of discrimination as being due to smaller numbers. For these metabolites both, group values from averaged and non-averaged data with corresponding significance are given in Table 1; otherwise values are from averaged data.
Statistical analysis was performed with Matlab (The Maths Works Inc., U.S.A.). Metabolite concentrations determined with different basis sets were compared using the Wilcoxon signed rank test as a paired, two-sided test. ANOVA with multiple comparisons was used to compare metabolite values.
Results
Associated MRI changes
Myelination was delayed and the pallidum homogeneously T2-hyperintense in all four younger high excretors (5 MRIs). White matter changes were detected in all four MRIs of younger high excretors examined ≥12 months. Not surprisingly considering the largely T2-hyperintense white matter in young infants, no white matter changes were detected in pat. 1 examined at 7 months. The two MRIs of the low excretor revealed a small T2-hyperintensity at the lateral border of the putamen (pat. 2, no metabolic crisis or dystonia) occupying a minuscule portion of the spectroscopy voxel, but were otherwise normal. Consequently subgroups based on MRI and biochemical phenotype were very similar in patients ≤36 months.
Myelination was complete in all older patients. White matter changes were present in five of seven high excretors (7/10 MRIs). We therefore not only compared low and high excretors with controls but also low excretors, high excretors without and high excretors with white matter changes. For the nine basal ganglia spectra of older patients there was an associated small area of pallidal T2-hyperintensity in the two high excretors with white matter changes (pat. 7, 9), whereas pallidum and putamen were normal in the remaining patients.
Detection of disease-specific metabolites
First, we observed that GA, 2-OH-GA, 3-OH-GA, 3-OH-But were fitted at low level and with large variability in individual control spectra. This was not unexpected due to the overlap of resonances with those of standard metabolites. The low levels in controls were accordingly used as a reference value for comparison with patient values.
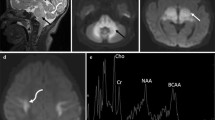
While patients’ GA was not increased in basal ganglia, it was elevated in white matter. GA was significantly increased in high excretors but not low excretors (Table 2), which might be partly due to the difficulty of detecting GA with its concurrent high variability. To reduce variability summed spectra were generated, which have less noise and consequently more reliable detection of metabolites and usually lower metabolite concentrations than individual spectra. In summed spectra, GA was detected in younger and older high excretors, but neither in younger or older controls nor older low excretors (Fig. 1). 3-OH-GA was highest and significantly increased in older high excretors with white matter changes. There was no correlation with lysine intake. For 2-OH-GA, 3-OH-But, and carnitine no significant differences between patient and control spectra were observed.

Individual and summed white matter spectra of older patients and controls. LCModel output with insertion of fitted GA. a, b: Spectra of a low (pat. 10) and high excretor (pat. 9). c-f: Summed spectra of controls (c), low excretors (d), high excretors without (e) and with (f) white matter changes. Note fitted GA in high excretors vs. lack of detection in controls and low excretors, reduced tNAA of high excretors and increased Cho in high excretors with white matter changes
Influence of GA on the standard metabolites of the basis set
As addition of any metabolite to the basis set will affect the fitting of the standard metabolites, we investigated how inclusion of GA affected these. The difference of metabolite concentrations determined with the two basis sets (std&GA-std) in all patient and control spectra correlated (p < 0.001) positively with GA for tNAA (r = 0.74; Suppl. Fig. 1) and lipids at 1.3 ppm (r = 0.64) and negatively for Glu (glutamate; r = −0.68), GABA (r = −0.45), and macromolecules&lipids at 2.0 ppm (r = −0.9). Inclusion of GA in the basis set apparently results in a concentration-dependent overestimation of tNAA. Conversely the standard basis set would underestimate tNAA in patients with elevated GA. We therefore used the std&GA basis set for analysis, preferring underestimation of potential tNAA reductions in patients to overestimation with the standard basis set.
Alterations of non disease-specific standard metabolites
White matter
In younger patients none of the standard metabolites differed significantly between controls, low, and high excreting patients. tNAA was mildly decreased in low and high excretors compared to controls (Table 2).
In older patients, tCr was significantly reduced in high excretors with white matter changes on MRI and normal in low excretors and high excretors without white matter changes. Low excretors were not significantly different from controls. In contrast high excretors had significantly reduced tNAA with similar values for high excretors with and without white matter changes. In addition Cho was increased in high excretors due to a significant increase in those with white matter changes, while those without white matter changes had normal Cho (Fig. 2). Glu was mildly decreased in high and low excretors. For the minor metabolites glutamine (Gln) and aspartate (Asp) we observed small, but significant increases: Gln was increased in high excretors with white matter changes. Asp was increased in high excretors without white matter changes compared to high excretors with white matter changes and controls and it was high normal in low excretors. No metabolite correlated with lysine intake.

White matter metabolites in patients and controls (boxplots). For older high excretors subgroups without and with white matter changes are depicted in addition to the entire group. tCr (a), GA/tCr (b), tNAA/tCr (c), Cho/tCr (d). contr: controls, low/high: low/high excretors, high_s/c: high excretors without and with white matter changes, </>36: younger and older age groups
Basal ganglia
In younger patients tNAA/tCr was reduced in the low excretor due to a non-significant increase of tCr and decrease of tNAA, and it was mildly, non-significantly reduced in high excretors. In older patients tCr was significantly increased in low excretors with a concurrent decrease of tNAA/tCr and mildly increased in high excretors. There was no correlation with lysine intake. No differences of other standard metabolites were observed.
Discussion
The aim of this study was to systematically evaluate brain metabolism in patients with GA1 using 1H-MRS. The major results are that (1) the disease-related metabolites GA and 3-OH-GA can be detected in treated patients in vivo and that (2) metabolic patterns differ depending on biochemical phenotype and age.
In vivo detection of GA, the name-giving metabolite of GA1
In GA1, GA and 3-OH-GA accumulate within the brain due to deficiency of GCDH and limited efflux of dicarboxylic acids across the blood–brain-barrier. Their concentrations exceed serum values 10- to 1000-fold (Sauer et al 2010). Intracerebrally accumulating GA and 3-OH-GA appear to be central for pathogenesis: Data point to neurotoxicity via synergistic effects of disrupted mitochondrial energy homeostasis, excitotoxicity, and increased susceptibility to oxidative stress. Neuronal apoptosis and oligodendrocyte damage secondary to astrocytosis have further been reported as well as injury of immature oligodendroglial precursor cells by GA and 3-OH-GA (Kölker et al 2004; Sauer et al 2005; Stellmer et al 2007; Zinnanti et al 2007; Lamp et al 2011; Olivera-Bravo et al 2011, 2014; Seminotti et al 2013; Gerstner et al 2005). Combined metabolic treatment with low lysine diet, carnitine supplementation, and carbohydrate-rich emergency treatment during catabolic state aims to reduce accumulation of GA and 3-OH-GA which has dramatically improved the neurological outcome (Heringer et al 2010; Kölker et al 2011). Monitoring by following serum/urine values is not reliable and as yet no reliable biomarker for biochemical follow-up monitoring or for predicting the disease course has been identified (Kölker et al 2011). We hypothesized that 1H-MRS might allow detection of the disease-related and neurotoxic metabolites GA and 3-OH-GA, furthering our understanding of GA1 and finally allowing biochemical follow-up monitoring.
Our results show that increased GA can be detected in vivo in GA1 within certain limitations. As expected for a metabolite with complex resonances overlapping resonances of other, much more highly concentrated metabolites, GA was fitted at low level in controls. This level was used as reference for comparison. Sensitivity was further limited by large variability of fitting of individual spectra and even in summed spectra of groups Cramer-Rao lower bounds were still approximately 60 %. Nevertheless, higher mean values of selected groups suggested that indeed concentrations were elevated and results of summed spectra were consistent with this. Detection could in future be enhanced by higher field strength, a larger VOI for higher SNR (e.g., supraventricular 30x30x40 mm containing white matter and cortex), or edited spectroscopy as used for in vivo detection of 2-OH-GA in glioma (Choi et al 2013).
We found significantly increased GA in patients’ white matter, but not basal ganglia, which is most likely at least partially due to the latter’s higher iron content and consecutively less favorable spectroscopy conditions. Elevated white matter GA is consistent with postmortem (Goodman et al 1977; Leibel et al 1980; Kölker et al 2003; Funk et al 2005), biopsy (Külkens et al 2005), and mouse model (Sauer et al 2006) findings. These have demonstrated elevated GA and 3-OH-GA throughout the brain with 3-OH-GA apparently not elevated to the same degree as GA (Funk et al 2005; Külkens et al 2005; Sauer et al 2006).
Patients with GA tend to increase their daily lysine intake after age 5–6 years (Boy et al 2013). Why some patients develop white matter changes with myelin splitting on histopathology (Bergman et al 1989; Soffer et al 1992) is not known. Intriguingly 3-OH-GA concentrations were highest in this group. Normally intramyelinic edema leading to myelin splitting is prevented by rapid siphoning and dispersion of potassium released on neural activity. This occurs via the panglial syncytium and is driven by electrical and osmotic gradients (Rash 2010). In GA1 flux through the citric acid cycle is reduced due to direct inhibition of 2-oxoglutarate dehydrogenase complex by glutaryl-CoA (Sauer et al 2005) and reduced uptake of anaplerotic substrates with higher GA and 3-OH-GA (Lamp et al 2011). A large and long-standing elevation of GA and 3-OH-GA might therefore result in chronically lower ATP generation for neuronal Na+/K+-ATPases and consequently slightly lower transmembrane potential and increased paranodal K+. This in turn might lead to overburdening of the siphoning system and decreased driving forces resulting in white matter changes.
Spectroscopic correlates of structural and maturational changes in GA1
While disease-related metabolites have not previously been investigated in GA1, there are reports on metabolite alterations related to assumed structural changes and the presence of lactate in a total of thirteen patients (Bähr et al 2002; Bodamer et al 2004; Elster 2004; Kurul et al 2004; Oguz et al 2005; Santos and Roach 2005; Sijens et al 2006; Sonmez et al 2007; Pérez-Duenas et al 2009; Cakmakci et al 2010) (Suppl.Table 1). In contrast to the reported cases our cohort of treated, metabolically stable patients without acute encephalopathic crisis was clinically homogeneous and the biochemical phenotype was known. We could therefore investigate alterations related to age, biochemical phenotype, and coexistent MRI changes. We realize that in spite of presenting the largest cohort of GA1 patients examined with spectroscopy so far, our results are still limited by the small number of patients in subgroups.
While different patterns were observed for white matter, basal ganglia changes in our patients were sparse: Increased tCr and decreased tNAA suggest slight astrogliosis and are consistent with reported patients without acute encephalopathic crisis (Bähr et al 2002; Oguz et al 2005; Sonmez et al 2007).
White matter in younger patients was statistically normal with mildly lower tNAA and without clear differences between high and low excretors. As tNAA normally increases after birth (Kreis et al 1993; Pouwels et al 1999) the mildly subnormal tNAA might be either evidence of retarded maturation, as suggested by delayed myelination on MRI, or of early white matter alterations. Elevated white matter Cho/Cr in two of three reported children up to 37 months (Kurul et al 2004; Cakmakci et al 2010) might reflect the more severe disease course in these two untreated children with striatal lesions.
White matter of older low excretors, similar to younger ones, was statistically normal. By contrast tNAA was significantly decreased in older high excretors indicating progressive neuroaxonal impairment and/or loss. Values did not clearly differ between high excretors with and without white matter changes. Other metabolic changes in high excretors were clearly related to coexistent white matter changes, namely the reduction of tCr and increase of Cho as well as the increase of Gln. While reduction of tCr in high excretors with white matter changes is consistent with the myelin splitting observed on histopathology (Bergman et al 1989; Soffer et al 1992), the elevated Cho suggests additional, most likely secondary, low grade demyelination in long standing GA1.
The increase of Asp in older patients without white matter changes is puzzling. Since Asp is formed from oxaloacetate, high Asp indicates a good flux through the citric acid cycle. This is consistent with lower GA and consecutively less impairment of the citric acid cycle. Being moreover a constituent of NAA, a potential donor of acetyl-CoA in brain lipid synthesis, there might be a link to myelination and myelin maintenance (D’Adamo et al 1968; Chakraborty et al 2001). Whether or not there is a link between increased Asp, near normal NAA, and absence of white matter changes is open to speculation.
Different metabolic profiles in high and low excretors
As yet, there is no known correlation between the biochemical phenotype, namely high or low excretors, and clinical phenotype. High and low excretors differ by GA excretion (≥/<100 mmol GA/mol creatinine (Baric et al 1999)) and complete loss of GCDH activity in high excretors vs. ≤30 % residual activity in low excretors (Kölker et al 2006, b). During the critical time for acute encephalopathic crises, e.g., until the age of six years, the frequency of dystonia, acute encephalopathic crises, and of morbidity is similar in both groups. As generalized dystonia is the predominant clinical feature (Gitiaux et al 2008) in older symptomatic patients and generally remains stable for many years, it is not surprising that no clinical difference has been reported for older patients. By contrast, brain metabolic profiles apparently differ: While low excretors were statistically normal, GA was significantly increased in high excretors, which fits well with the differing enzyme (in)activity. Elevated 3-OH-GA together with high GA in older high excretors with white matter changes is consistent with increased 3-OH-GA and GA as risk factors for the development of white matter changes, although we can exclude neither pooling in altered white matter nor additional factors. Moreover high excretors had significantly reduced tNAA, which was similar in subgroups with and without white matter changes. Our results therefore imply that — in spite of the similar clinical course in younger patients — a high excreting biochemical phenotype is a risk factor for overall lower neuronal marker tNAA and greater accumulation of GA as well as the development of white matter changes associated with and potentially caused by elevated 3-OH-GA and GA in a subgroup of high excretors.
To summarize, we have shown that 1H-MRS can detect the neurotoxic metabolites GA and 3-OH-GA in white matter of treated GA1 patients. 1H-MRS might therefore finally present a tool for intracerebral monitoring of metabolic maintenance treatment and its efficacy in reducing the accumulation of GA and 3-OH-GA in the brain.
Our results indicate that a high excreting phenotype is a risk factor for GA accumulation and progressive neuroaxonal compromise despite the similar clinical course in younger high and low excreting patients. This might have consequences for the long-term outcome and should be carefully evaluated in follow-up studies. 1H-MRS in signal-altered white matter of older patients suggests additional low grade demyelination in addition to tissue rarefaction and myelin splitting known from histopathology.
References
Bähr O, Mader I, Zschocke J, Dichgans J, Schulz J (2002) Adult onset glutaric aciduria type I presenting with a leukoencephalopathy. Neurology 59:1802–1804
Bal D, Gryff-Keller A (2002) 1H and 13C NMR study of 2-hydroxyglutaric acid and its lactone. Magn Reson Chem 40:533–536
Baric I, Wagner L, Feyh P, Liesert M, Buckel W, Hoffmann G (1999) Sensitivity and specificity of free and total glutaric and 3-hydroxyglutaric acid measurements of stable-sotope dilution assays for the diagnosis of glutaric aciduria type I. J Inherit Metab Dis 22:867–881
Barry M, VanSwearingen J, Albright A (1999) Reliability and responsiveness of the Barry-Albright dystonia scale. Dev Med Child Neurol 41:404–411
Bergman I, Finegold D, Gartner J, Zitelli B, Claassen D, Scarano J et al (1989) Acute profound dystonia in infants with glutaric acidemia. Pediatrics 83:228–234
Bodamer OA, Gruber S, Stöckler-Ipsiroglu S (2004) Nuclear magnetic resonance spectroscopy in glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis 27:877–883
Boy N, Haege G, Heringer J, Assmann B, Mühlhausen C, Ensenauer R et al (2013) Low lysine diet in glutaric aciduria type I–effect on anthropometric and biochemical follow-up parameters. J Inherit Metab Dis 36:525–533
Cakmakci H, Pekcevik Y, Yis U, Unalp A, Kurul S (2010) Diagnostic value of proton MR spectroscopy and diffusion-weighted MR imaging in childhood inherited neurometabolic brain diseases and review of the literature. Eur J Radiol 74:e161–e171
Chakraborty G, Mekala P, Yahya D, Wu G, Ledeen R (2001) Intraneuronal N-acetylaspartate supplies acetyl groups for myelin lipid synthesis: evidence for myelin-associated aspartoacylase. J Neurochem 78:736–745
Choi C, Ganji S, Hulsey K, Madan A, Kovacs Z, Dimitrov I et al (2013) A comparative study of short- and long-TE 1H MRS at 3 T for in vivo detection of 2-hydroxyglutarate in brain tumors. NMR Biomed 26:1242–1250
Christensen E, Ribes A, Merinero B, Zschocke J (2004) Correlation of genotype and phenotype in glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis 27:861–868
D’Adamo A, Gidez L, Yatsu F (1968) Acetyl transport mechanisms. Involvement of N-Acetyl aspartic acid in de novo fatty acid biosynthesis in the developing Rat brain. Exp Brain Res 5:267–273
Elster A (2004) Glutaric aciduria type I: value of diffusion-weighted magnetic resonance imaging for diagnosing acute striatal necrosis. J Comput Assist Tomogr 28:98–100
Funk CBR, Prasad AN, Frosk P, Sauer S, Kölker S, Greenberg CRDB, Marc R (2005) Neuropathological, biochemical and molecular findings in a glutaric acidemia type 1 cohort. Brain 128:711–722
Gerstner B, Gratopp A, Marcinkowski M, Sifringer M, Obladen M, Buhrer C (2005) Glutaric acid and its metabolites cause apoptosis in immature oligodendrocytes: a novel mechanism of white matter degeneration in glutaryl-CoA dehydrogenase deficiency. Pediatr Res 57:771–776
Gitiaux C, Roze E, Kinugawa K, Flamand-Rouvière C, Boddaert N, Apartis E et al (2008) Spectrum of movement disorders associated with glutaric aciduria type 1: a study of 16 patients. Mov Disord 23:2392–2397
Goodman S, Norenberg M, Shikes R, Breslich D, Moe P (1977) Glutaric aciduria: biochemical and morphologic considerations. J Pediatr 90:746–750
Harting I, Neumaier-Probst E, Seitz A, Maier EM, Assmann B, Baric I et al (2009) Dynamic changes of striatal and extrastriatal abnormalities in glutaric aciduria type I. Brain 132:1764–1782
Heringer J, Boy S, Ensenauer R, Assmann B, Zschocke J, Harting I et al (2010) Use of guidelines improves the neurological outcome in glutaric aciduria type I. Ann Neurol 68:743–752
HMDB: Human Metabolome Database. http://www.hmdb.ca., access year 2013
Kölker S, Hoffmann G, Schor D, Feyh P, Wagner L, Jeffrey I et al (2003) Glutaryl-CoA dehydrogenase deficiency: region-specific analysis of organic acids and acylcarnitines in post mortem brain predicts vulnerability of the putamen. Neuropediatrics 34:253–260
Kölker S, Koeller D, Sauer S, Hörster F, Schwab M, Hoffmann G et al (2004) Excitotoxicity and bioenergetics in glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis 27:805–812
Kölker S, Garbade S, Greenberg C, Leonard J, Saudubray J, Ribes A et al (2006) Natural history, outcome and therapeutic efficacy in children and adults with glutaryl-CoA dehydrogenase deficiency. Pediatr Res 59:840–847
Kölker S, Christensen E, Leonard J, Greenberg C, Burlina A, Burlina A et al (2007a) Guideline for the diagnosis and management of glutaryl-CoA dehydrogenase deficiency (glutaric aciduria type I). J Inherit Metab Dis 30:5–22
Kölker S, Garbade S, Boy N, Maier E, Meissner T, Mühlhausen C et al (2007b) Decline of acute encephalopathic crises in children with glutaryl-CoA dehydrogenase deficiency identified by newborn screening in Germany. Pediatr Res 62:357–363
Kölker S, Christensen E, Leonard J, Greenberg C, Boneh A, Burlina A et al (2011) Diagnosis and management of glutaric aciduria type I - revised recommendations. J Inherit Metab Dis 34:677–694
Kreis R, Emst T, Ross BD (1993) Development of the human brain: in vivo quantification of metabolite and water content with proton magnetic resonance spectroscopy. Magn Reson Med 30:424–437
Külkens S, Harting I, Sauer S, Zschocke J, Hoffmann GF, Gruber S et al (2005) Late-onset neurologic disease in glutaryl-CoA dehydrogenase deficiency. Neurology 64:2142–2144
Kurul S, Çakmakçi H, Dırık E (2004) Glutaric aciduria type 1: proton magnetic resonance spectroscopy findings. Pediatr Neurol 31:228–231
Lamp J, Keyser B, Koeller D, Ullrich K, Braulke T, Mühlhausen C (2011) Glutaric aciduria type 1 metabolites impair the succinate transport from astrocytic to neuronal cells. J Biol Chem 286:17777–17784
Leibel R, Shih V, Goodman S et al (1980) Glutaric acidemia: a metabolic disorder causing progressive choreoathetosis. Neurology 30:1163–1168
Oguz K, Ozturk A, Cila A (2005) Diffusion-weighted MR imaging and MR spectroscopy in glutaric aciduria type 1. Neuroradiology 47:229–234
Olivera-Bravo S, Fernández A, Sarlabós M, Rosillo J, Casanova G, Jiménez M et al (2011) Neonatal astrocyte damage is sufficient to trigger progressive striatal degeneration in a rat model of glutaric acidemia-I. PLoS One 6:e20831
Olivera-Bravo S, Isasi E, Fernández A, Rosillo J, Jiménez M, Casanova G et al (2014) White matter injury induced by perinatal exposure to glutaric acid. Neurotox Res 25:381–391
Pérez-Duenas B, De La Osaa A, Capdevilab A, Navarro-Sastred A, Leistc A, Ribesd A et al (2009) Brain injury in glutaric aciduria type I: the value of functional techniques in magnetic resonance imaging. Eur J Paediatr Neurol 13:534–540
Pouwels PJW, Brockmann K, Kruse B, Wilken B, Wick M, Hanefeld F et al (1999) Regional age dependence of human brain metabolites from infancy to adulthood as detected by quantitative localized proton MRS. Pediatr Res 46:474–485
Provencher S (1993) Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn Reson Med 30:672–679
Rash J (2010) Molecular disruption of the panglialsyncytium block potassium siphoning and axonal saltatory conduction: pertinence to neuromyelitis optica and other demyelinating diseases of the central nervous system. Neuroscience 168:982–1008
Santos C, Roach E (2005) Glutaric aciduria type I: a neuroimaging diagnosis? J Child Neurol 20:588–590
Sauer S, Okun J, Schwab M, Crnic L, Hoffmann G, Goodman S et al (2005) Bioenergetics in glutaryl-coenzyme a dehydrogenase deficiency: a role for glutaryl-coenzyme a. J Biol Chem 280:21830–21836
Sauer S, Okun J, Fricker G, Mahringer A, Crnic L, Mühlhausen C et al (2006) Intracerebral accumulation of glutaric and 3-hydroxyglutaric acids in glutaryl-coenzyme a dehydrogenase deficiency, a biochemical risk factor for neurodegeneration. J Neurochem 97:899–910
Sauer S, Opp S, Mahringer A, Kaminski M, Thiel C, Okun J et al (2010) Glutaric aciduria type I and methylmalonic aciduria: simulation of cerebral import and export of accumulating neurotoxic dicarboxylic acids in in vitro models of the blood–brain barrier and the choroid plexus. Biochim Biophys Acta 1802:552–560
Seminotti B, Amaral A, da Rosa M, Fernandes C, Leipnitz G, Olivera-Bravo S et al (2013) Disruption of brain redox homeostasis in glutaryl-CoA dehydrogenase deficient mice treated with high dietary lysine supplementation. Mol Genet Metab 108:30–39
Sijens P, Smit G, Meiners L, Oudkerk M, van Spronsen F (2006) Cerebral 1H MR spectroscopy evealing white matter NAA decreases in glutaric aciduria type I. Mol Genet Metab 88:285–289
Soffer D, Amir N, Elpeleg O, Gomori J, Shalev R, Gottschalk-Sabag S (1992) Striatal degeneration and spongy myelinopathy in glutaric acidemia. J Neurol Sci 107:199–204
Sonmez G, Mutlu H, Ozturk E, Sildiroglu H, Keskin A, Basekim C et al (2007) Magnetic resonance imaging findings of adult-onset glutaric aciduria type I. Acta Radiol 48:557–559
Stellmer F, Keyser B, Burckhardt B, Koepsell H, Streichert T, Glatzel M et al (2007) 3-Hydroxyglutaric acid is transported via the sodium-dependent dicarboxylate transporter NaDC3. J Mol Med 85:763–770
VeSPA: Versatile Simulation, Pulses, and Analysis for Magnetic Resonance Spectroscopy. http://scion.duhs.duke.edu/vespa/project
Zinnanti W, Lazovic J, Housman C, LaNoue K, O’Callaghan J, Simpson I et al (2007) Mechanism of age-dependent susceptibility and novel treatment strategy in glutaric acidemia type I. J Clin Invest 117:3258–3270
Acknowledgments
We thank the patients with GA1 and their parents for participation in this study. The study was supported by a grant from the Kindness for Kids Foundation, Munich, Germany, to IH.
Conflict of Interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Jutta Gaertner
Electronic supplementary material
Below is the link to the electronic supplementary material.
Suppl.Table 1
Previously reported 1H-MRS of cerebral white matter and basal ganglia. (n: number of patients with MRS; AEC: acute encephalopathic crisis; bgl: basal ganglia; d: day(s); mo: months; yrs: years; TE: echo time) (DOCX 66 kb)
Suppl. Fig. 1
Concentration differences of tNAA and MMLip20 due to inclusion of GA in the basis set (std&GA -std basis sets) plotted against corresponding GA values together with results of linear fitting (tNAA: y=0.16*x+0.023; MMLip20: y=-1.09*x+-0.08). (GIF 7 kb)
Rights and permissions
About this article

Cite this article
Harting, I., Boy, N., Heringer, J. et al. 1H-MRS in glutaric aciduria type 1: impact of biochemical phenotype and age on the cerebral accumulation of neurotoxic metabolites. J Inherit Metab Dis 38, 829–838 (2015). https://doi.org/10.1007/s10545-015-9826-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-015-9826-8