
Abstract
The advent of microfluidic technologies has enabled a better recapitulation of in vitro tumor model with higher biological relevance over conventional monolayer assays. This work built upon a microfluidic system that supported the spontaneous aggregate formation of tumoral cells under flow-induced dynamic physical forces in a confined microchamber without additional matrix materials. Our findings indicated that fluidic streams significantly modulated the biological and architectural features of human breast adenocarcinoma cell (MCF-7), human hepatocarcinoma cell (HepG2), and human cervix adenocarcinoma cell (HeLa) with cell-type-dependent variation. The microfluidic platform was further integrated with a fluorescence detection and imaging system, allowing for non-invasive monitoring of cellular accumulation and spatial distribution of a chemotherapeutic agent, doxorubicin (DOX). The cytotoxic effects of DOX of various concentrations were determined and compared in MCF-7 cells in conventional two-dimensional (2D) static and microfluidic culture conditions. Dose-dependent response to DOX was noticed in both cultures, whereas tumor micronodules grown in microfluidic devices demonstrated significantly lower sensitivity to DOX at increased concentration. Our platform owns promising potentials as a universal modality for bridging traditional 2D cell cultures and in vivo experimentation for preclinical anticancer drug screening.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
In recognition of the limitations of conventional two-dimensional (2D) static cell culture platform for recapitulating native tumor biology, innovative three-dimensional (3D) cell culture systems have been developed for better mimicking the physiological complexity of in vivo tumor phenotype and microenvironment (Edmondson et al. 2014; Li and Kumacheva 2018; Monteiro et al. 2020). Recent advances in ex vivo assembly of biomimetic 3D tumor models for anticancer drug screening have been focused on multicellular tumor spheroid formation with the assistance of biologically-derived matrix components (e.g. hydrogel, Matrigel) or scaffold (Li and Kumacheva 2018; Monteiro et al. 2020). However, limited throughput, low reproducibility, non-uniform spheroid size, and unmet long-term viability remained the major challenges for the use of these tumor models to test drug efficacy (Chaicharoenaudomrung et al. 2019; Verjans et al. 2018). In addition, aside from providing structural support and regulating cellular behavior, the absorptive nature of the 3D matrix materials may affect cell viability as a result of medium depletion. The interaction between the matrix composition and the chemotherapeutic agents will also possibly influence the effective concentration of drugs delivered to cells and lead to inaccurate assessment of the therapeutic efficacy (Casey et al. 2016).
Tumor cells experienced a range of dynamic mechanical forces associated with the host environment, including shear stress, compressive forces, tensile stress, and matrix stiffness (Santoro et al. 2015). These biomechanical stimulations not only modulate tumor cell behavior via cellular signaling changes known as mechanotransduction but also facilitate tumor cell clustering and aggregate formation by overcoming mass transfer limitations (Novak et al. 2018). Moreover, a number of mechanical factors also affect drug penetration and enhance the tumor’s chemoresistance to anticancer drugs (Santoro et al. 2015; Senthebane et al. 2017). Therefore, physiologically relevant biomechanical stimuli must be involved in tumor model recapitulation toward more accurate preclinical screening of antineoplastic activity of therapeutic agents.
Microfluidic techniques have gained wide applicability for in vitro 3D cancer cell studies and drug evaluation (Han et al. 2016; Shang et al. 2019; Trujillo-de Santiago et al. 2019; Valente et al. 2017; Zhang et al. 2018). One of the advantages of the microfluidic system over conventional static spheroid-based tumor models is the ability to reflect the spatial and hydrodynamic complexities by tailoring microchannel design and experimentally regulating flow rate (Ip et al. 2016; Rizvi et al. 2013; Wu et al. 2008). The dynamic fluidic microenvironment and physical forces can be reproduced in the microfluidic system within a physiological range. In our initial attempt, we have reconstructed an in vitro 3D breast tumor tissue model based on a microfluidic system, which then successfully demonstrated its utilization in evaluating the therapeutic efficacy of photodynamic therapy (Yang et al. 2015). In this research, we aimed to further explore the potentials of the microfluidic device in supporting long-term growth and fostering self-organized tumor micronodule formation across a variety of tumor cell types. The microfluidic platform was further integrated with an on-line fluorescence detection and imaging system that allowed in situ monitoring of the tumor cell-drug interaction in a non-invasive way. We investigated how flow-induced mechanical stress influenced tumor cell morphology, proliferation, and drug sensitivity. The commercially available reagent doxorubicin (DOX) was chosen as a model chemotherapeutic compound, of which diffusion, localization, and dose-dependent cytotoxicity were analyzed in the microfluidic-based tumor model and compared with monolayer cells cultured in static conditions. Our integrated microfluidic platform is not only useful for the mass production of tumor micronodules but also efficient for analyzing the therapeutic effects of anticancer drugs with real-time fluorescence intensity detection and quantitative cytotoxicity output.
2 Materials and methods
2.1 Microfluidic device design and fabrication
The design of microfluidic device was followed by our previous study (Yang et al. 2015, 2019), where successful establishment of an in vitro breast tumor tissue model was achieved. The elongated hexagonal prism shape (12 mm long, 6 mm wide, and 200 μm deep) allows the stabilization and uniform distribution of medium flow over the majority of cells. The microfluidic device was fabricated using polydimethylsiloxane (PDMS) based on soft lithography technique and bonded with a glass slide. The detailed process was described elsewhere previously (Yang et al. 2015, 2019).
2.2 Numerical simulation
The flow velocity and associated shear stress value in microchambers were estimated using numerical simulation software to ensure their physiological relevance. The simulations were performed using numerical grids composed of tetrahedron elements generated by ANSYS package ICEM Computational Fluid Dynamics (CFD) 18.0. Following the Navier-Stokes equation, the velocity of the fluid was calculated by Fluent 18.0. Incompressible, single-phase flow under laminar conditions was used. Four flow rates, 0.8, 5, 50, and 500 μL/min, were specified at the inlet of the channel, respectively. Zero-gauge pressure was applied at the outlet of the channel.
2.3 Cell culture
The human breast adenocarcinoma cell line (MCF-7), human hepatocarcinoma cell line (HepG2), and human cervix adenocarcinoma cell line (HeLa) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, 100 mg/ml streptomycin, 1% L-glutamine, 0.14% sodium bicarbonate, and 0.1 mM sodium pyruvate. The cell lines were maintained in a humidified atmosphere containing 5% CO2 at 37 °C. Cells were cultured until they reached approximately 75% confluency before preparing the plates for the cytotoxicity assay or microfluidic culture.
2.4 Setup of the microfluidic system
Prior to cell culture, the chambers of the microfluidic device were exposed to UV irradiation for 30 min and sequentially washed with 70% (v/v) ethyl alcohol, rinsed with phosphate-buffered saline (PBS, pH 7.4), and complete cell culture medium for three times. For cell seeding in the microfluidic chamber, the cell-loaded syringes fitted with 18-G blunt needles were attached to the inlet tubing and were pumped into each chamber. The microfluidic device was then kept at 37 °C and 5% CO2 incubator for 30 min, allowing for the attachment of cells to the substrate. Afterward, a fresh culture medium was continuously introduced into the chambers at a flow rate of 0.8 μL/min controlled by a syringe pump (NE 1000 New Era Pump Systems Inc., Farmingdale, NY).
2.5 Integration of the fluorescence detection system and the microfluidic platform
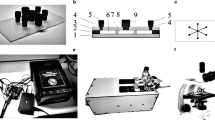
In order to continuously detect the emission intensity of different fluorescent drug substances and quantify their accumulation in cells, a real-time fluorescence detection system was designed based on LabWindows programming and integrated with the microfluidic system. Figure 1 illustrates the systematic setup of the integrated microfluidic-fluorescence intensity detection system, which is consisted of two laser sources (488 nm and 650 nm, 15 mW, Thorlabs), one portable QE Pro Fiber Optic Spectrometer, and a detection probe. The probe has a single 400 μm excitation fiber at its center and is surrounded by nine 200 μm collection fibers, which enables efficient excitation from the laser sources and collection of fluorescent signals from the samples in the microfluidic device. SMA 905 connectors were used for the probe connection between the spectrometer and laser sources. LabWindows-based software was designed for laser source control, spectrometer control, fluorescence acquisition, signal noise reduction, data storage, result analysis, and display. Noted here that two laser sources and corresponding control unit were involved in our system design towards future applications by providing multiple excitation wavelength. System performance and accuracy were firstly tested by measuring the fluorescence intensity of fluorescein isothiocyanate (FITC) with known concentration (Figure S1).

Schematic diagram of the microfluidic platform integrated with in situ fluorescence detection and imaging system. Each chamber of the microfluidic device is connected with a syringe pump for continuous medium supply and a waste container for waste collection. A stereomicroscope equipped with a CCD camera was mounted at the top of the microfluidic chamber which allows real-time observation. Excitation light at the wavelength of 488 nm and 650 nm controlled by the laser shutter (SH) is brought to the platform via a multimode optical fiber. The mirror (M) placed at 45° to the beam bends the excitation light through 90°. The dichroic mirror (DM) and lens (L) are used to couple light into a fiber bundle. The emission light is collected using the same probe connected with the multimode optical fiber with standard SMA 905 connector. This fiber then transmits the light to a QF Pro spectrometer
2.6 DOX exposure
DOX hydrochloride was purchased from Aladdin Reagent Co. (Shanghai, China). All DOX solutions were prepared fresh immediately prior to cellular exposure. A stock solution of 1 mM DOX was prepared in dimethyl sulfoxide (DMSO) then serially diluted with complete DMEM media to reach the final concentration of 1, 5, 10, 50, and 100 μM. The calibration experiment was performed using above known concentrations of DOX, and the derived standard curve was obtained by measuring the absorbance at 485 nm using a UV-vis spectrophotometer (Molecular Devices) (Figure S2). After a preliminary test of cell response to DOX of various concentrations (data not shown), we then chose DOX concentrations of 1 and 10 μM for the following experiments to cover a range from no significant cytotoxicity to acute cytotoxicity and to fall within the range of previous toxicological and clinical studies on DOX (Ravizza et al. 2004; Yokochi and Robertson 2004). For 2D conditions, the cell culture medium was removed, and cells were exposed to DOX containing medium of various DOX concentrations. For microfluidic conditions, after confirming 3D tumor construct formation in each chamber following 10 days of cell culture, DOX at the concentrations of 1 and 10 μM in cell culture medium were loaded in 1 mL syringes and continuously introduced into microfluidic chambers through the inlets at the flow rate of 0.8 μL/min by a digital pump.
2.7 Morphological assessment of cells
For morphological comparison between cells grown in different conditions, MCF-7, Hela, and HepG2 cells in monolayer culture were seeded respectively in 6-well cell culture plates and microfluidic culture chambers at the same cell density of 3 × 105/mL and kept in culture at 37 °C in CO2 incubator. Time-resolved images of morphological changes in cells at different culture conditions were continuously recorded under bright-field microscopy for up to 10 days. To monitor the morphological changes of cells in response to DOX exposures, monolayer and microfluidic cultures of MCF-7 cells were treated with DOX at the concentrations of 1 and 10 μM, respectively. After 24 h incubation, bright-field images were taken using an inverted phase-contrast microscope (Leica). Cells without any treatment were used as controls. Cell area and integral density was calculated using the cellular analysis feature of the software Image J after automated recognition and manual segmentation.
2.8 DOX uptake and distribution
To observe the cellular uptake and distribution of DOX in monolayer cells and microfluidic cultures, after incubating with DOX for 24 h, fluorescent images were respectively obtained in situ at monolayer cells seeded in 6 well-plate and cultured in microfluidic chambers under the same exposure settings. The absorbance of DOX at 485 nm was measured by a UV-vis spectrophotometer (Molecular Devices) to determine the DOX amount using a previously established calibration curve (Figure S2). For 2D conditions in 96 well-plates, the DOX accumulated in cells was calculated by subtracting the free DOX in the supernatant from the total DOX amount in the initial loading medium. To compare the DOX accumulation in 2D and microfluidic conditions, the relative change of DOX concentration as compared to original DOX concentration was calculated as: (DOXbefore- DOXafter)/DOXbefore × 100%, where DOXbefore refers to the initial DOX concentration before cell incubation and DOXafter means the DOX concentration in the collected solution from supernatant or from the microchamber outlet after treatment. Fluorescence intensity in the microfluidic cultures after DOX perfusion was also detected with the integrated fluorescence detection system. Before the measurement, extra DOX remaining in the culture chamber was washed away by rinsing with PBS solution for three times. Fluorescence intensity was measured at five different locations in the DOX-treated tissues after 4 h and 24 h perfusion, respectively. DOX was excited by 488 nm, and its emission at 590 nm was recorded and analyzed.
2.9 In vitro cytotoxicity test
Cell viability in 2D static culture after 24 h treatment with DOX at different concentrations was determined by MTT assay. Briefly, MCF-7, HepG2, and HeLa cells were seeded in 96-well plates at a density of 5 × 103 cells/well in 200 μL culture medium. Following 24 h incubation and attachment, the cells were treated with different concentrations of DOX for further 24 h. After treatment, the cell culture medium was removed, and 0.5 mg/mL MTT solution diluted in fresh cell culture medium was added to each well and incubated for 2 h. The MTT dye was reduced by succinate dehydrogenase in the mitochondria of viable cells to purple formazan crystals. After that, the medium was removed, and DMSO was added, and the absorbance of dissolved formazan crystals was determined by a microplate reader (Molecular Devices). The plates were gently shaken for 1 min, and absorbance was measured at 492 nm. The percentage of cell viability was calculated as: (mean optical density of treated cells-background)/(mean optical density of control cells-background) × 100%, where untreated cells regarded as 100%. The data were obtained from three independent assays.
2.10 Live/dead assay
Cell viability was measured using a Live/Dead Viability/Cytotoxicity Kit (calcein acetoxymethyl ester, calcein-AM, and propidium iodide, PI, Aladdin Reagent Co. Shanghai, China) following vendor’s protocols. Briefly, 5 μL of calcein-AM and 15 μL of PI were mixed well in 5 mL of 1 × Assay Buffer. For 2D culture, cells cultured in 6 well-plate after various DOX treatments were incubated in 500 μL of working Live/Dead stains. For microfluidic cultures, Live/Dead staining solution was loaded into the treated tumor tissues from the inlet. After 15 min incubation, cells were washed twice with PBS and immediately imaged with the inverted fluorescence microscope (Nanjing Jiangnan Novel Optics Co., Ltd.). Samples were excited at 488 nm, and green fluorescence emission for calcein-AM and red fluorescence emission for PI were recorded. Cultures without any treatment were stained as controls.
2.11 CellTiter-Blue assay
Cell viability was analyzed using the CellTiter-Blue assay kit (Promega Corporation, Madison, WI) following the manufacturer’s protocol. Briefly, the CellTiter-Blue reagent was mixed with cell culture medium at a volume ratio of 1:5 to obtain the working solution. For 2D culture, cells cultured in 96-well plates after various treatments were treated with 100 μL of the working solution per well and incubated for 2 h at 37 °C. For microfluidic cultures, chamber tissues after various DOX treatments continuously flowed with the CellTiter-Blue working solution for 24 h. The supernatant solution in 2D well-plate and metabolic waste from each outlet of the microfluidic chambers were collected and then measured for their fluorescence intensity at 560 nm (Ex)/590 nm (Em). Cultures without any treatment were used as controls.
2.12 Statistical analysis
At least three independent experiments were conducted for all experiments. Data were expressed as the mean ± SD. For comparisons, the student t-test was performed between two groups, while the one-way ANOVA was used for comparisons of more than two groups. A value of p < 0.05 was considered statistically significant.
3 Results
3.1 Growth of tumor cells in monolayer and in the microfluidic device
Prior to applying dynamic perfusion in the microfluidic device, numerical simulation was firstly performed to confirm that the flow velocity, shear stress, and pressure are biologically relevant in the microchamber. As shown in Fig. 2, at the inlet flow rates of 0.8, 5, 50, and 500 μL/min, the averaged shear stresses at the center area of the microfluidic chamber were 0.000192, 0.0012, 0.012, and 0.12 dyne/cm2, respectively, which were within the range of reported fluid shear stress generated by the interstitial flows in the tumor microenvironment (approximately 10−6 to 0.1 dyne/cm2) (Mitchell and King 2013a, b). To assure this biological relevance and allow the uniform flow distribution over the microchamber area where most cells were grown and morphological characterization was performed, the flow rate of 0.8 μL/min was used for the following experiments.

CFD simulation of flow velocity field, shear stress, and pressure distribution (from left to right) in the microfluidic chamber fitting at inlet fluid flow rates of 0.8, 5, 50, and 500 μL/min (from top to bottom). The simulation was performed by FLUENT 18.0
As shown in Fig. 3, after initial cell seeding and attachment following the first 24 h, no apparent morphological difference was found between cells grown in 2D well-plate compared to those in the microfluidic device, indicating that cellular growth characteristics did not significantly alter. When initially grown with an identical seeding density, cells cultured in microfluidic chambers were at lower confluency as compared to static well plate, which could be attributed to the weaker cell adhesion onto glass substrate. However, after 5 days of growth, in 2D culture conditions, although the culture medium was changed every day manually, the majority of cells were detached from the substrate and easily removed during medium change. Conversely, under continuous flow, all three types of cells proliferated at a certain growth rate with high viability, merged as a confluent flat layer, which fully spread across the area of the substrate in the microchamber (Fig. 3a). In contrast to the monolayer cultures where cells were flat and stretched, in the microfluidic culture, cells tended to form cell-cell interaction and appeared spherical morphology, leading to diminished individual cell size. After reaching the confluence on the substrate, proliferating cells tended to grow in the vertical direction and fuse together by forming aggregates.

Morphological changes in MCF-7, Hela, and HepG2 cells grown in monolayer culture after attachment for 24 h and in microfluidic culture for up to 10 days. a Representative images were captured in 2D conditions and in microchambers after 24 h, 5 days, and 10 days. Images were taken using an inverted phase-contrast microscope. Scale bar: 100 μm. The changes of individual cell size and integral contrast intensity were calculated in b MCF-7, c Hela, and d HepG2 grown in different conditions over the cultivation period. The average cell areas and contrast intensity were normalized by comparing with the cells cultured in 2D conditions. Five regions of interest were selected based on automated area recognition and manually segmentation, and the analysis was performed in five replicate images in each group using Image J
Based on automated area recognition and manually segmentation, we further analyzed the changes of individual cell size and the contrast of brightfield images in corresponding areas of cell images. We randomly selected five regions of interest in five replicate images, then calculated the cell areas and the integral density. As shown in Fig. 3b–d, decreased average size for individual cell was observed for each cell type as compared to 2D cultures over the entire cultivation period. As the intensity of the contrast measured by the microscope is approximately proportional to the sum of the overlapping cell contributions, the significant enhanced contrast found in microchamber cultures suggested higher cell density for the overlapping regions. The detailed image analysis revealed different cell behavior across cell types in long-term cultivation. As shown in Fig. 3b–d, among the three types of cells, the most dramatic intensity variation of contrast was found in MCF-7 cells, where firm and stable micronodules were generated. Therefore, for the following drug evaluation, we mainly focused on MCF-7-derived tumor tissue model for better comparison. The overall growth of MCF-7 cells in one microchamber was recorded under the phase-contrast microscopy and the stereo microscopy, and the cell viability was verified using a Live/Dead assay (Fig. S3). To determine the integrity of the formed tumor tissue, tissue samples from the microchamber were collected, fixed, and sectioned into thin slices for Hematoxylin & Eosin (H&E) staining. A compact structure composed of ~10 layers of stacked cells was observed (Fig. S3), which shared morphological similarities with in vivo tumor (Li et al. 2017).
3.2 Cellular uptake of DOX and its distribution in 2D and microfluidic cultures
DOX is one of the most commonly used chemotherapeutics commercially available to treat a wide range of cancers. Owning to its intrinsic fluorescence (Farhane et al. 2015; Ozcelikkale et al. 2017), we firstly investigated the cellular uptake behavior in different tumor cell lines and compared its distribution pattern in monolayer cells and microfluidic tumor micronodules. Figure 4a shows the representative fluorescence microscopy images of Hela and HepG2 cells grown on 2D conditions exposed to DOX for 24 h (MCF-7 results were presented later with the comparison with microfluidic results). In 2D static culture, monolayer cells were exposed to a homogenous amount of DOX from the medium and absorbed DOX was clearly traceable in the cytoplasm in every cell with a localization in the nuclei. For quantitative comparison of DOX consumption in different conditions, free DOX remaining in the supernatant solution of monolayer culture and in the waste collected from the microchamber outlet were determined by measuring DOX absorbance and assigning to the DOX standard curve. As shown in Fig. 4b, the cellular uptake of DOX was comparable between different tumor cell lines, where no significant difference was found. As the overall cell number in monolayer and microfluidic culture is difficult to quantify, we compared the relative change of DOX concentration before and after treatment to estimate the amount of DOX consumed by cells. As shown in Fig. 5a, although DOX was delivered to cells at the same dosage, more noticeable relative change of DOX concentration was found in monolayer culture than that in 3D tumor micronodules after 24 h incubation. Specifically, at lower DOX loading dosage, ~40% of DOX was uptaken by monolayer cells, while merely 10% of DOX was accumulated in cells in microfluidic culture via dynamic perfusion. With the increase of DOX loading concentration to 10 μM, the relative change of DOX concentration in monolayer culture (~76%) was still significantly higher than that in 3D microfluidic culture (28%), suggesting that dynamic perfusion was less effective in delivering DOX to cells than homogenous exposure.

a Representative fluorescence microscopy images showing intracellular fluorescence of DOX in Hela and HepG2 cells. Scale bar: 100 μm. b Calculated cellular accumulation of DOX at initial loading concentrations of 1 and 10 μM after incubation with various tumor cell lines for 24 h. c Cell viability of MCF-7, Hela, and HepG2 cells after exposure to DOX at different concentrations for 24 h as determined by MTT assay. All the measurements were done in at least three replicates and the results are expressed as standard error on the mean

a Relative change of DOX concentration after 24 h treatments in 2D static and microfluidic systems as compared to initial loading concentration. b Fluorescence intensity of DOX at different concentration in microfluidic cultures after perfusion for 4 h and 24 h. DOX was excited by 488 nm and its emission was recorded at 590 nm. Data are mean ± standard deviation. c Representative fluorescence microscopy images showing intracellular fluorescence of DOX in MCF-7 cells cultured in monolayer and microfluidic conditions. The cells were exposed to DOX of 1 and 10 μM for 24 h. Scale bar: 200 μm
Our integrated fluorescence detection system (Fig. 1) enabled real-time quantification of DOX concentration by measuring the emission of DOX under laser excitation at a given time. As indicated in Fig. 5b and c, we compared the penetration of DOX in microfluidic micronodules through dynamic perfusion versus static interaction with monolayer cells. We found that the increase of dosage significantly affected the DOX accumulation in both the monolayer condition and tumor micronodules. To our interest, in microfluidic cultures, at the same incubation time (4 h and 24 h), increased loading concentration of DOX from 1 to 10 μM resulted in an approximately two-fold increase of DOX uptaken by MCF-7 cells. With the same loading concentration of DOX, prolonged incubation time from 4 h to 24 h significantly enhanced the intracellular accumulation of DOX with an approximately three-fold increase.
3.3 Cytotoxicity of DOX in monolayer and microfluidic culture
Dose-dependent cytotoxicity assay of DOX was firstly carried out in 2D cell culture systems in multiple tumor cell lines. Figure 4c shows the cellular viability of MCF-7, HeLa, and HepG2 cells to DOX exposure at concentrations of 1 and 10 μM at time points of 24 h, as measured using MTT assay. After treating the monolayer cells with DOX at lower concentration (1 μM), MCF-7 and HepG2 cells showed similar behavior with cell viabilities diminished to 67% and 79%, respectively. Likewise, after DOX treatment at higher concentration (10 μM), similar trends were observed in these two cell lines with cell viabilities of 38% and 26%, respectively. Intriguingly, although the difference of cellular uptake of DOX at both concentrations is not in evidence for the three cell lines (Fig. 4b), DOX is less toxic to the HeLa cells at the lower dosage. However, when increasing the DOX concentration to 10 μM, a significant decrease in cell viability was noticed, leading to 90% cell destruction in Hela cells after treatment.
As the non-invasive MCF-7 cells demonstrated the maximum effectiveness in forming 3D-nodule-like tumor tissue structure among the tested three types of cells, we mainly investigated their response to DOX treatment for better comparison with monolayer cultures. As shown in Fig. 6, after treatment with DOX, morphological changes in MCF-7 cells cultured in different conditions were observed and demonstrated a concentration-dependence. In 2D cultures, cells exposed to the higher concentration of DOX at 10 μM lost their normal morphology and shape, became more rounded and less adherent than the control. Similarly, in the microfluidic culture perfused with DOX continuously, a loosening of tumor mass and interconnections with many disseminating or dissociated cells was noticed, which was associated with the increased DOX concentration. We further calculated the integral density in five randomly chosen areas to semi-quantitatively identify the morphological changes by combining the changes of cell size and contrast intensity in image analysis. From Fig. 6c, a significant difference was noticed in the monolayer and microfluidic cultures after 10 μM DOX treatment. Clearly, under the same exposure conditions, more distinguishable morphological changes were found in 2D cultures, suggesting that static exposure to the therapeutic reagents could lead to more obvious cell destruction than dynamic perfusion.

Representative bright-field images of morphological changes in MCF-7 cells in a monolayer culture and b microfluidic culture. The cells were exposed to different concentrations of DOX at 1 and 10 μM for 24 h, respectively. Cells without any treatment were used as controls. c Morphological changes were further analyzed by calculating the average cell size and integral contrast intensity in selected areas of interests. The data were normalized by comparing with the controls. Images were taken using an inverted phase-contrast microscope and were analyzed using Image J. Scale bar: 100 μm
We then performed the cytotoxicity test in the microfluidic culture and compared that with 2D monolayer results under the same DOX dosage. The changes of cell viability induced by DOX was firstly confirmed using the Live/Dead apoptotic staining. As shown in Fig. 7a, the majority of cells exhibited green fluorescence and remained alive after exposure to 1 μM DOX, whereas a significant number of dead cells was observed after 10 μM DOX treatment. As the intracellular DOX might still emit red fluorescence, it was difficult to distinguish them from dead cells in microfluidic cultures. Therefore, the quantitative comparison of cell viability was further determined by the CellTiter-Blue assay. As shown in Fig. 7b, when treating with a lower concentration of DOX at 1 μM, MCF-7 cell viability was comparable in micronodules (74%) and in the monolayer (84%). However, for the higher concentration of DOX at 10 μM, the results were significantly distinct, where 84% of the cells were alive in the microfluidic culture, and only approximately 46% of the cells had survived in the monolayer after treatment. These findings were also well correlated with above morphological analysis results.

Representative Live/Dead fluorescence staining images of a 2D monolayer and b microfluidic cell culture after various DOX treatments. Live cells were stained fluorescence green with calcein-AM and dead cells were stained fluorescence red with PI. Scale bar: 200 μm. c Cell viability was measured on 2D static and 3D microfluidic tissue culture after 24 h treatment with DOX at the concentration of 1 and 10 μM as compared to control group without any treatment, determined by the CellTiter-Blue assay. *p < 0.01
4 Discussion
Consistent with previous studies (Rizvi et al. 2013), our findings indicated that biologically relevant fluidic flow significantly promoted the long-term growth of tumor cells while maintained cell viability across a variety of tumor cell types, including human breast adenocarcinoma, hepatocarcinoma, and cervix adenocarcinoma cells. Dynamic flow perfusion promoted nutrient delivery within the construct and consequently improved cell survival, growth, and proliferation, which cannot be achieved under conventional 2D static culture conditions (Santoro et al. 2015). The biophysical forces played an important role in fostering the spontaneous aggregation of tumor cells towards the formation of 3D multicellular tumor micronodules through resembling clustered buds (Agosti et al. 2019). The confined culture conditions and fluid shear stress experienced by the micronodules maintained their size consistency and viability versus conventional 2D static methods and previous matrix-dependent 3D tumor spheroids.
Moreover, we have also noticed that the capacity for the tumor cells to develop into a 3D in vivo-like cytoarchitecture via self-organization could be highly relying on the specific cell type. Albeit the same initial seeding density and identical culture conditions, among the three types of cell lines tested in our study, in comparison to MCF-7 cells, Hela and HepG2 cells revealed less significant changes of morphological features and growth behavior with respect to culture conditions. Rather than the non-invasive MCF-7 cells, Hela and HepG2 cells were reported with higher metastatic potentials and invasive abilities, which were closely associated with lower interconnection force and weak attachment properties (Nath et al. 2018; Senthebane et al. 2017). Therefore, a loosely bound organization was noticed in Hela and HepG2 cells cultured in microchamber, whereas MCF-7 cells demonstrated a more compact formation of tumor tissue with higher in vivo similarity.
As one of the most celebrated anti-cancer therapeutics in clinical applications, dosage-dependence of DOX cytotoxicity is well recognized in literatures (Lüpertz et al. 2010; Ravizza et al. 2004; Yokochi and Robertson 2004) and was consistently found in monolayer cultures across different cell lines in our study. Our microfluidic-based in vitro tumor models are readily available for DOX evaluation while providing with non-destructive monitoring of the drug consumption with the integration of on-line detection function.
Our findings suggested significant variations of cellular behaviors in response to DOX in 2D and microfluidic conditions, which were in good agreement with previous studies using 3D tumor spheroids (Casey et al. 2016; Lovitt et al. 2018). The less sensitivity to DOX found in microfluidic culture as compared to 2D conditions could be mainly attributed to 1) less efficient drug delivery into tumor tissues via dynamic perfusion; and 2) drug resistance developed in the tumor microenvironment in the microfluidic culture. From both on-line detection of DOX accumulation and off-line quantitative measurement of consumed DOX, our results suggested that dynamic perfusion was less effective in delivering DOX against tumor micronodules than static interaction. Unlike traditional monolayer conditions that allow a more uniform drug accumulation upon homogenous exposure, uneven DOX distribution in the microfluidic culture would occur during dynamic perfusion when DOX was penetrating the 3D architecture of tumor construct. Reduced DOX transport into the inner part of the micronodules could thus be associated with less cell destruction observed in the microfluidic culture.
Although increased DOX injection concentration from 1 to 10 μM indeed resulted in higher overall drug accumulation in tumor nodules, as reflective of fluorescence imaging and intensity measurement, the elevated cellular DOX level still did not lead to significant enhanced cell killing as expected. Aside from inefficient drug delivery into the center of tumor cells through diffusion, the changes of tumor microenvironment factors may also influence the efficacy of anticancer drugs (Jo et al. 2018). Noticed that monolayer cells grown in static conditions were lacking of essential biomechanical stimuli as occurred in vivo, the cells may thus bypass the hyper-activated signaling pathways associated with drug resistance activation (e.g. extracellular signal-regulated kinase pathway) and present higher sensitivity to therapeutics (Nwosu et al. 2020). On the other hand, tumor cells exposed to a range of dynamic mechanical forces could evoke key proteins, such as Caveolin-1/caveolae, involved in transmembrane signaling systems and further mediate the chemoresistance (Hehlgans and Cordes 2011; Qian et al. 2019). The chronic exposure to acidic microenvironment would also possibly increase the expression level of other proteins relevant to chemoresistance such as heat shock protein HSP27 (Wachsberger et al. 1997). In addition, DOX is prone to protonation in the acidic extracellular pH and suffers from lower cellular uptake due to ‘ion trapping’ (Jo et al. 2018; Raghunand and Gillies 2000). As we did observe the development of a progressive acidic environment in the microfluidic culture with continuously decreasing pH value found in the collected metabolic waste (data not shown), the microfluidic conditions could support the crucial role of tumor microenvironment in modulating cellular resistance to chemotherapeutics.
Previous works have verified the predictive value of various 3D in vitro tumor models for drug efficacy analysis, from which a close resemblance to the in vivo organotypic tumors and similar responses to therapeutics were found as those obtained in vivo (Gunness et al. 2013; Nunes et al. 2019). Previous cytotoxicity studies also reported similar degree of sensitivity to DOX treatment and DOX toxicity mechanism between the in vitro tumor spheroid models and the in vivo animal tests (Baek et al. 2016; Verheijen et al. 2018). Therefore, we envision that similar DOX efficacy and the cellular response observed in our study is likely to occur in solid tumors in vivo, where chemotherapeutic effects not only depend on drug dosage but also the extent of mass transport, local biomechanical forces, distribution patterns, and biochemical cues in the tumor microenvironment. Furthermore, although prospective clinical validations have confirmed the ex vivo patient-derived 3D tumor model as a powerful tool for personalized response predictions in cancer care (Shuford et al. 2019), keeping in mind the much higher complexity in vivo, substantial works are still needed to unravel the correlation between therapeutics efficacy in in vitro 3D tumor models and the results obtained during clinical trials.
5 Conclusion
The present study developed a microfluidic platform that highlighted the value of biomechanical stimuli and sustained medium flow perfusion on tumor cell viability, proliferation, and micronodule formation. The microfluidic device was not only capable of long-term cultivation across a variety of tumor types, but also had the ability to perform cytotoxicity tests with non-invasive monitoring of cell-drug interaction. The results demonstrated the cell-type dependent capacity for tumor micronodule formation via self-organization and the dose-dependent drug resistance in tumor tissues, indicating the importance of mimicking physiologically relevant biochemical and biomechanical conditions during in vitro cytotoxicity study. The use of microfluidic-based experimental system is expected to greatly improve our understanding of the interplay between tumor microenvironment, tumor progression, and chemoresistance to anticancer drugs.
References
A. Agosti, S. Marchesi, G. Scita, P. Ciarletta, ArXiv, 1905.08074 1 (2019)
N.H. Baek, O.W. Seo, M.S. Kim, J. Hulme, S.S.A. An, Onco. Targets. Ther. 9, 7207 (2016)
A. Casey, M. Gargotti, F. Bonnier, H.J. Byrne, Toxicol. Vitr. 33, 99 (2016)
N. Chaicharoenaudomrung, P. Kunhorm, P. Noisa, World J. Stem Cells 11, 1065 (2019)
R. Edmondson, J.J. Broglie, A.F. Adcock, L. Yang, Assay Drug Dev. Technol. 12, 207 (2014)
Z. Farhane, F. Bonnier, A. Casey, H.J. Byrne, Analyst 140, 4212 (2015)
P. Gunness, D. Mueller, V. Shevchenko, E. Heinzle, M. Ingelman-Sundberg, F. Noor, Toxicol. Sci. 133, 67 (2013)
B. Han, C. Qu, K. Park, S.F. Konieczny, M. Korc, Cancer Lett. 380, 319 (2016)
S. Hehlgans, N. Cordes, Am. J. Cancer Res. 1, 521 (2011)
C.K.M. Ip, S.S. Li, M.Y.H. Tang, S.K.H. Sy, Y. Ren, H.C. Shum, A.S.T. Wong, Sci. Rep. 6, 1 (2016)
Y. Jo, N. Choi, K. Kim, H.J. Koo, J. Choi, H.N. Kim, Theranostics 8, 5259 (2018)
Y. Li, E. Kumacheva, Sci. Adv. 4, eaas8998 (2018)
W. Li, Y. Liu, Z. Qian, Y. Yang, Sci. Rep. 7, 1 (2017)
C.J. Lovitt, T.B. Shelper, V.M. Avery, BMC Cancer 18, 1 (2018)
R. Lüpertz, W. Wätjen, R. Kahl, Y. Chovolou, Toxicology 271, 115 (2010)
M.J. Mitchell, M.R. King, Front. Oncol. 3, 1 (2013a)
M.J. Mitchell, M.R. King, New J. Phys. 18, 015008 (2013b)
M.V. Monteiro, V.M. Gaspar, L.P. Ferreira, J.F. Mano, Biomater. Sci. (2020)
B. Nath, A. Raza, V. Sethi, A. Dalal, S.S. Ghosh, G. Biswas, Sci. Rep. 8, 1 (2018)
C. Novak, E. Horst, G. Mehta, APL Bioeng. 2, 031701 (2018)
A.S. Nunes, A.S. Barros, E.C. Costa, A.F. Moreira, I.J. Correia, Biotechnol. Bioeng. 116, 206 (2019)
Z.C. Nwosu, W. Piorońska, N. Battello, A.D. Zimmer, B. Dewidar, M. Han, S. Pereira, B. Blagojevic, D. Castven, V. Charlestin, P. Holenya, J. Lochead, C. De La Torre, N. Gretz, P. Sajjakulnukit, L. Zhang, M.H. Ward, J.U. Marquardt, M.P. di Magliano, C.A. Lyssiotis, J. Sleeman, S. Wölfl, M.P. Ebert, C. Meyer, U. Hofmann, S. Dooley, EBioMedicine 54, 102699 (2020)
A. Ozcelikkale, K. Shin, V. Noe-Kim, B.D. Elzey, Z. Dong, J.T. Zhang, K. Kim, I.C. Kwon, K. Park, B. Han, J. Control. Release 266, 129 (2017)
X.L. Qian, Y.H. Pan, Q.Y. Huang, Y.B. Shi, Q.Y. Huang, Z.Z. Hu, L.X. Xiong, Onco. Targets Ther. 12, 1539 (2019)
N. Raghunand, R.J. Gillies, Drug Resist. Updat. 3, 39 (2000)
R. Ravizza, M.B. Gariboldi, L. Passarelli, E. Monti, BMC Cancer 4, 1 (2004)
I. Rizvi, U.A. Gurkan, S. Tasoglu, N. Alagic, J.P. Celli, L.B. Mensah, Z. Mai, U. Demirci, T. Hasan, Proc. Natl. Acad. Sci. U. S. A. 110, E1974 (2013)
M. Santoro, S.E. Lamhamedi-Cherradi, B.A. Menegaz, J.A. Ludwig, A.G. Mikos, Proc. Natl. Acad. Sci. U. S. A. 112, 10304 (2015)
D.A. Senthebane, A. Rowe, N.E. Thomford, H. Shipanga, D. Munro, M.A.M. Al Mazeedi, H.A.M. Almazyadi, K. Kallmeyer, C. Dandara, M.S. Pepper, M.I. Parker, K. Dzobo, Int. J. Mol. Sci. 18 (2017)
M. Shang, R.H. Soon, C.T. Lim, B.L. Khoo, J. Han, Lab Chip 19, 369 (2019)
S. Shuford, C. Wilhelm, M. Rayner, A. Elrod, M. Millard, C. Mattingly, A. Lotstein, A.M. Smith, Q.J. Guo, L. O’Donnell, J. Elder, L. Puls, S.J. Weroha, X. Hou, V. Zanfagnin, A. Nick, M.P. Stany, G.L. Maxwell, T. Conrads, A.K. Sood, D. Orr, L.M. Holmes, M. Gevaert, H.E. Crosswell, T.M. DesRochers, Sci. Rep. 9, 1 (2019)
G. Trujillo-de Santiago, B.G. Flores-Garza, J.A. Tavares-Negrete, I.M. Lara-Mayorga, I. González-Gamboa, Y.S. Zhang, A. Rojas-Martínez, R. Ortiz-López, M.M. Álvarez, Materials (Basel) 12 (2019)
K.P. Valente, S. Khetani, A.R. Kolahchi, A. Sanati-Nezhad, A. Suleman, M. Akbari, Drug Discov. Today 22, 1654 (2017)
M. Verheijen, Y. Schrooders, H. Gmuender, R. Nudischer, O. Clayton, J. Hynes, S. Niederer, H. Cordes, L. Kuepfer, J. Kleinjans, F. Caiment, Toxicol. Lett. 294, 184 (2018)
E.T. Verjans, J. Doijen, W. Luyten, B. Landuyt, L. Schoofs, J. Cell. Physiol. 233, 2993 (2018)
P.R. Wachsberger, J. Landry, C. Storck, K. Davis, M.D. O’Hara, C.S. Owen, D.B. Leeper, R.A. Coss, Int. J. Hyperth. 13, 251 (1997)
L.Y. Wu, D. Di Carlo, L.P. Lee, Biomed. Microdevices 10, 197 (2008)
Y. Yang, X. Yang, J. Zou, C. Jia, Y. Hu, H. Du, H. Wang, Lab a Chip - Miniaturisation Chem. Biol. 15, 735 (2015)
Y. Yang, A. Lü, W. Li, Z. Qian, AIP Adv. 9 (2019)
T. Yokochi, K.D. Robertson, Mol. Pharmacol. 66, 1415 (2004)
H. Zhang, L. Xiao, Q. Li, X. Qi, A. Zhou, Biomicrofluidics 12, 024119 (2018)
Acknowledgments
This work was supported by the National Natural Science Foundation of China (61875085, 81727804, 81827803). The authors thank Dr. Yangyang Liu for her technical assistance in manuscript revision.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 1293 kb)
Rights and permissions
About this article

Cite this article
Yang, Y., Liu, S., Chen, C. et al. Microfluidic-enabled self-organized tumor model for in vitro cytotoxicity assessment of doxorubicin. Biomed Microdevices 22, 70 (2020). https://doi.org/10.1007/s10544-020-00523-2
Accepted:
Published:
DOI: https://doi.org/10.1007/s10544-020-00523-2