
Abstract
Microfluidics-based production of stable microbubbles for ultrasound contrast enhancement or drug/gene delivery allows for precise control over microbubble diameter but at the cost of a low production rate. In situ microfluidic production of microbubbles directly in the vasculature may eliminate the necessity for high microbubble production rates, long stability, or small diameters. Towards this goal, we investigated whether microfluidic-produced microbubbles directly administered into a mouse tail vein could provide sufficient ultrasound contrast. Microbubbles composed of nitrogen gas and stabilized with 3 % bovine serum albumin and 10 % dextrose were injected for 10 seconds into wild type C57BL/6 mice, via a tail-vein catheter. Short-axis images of the right and left ventricle were acquired at 12.5 MHz and image intensity over time was analyzed. Microbubbles were produced on the order of 105 microbubbles/s and were observed in both the right and left ventricles. The median rise time, duration, and decay time within the right ventricle were 2.9, 21.3, and 14.3 s, respectively. All mice survived the procedure with no observable respiratory or heart rate distress despite microbubble diameters as large as 19 μm.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Shell-stabilized gas microbubbles are a versatile ultrasound contrast agent and drug delivery vehicle (Klibanov 2002; Ferrara et al. 2007; Hernot and Klibanov 2008; Phillips et al. 2011). Microbubbles are most often produced via high-shear mixing (e.g. sonication or agitation) of a gas phase with a liquid phase containing a shell stabilizer (Feinstein et al. 1984; Keller et al. 1987; Klibanov 2002; Stride and Edirisinghe 2009). This bulk mixing method is a stochastic process, resulting in the rapid production (up to 1010 / batch) of highly polydisperse microbubbles (diameter standard deviation up to 150 %) (Feshitan et al. 2009; Stride and Edirisinghe 2009). Recently, microbubble production via microfluidic devices has been investigated, as microfluidics enables precise control over microbubble diameter and production rate (Gañán-Calvo and Gordillo 2001; Gordillo et al. 2004; Garstecki et al. 2004). The three main microfluidic device designs are T-junctions (Xu et al. 2006; Garstecki et al. 2006; Pancholi et al. 2008), flow focusing microfluidic devices (FFMDs) (Garstecki et al. 2004; Talu et al. 2006; Hettiarachchi et al. 2007; Dhanaliwala et al. 2013), and co-flow microfluidic devices (Castro-Hernández et al. 2011). Microfluidic methods, however, exhibit production rates several orders of magnitude less than those achieved using agitation methods, especially when attempting to produce stable monodisperse microbubbles (Hettiarachchi et al. 2007; Stride and Edirisinghe 2009; Kaya et al. 2010; Shih et al. 2013).
Unlike bulk mixing methods, microfluidic devices have the potential to be miniaturized towards dimensions compatible with placement within a catheter (Dhanaliwala et al. 2013). Administration of stabilized microbubbles produced via microfluidics through a catheter placed directly within a vessel (i.e. in situ production) could provide several advantages. First, microbubble loss secondary to systemic injection may be reduced (Butler and Hills 1979; Lim et al. 2004; Talu et al. 2008). Second, in situ production could enable investigation of transiently-stable microbubbles since the microbubbles would already be near the target of interest and need only survive a few seconds post-production. Access to transiently-stable microbubbles would then allow the investigation of larger diameters, as microbubble dissolution could be relied upon to reduce the risk of emboli formation. Large diameter (10–20 μm) microbubbles are of interest because even when insonated off-resonance, they provide a large acoustic response. As a result, large diameter microbubbles can provide both enhanced contrast for imaging studies (Soetanto and Chan 2000; Gorce et al. 2000), as well as increased therapeutic effects (Choi et al. 2010; Tung et al. 2011).
Another advantage of microfluidics is the precise control of production rate and microbubble diameter through the interaction of the gas pressure, liquid flow rate, and device geometry (Gañán-Calvo 2004; Garstecki et al. 2005; Tan et al. 2006; Wang et al. 2013). The deterministic nature of microbubble production within microfluidic devices offers the possibility of real-time control of microbubble size and concentration and thus optimization of the contrast dose for the intended application.
As a step towards in situ microbubble production, we have previously demonstrated that microfluidics can produce a sufficient quantity of microbubbles to effect drug delivery under flow (i.e. cells are not in static contact with the microbubbles) (Dixon et al. 2013). In addition, we demonstrated microfluidic-produced microbubbles stabilized with albumin that have a half-life under 10 s, yet still produce a strong acoustic signal when imaged with a clinical ultrasound scanner (Chen et al. 2014). In this paper we report on the administration of microbubbles directly from the output of a microfluidic device, through a tail-vein catheter, and into the mouse vasculature along with the simultaneous in vivo ultrasound imaging of the injected microbubbles. We sought to investigate whether transiently-stable microbubbles produced by a flow-focusing microfluidic device and directly administered into the vasculature could provide sufficient acoustic contrast; and whether transient-stability would abrogate any acute health effects of large diameter microbubbles.
2 Materials and methods
2.1 Microfluidic production of microbubbles
Microbubbles were fabricated using a flow-focusing microfluidic device (FFMD) as described previously (Dhanaliwala et al. 2013). An expanding-nozzle flow-focusing design was selected as it is easier to fabricate smaller microbubbles with this design as compared to T-junction designs (Tan et al. 2006; Tumarkin and Kumacheva 2009; Tran et al. 2013). The FFMD was fabricated by casting poly-dimethyl-siloxane (PDMS) (Sylgard 184, Dow Corning, Midland, MI) onto a custom designed SU-8 (3025, Microchem, Newton, MA) mold (Fig. 1). The FFMD was designed with: support posts in the liquid and gas channels to prevent the PDMS from collapsing; a 25∘ flare expanding nozzle to increase the pressure difference across the nozzle orifice while minimizing flow disruption (Fig. 1a-3); and filters to catch debris between the inlets and the nozzle (Fig. 1a-4). The final dimensions of the device were: 35 μm gas channel width, 50 μm liquid channel width, 22 μm channel height, and 7 μm nozzle orifice width. Polytetrafluoroethylene (PTFE) tubing (Cole Parmer, Vernon Hills, IL) was used to convey the gas and liquid phase to their respective inlet. The liquid phase consisted of 3 % bovine serum albumin (BSA) and 10 % dextrose (Sigma-Aldrich, St. Louis, MO) in isotonic saline. The gas phase consisted of 99.998 % pure nitrogen (GTS Welco, Richmond, VA). To produce microbubbles, the pressure of the gas phase was set and the flow rate of the liquid phase adjusted until the microbubbles were of the desired size.

a Schematic of the flow-focusing microfluidic device. Maximum dimensions of the device were 6.6 mm × 7 mm. Black circles represent support posts. Width of the liquid channel (1), gas channel (2), and nozzle orifice (3) were 50, 35, and 7 μm, respectively. Filter channel widths (4) were 5 μm. b Single frame from a high speed video showing microbubble production. The gas cone in the center is sheared at the nozzle orifice by the liquid phase to produce microbubbles. Scale bar is 10 μm
A separate population of albumin-shelled fluorocarbon microbubbles were fabricated via sonication, as described previously (Chappell et al. 2008), and served as a control. A 1 % solution of serum albumin in isotonic saline was purged with octafluoropropane gas (C3 F 8, Scott Medical Products, Plumsteadville, PA). Octafluoropropane was then layered above the solution in a flask and the solution was sonicated for 30 s with an ultrasonic tip sonicator (XL2020, Misonix, Farmingdale, NY) equipped with an extended 1/2” titanium probe. Prepared microbubbles were stored in a sealed vial with a octafluoropropane headspace at 4 ∘C.
2.2 In vivo imaging
All animal imaging was in compliance with a University of Virginia Animal Care and Use Committee approved protocol. C57BL/6 mice were placed on an animal handling platform (VisualSonics, Toronto, ON) that enabled warming and monitoring of heart rate, respiration rate, and temperature. The mice were anesthetized with 2 % isofluorane and 98 % room air for the duration of the study. One end of an intravenous catheter was inserted into the tail vein of a mouse, while the other end was inserted directly into the outlet of the FFMD. A high frequency, small animal ultrasound scanner (Vevo 2100, VisualSonics, Toronto, ON) was used to image the microbubbles in the right and left ventricles via a short-axis view of the heart. The microbubbles were imaged at 12.5 MHz and 10 % transmit power, and both B-mode (1 pulse, 1 cycle, 15 dB gain) and nonlinear contrast mode (2 pulse, 4 cycle, amplitude modulation, 27 dB gain) images were acquired. The FFMD was affixed to a microscope to enable continuous monitoring of microbubble production rate and diameter using a high speed camera (SIMD24, Specialised Imaging, Temecula, CA) operating at a minimum of 1 × 106 frames per second (Dhanaliwala et al. 2013).
A total of six mice were each imaged three times yielding a total of 18 imaging sessions. For each imaging session, the outlet of the FFMD was directly connected to the tail vein catheter for 10 s, and a short-axis cine of the heart was acquired from just prior to microbubble injection until the image contrast in the ventricles returned to baseline. The first two imaging sessions were separated by an hour. The final imaging session was performed one week later.
For comparison, five imaging sessions were acquired using the agitation-produced microbubbles. Prior to use, diameter and concentration of the agitation-produced microbubbles were measured with a Coulter counter (Multisizer III, Beckman Coulter, Brea, CA). The microbubbles were then diluted in isotonic saline such that an equivalent number of microbubbles were injected over 10 s as were used in the microfluidic-microbubble imaging studies.
Beamformed, envelope-detected, 8-bit, B-mode images were exported from the ultrasound scanner and analyzed in MATLAB (Mathworks, Natick, MA). Regions of interest (ROIs) were placed in the right and left ventricle to calculate mean image intensity over the duration of the B-mode cine. Intensity curves were background subtracted using the mean of the first 0.33 s of the image prior to contrast injection, and normalized to the peak intensity in the right ventricle. Rise times were calculated as the time from 25 % to 75 % image intensity. Decay times were calculated as the time from 75 % to 25 % image intensity after peak intensity. Duration was calculated as the time between 50 % image intensity before and after peak intensity. Microbubble diameter and production rate were determined from high speed camera stills using ImageJ (NIH, Bethesda. MD).
3 Results
3.1 In vivo imaging
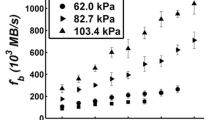
Microbubbles with diameters between 9.1 and 19.8 μm at production rates between 250,000 and 830,000 microbubble/s (Fig. 2) were administered to the mice. The median polydispersity index (standard deviation of the diameter / mean of the diameter) of the microbubbles within the microfluidic device was 7.9 %, and 85 % of the trials had a polydispersity index less than 10 %.

a Microfluidic-produced microbubble characteristics across all imaging procedures. Total injected gas is the product of microbubble volume and production rate. Filled box represents 25th – 75th percentile and circle with dot (⊙) is the median. b Image of microbubbles within the FFMD outlet channel just prior to entering the tail vein catheter
Following injection of the microfluidic-produced microbubbles, image intesnsity in the right ventricle, but not the left ventricle, was clearly enhanced (Fig. 3). In only 63 % of the trials was the image intensity in the left ventricle greater than 10 a.u. on the 8-bit scale, and on average the peak image intensity in the left ventricle was only 18 % of the peak image intensity of the right ventricle (Fig. 5).

(Top) Pre-contrast (Bottom) post-contrast images of the right (RV) and left (LV) ventricles following injection of microfluidic-produced microbubbles. B-Mode (Left) and B-Mode with nonlinear contrast overlay (Right). The injected microbubbles had a diameter of 14.4 ± 1 μm and were produced at a rate of 435,000 microbubble/s. Peak B-Mode image intensity in the right ventricle and left ventricle were 108 and 10 a.u., respectively. Red rectangles indicate region of interest (ROI) within which image intensity was extracted. Arrowhead indicates image dropout due to sternal artifact. Ultrasound parameters: 12.5 MHz center frequency, 10 % transmit power, 15 dB B-Mode gain, 27 dB contrast-mode gain, 35 dB dynamic range
For comparison, when 20 μL of 1.8 μm diameter agitation-produced microbubbles at a concentration of 250 × 106 microbubbles/mL were administered over 10 s into the mouse, contrast was clearly observed in both the right and left ventricle (Fig. 4). The median contrast level in the right ventricle with the agitation-produced microbubbles was only 70 % of the signal observed with the microfluidic-produced microbubbles. The median contrast level in the left ventricle was four-fold higher as compared to the microfluidic-produced microbubbles.

a Size distribution of agitation-produced microbubbles. Light and dark gray bars represent the median (1.8 μm) and mean (2.1 μm) diameter, respectively. Filled black box represents 25th – 75th percentile. Dash line represents cumulative percentage (90 % < 3.1 μm). b Pre-contrast (Top) post-contrast (Bottom) images of the right (RV) and left (LV) ventricles following injection of agitation-produced microbubbles. B-Mode (Left) and B-Mode with nonlinear contrast overlay (Right). Peak B-Mode image intensity in the right ventricle and left ventricle were 65, and 63 a.u., respectively. Red rectangles indicate region of interest (ROI) within which image intensity was extracted. Arrowhead indicates image dropout due to sternal artifact. Ultrasound parameters: 12.5 MHz center frequency, 10 % transmit power, 15 dB B-Mode gain, 27 dB contrast-mode gain, 35 dB dynamic range
Across all trials, the median rise time, duration, and decay time for the microfluidic-produced microbubbles were 2.9, 21.3, and 14.3 s, respectively. Across all trials, the median rise time, duration, and decay time for the agitation-produced microbubbles were 2.4, 35.8, and 16.5 s, respectively. Median peak intensity for microfluidic-produced and agitation-produced microbubbles in the right ventricle was 80 and 56 a.u., respectively (Fig. 5). The median duration of contrast with the agitation-produced microbubbles was 70 % longer than the microfluidic-produced microbubbles. No correlation was observed between peak intensity and microbubble diameter, production rate, or volume-production rate (Fig. 6). Similarly, no correlation was observed between rise time, duration, and decay time, and microbubble diameter or production rate, or volume-production rate.

(Left) Rise time, decay time, and duration of image contrast in the right ventricle across all imaging trials. (Right) Peak video intensity in the right (RV) and left (LV) ventricle. Agitation-produced microbubbles exhibited a longer duration and a higher LV intensity. Microfluidic-produced microbubbles were observed to have the overall highest peak intensity. Filled box represents 25th – 75th percentile and circle with dot(⊙) is the median

No significant correlation was observed between maximum image intensity in the right ventricle (RV) and either microbubble diameter, production rate or the product of microbubble volume and production rate (i.e. total gas). Lines represent a least squares linear regression with their respective R2 values
All animals survived all imaging procedures. During imaging, median heart rate across all trials was 460 beats per minute and median respiration rate across all trials was 160 breaths per minute. We have observed that for our mice under isofluorane anesthesia, the normal bounds for the heart rate and the respiratory rate are 400-500 beats per minute and 90-180 breaths per minute, respectively, at 37 ∘C. One week post-imaging, all animals were observed to be in good health.
4 Discussion
4.1 In vivo imaging
We present the first report of direct and immediate in-line intravascular administration of microbubbles produced from a microfluidic device. Several notable achievements were observed. First, no microbubble post-production steps were necessary. The microbubbles did not have to be washed or concentrated prior to injection to achieve an acoustic signal. The excess shell material did not appear to affect the animal, probably a result of the shell being composed of only biocompatible materials (i.e. albumin and dextrose). Furthermore, the animals did not appear to become sensitized to the shell material as no allergic response was observed during the final imaging session. Second, microbubbles greater than 10 μm in diameter were successfully injected without any gross adverse events to the animal. The safety profile of these larger microbubbles is probably related to the high solubility of the nitrogen gas used for the microbubble core. By quickly dissolving, the microbubbles have limited opportunities to coalesce, form gas emboli, and cause ischemic damage (Appendix B). Third, and most importantly, acoustic contrast was achieved despite production rates that are orders of magnitude lower than those achieved with agitation methods. Microfluidic production methods have long been plagued by low production rates when trying to produce stable microbubbles. These results suggest that by eliminating the need for stability, the production rate can be increased, and that it is not necessary to produce microbubbles at the same rate as agitation methods to achieve acoustic contrast.
Improved acoustic contrast can be achieved by increasing the microbubble diameter or by insonating a microbubble at its resonance frequency. Consequently, the choice between large diameter microbubbles versus smaller microbubbles optimized to resonate at the imaging frequency is not obvious. Analysis of spherically symmetric microbubble oscillations suggests that within the range of clinical ultrasound frequencies and microbubble diameters between 1 - 20 μm, the improvement in acoustic scattering from large diameter microbubbles outweighs the gains achieved when microbubbles are insonated at resonance (Appendix A). As such, although the microfluidic device optimally produces microbubbles with large diameters (i.e. highest production rate), it was selected intentionally to produce large diameter microbubbles, given the improvement in acoustic scattering that can be achieved with large diameter microbubbles.
While a strong acoustic signal was observed in the right ventricle, little to no signal was observed in the left ventricle when imaging with the microfluidic-produced microbubbles. This lack of contrast in the left ventricle is probably due to both the “sieve” effect in which the lung capillaries filter out large microbubbles (Meltzer et al. 1980), and increased dissolution rates as the microbubbles pass through the lungs (Wible et al. 1996; Kabalnov et al. 1998). As a result, few microbubbles survive the transit through the lung vasculature to provide contrast in the left ventricle. This behavior is not unexpected as these microbubbles were designed to be unstable by using highly soluble nitrogen gas and non-crosslinked albumin (Chen et al. 2014). Furthermore, passage through the lungs is not a requirement as the intended application of these microbubbles would be local production and administration from a catheter tip, thus eliminating the need for systemic circulation.
Unlike the microfluidic-produced microbubbles, a strong signal was immediately observed in the left ventricle when the agitation-produced microbubbles were administered. The low solubility gas and the smaller mean diameter of the agitation-produced microbubbles likely enabled the microbubbles to survive the passage through the lungs and multiple passes through the circulation. While the agitation-produced microbubbles exhibited a higher intensity in the left ventricle, the image intensity in the right ventricle was only 70 % of the image intensity achieved with the large diameter microfluidic-produced microbubbles.
In general, the contrast enhancement for both microfluidic-produced and agitation-produced microbubbles within the right ventricle followed a similar pattern, a sharp rise to peak intensity followed by a slower decrease back to baseline, with a median duration of double the injection time (Fig. 5). Two patterns for the image peak were also observed. The intensity curves either exhibited a single peak before decaying (Fig. 7) or a short plateau before signal decay. These results suggest that the microbubbles may have dispersed upon entering the bloodstream, causing a widening of the width of the intensity curve. The larger the dispersion of the microbubbles the wider the observed plateau.
Interestingly, no correlation was observed between microbubble diameter or production rate and image intensity, rise time, decay time, or duration (Fig. 6). Given the high rate of gas dissolution out of the microfluidic-produced microbubbles, the distribution of microbubbles measured just prior to injection may not be representative of the distribution of microbubbles within the right ventricle, potentially obscuring any correlation between image intensity and microbubble diameter. This uncertainty of microbubble diameter within the vasculature also makes it difficult to directly compare simulation results with experimental results.

Representative B-mode image intensity in the right (RV) and left (LV) ventricle over time. Curves were normalized to the peak image intensity in the right ventricle. Rise time, duration, and decay time were calculated for the right ventricle only. a 14.4 ± 1 μm diameter microfluidic-produced microbubbles produced at a rate of 435,000 microbubble/s. Peak image intensity in the right ventricle was 108 a.u. b 5 × 106 agitation-produced microbubbles with a median microbubble diameter of 1.8 μm diameter. Peak image intensity in the right ventricle was 65 a.u
4.2 Limitations
As stated above, in the current implementation, the microfluidic-microbubbles are directly infused into the vasculature without any prior washing steps. While this production mode may be acceptable for general contrast or drug delivery applications, administering excess shell material into the vasculature may inhibit the ability to perform molecular imaging. To produce molecularly targeted microbubbles, the shell material must contain targeting ligands. In this case, the excess shell material may saturate the targeted ligands, thus preventing the necessary microbubble binding. Redesigned microfluidic devices that can filter the liquid phase post-microbubble fabrication (Yamada and Seki 2005; Kobayashi et al. 2010) may enable molecularly targeted microbubbles to be produced in situ.
Although video intensity of the B-mode images may be the most straightforward, and potentially most clinically relevant, method for comparing microfluidic-produced and agitation-produced microbubbles, video intensity is affected by both the concentration of the agent and the scattering cross section. As a result, differences in video intensity may be due to either differences in concentration, differences in scattering cross section, or some combination of both. Even though we attempted to administer an equal quantity of both microfluidic-produced and agitation produced microbubbles (5 × 106) over an equal time span (10 s), the absolute concentration of each population within the right ventricle is impossible to know. Thus the increase in video intensity cannot be directly traced to the increased scattering cross section of the microfluidic-produced microbubbles.
Although no adverse events were observed, this may be due to the excellent ability of the lungs to filter gas from the bloodstream (Butler and Hills 1979). Consequently, additional studies will be required to determine whether transiently-stable microbubbles administered on the arterial side maintain the same safety profile as observed in the current study.
Validating the feasibility of imaging microbubbles directly injected from the output of a microfluidic device in vivo is an important first step towards the ultimate goal of microbubble production directly in the vasculature via a microfluidic device within a catheter. This paradigm would potentially allow improved control and optimization of microbubble distribution and concentration for both image enhancement and drug delivery. The ability to change production rate and microbubble diameter in real-time could, for example, enable small targeted microbubbles at low concentrations to be used for molecular imaging or to “paint” vasculature to be treated, while large microbubbles at high concentrations could be used to deliver a therapeutic. Furthermore, microbubble loss due to storage, administration, and circulation would be limited. Finally, by producing microbubbles near the site of interest, the microbubbles can be designed to be short-lived, thus enabling the utilization of large diameter microbubbles, and their concomitant advantages, with minimal risk of emboli formation.
5 Conclusion
Large diameter, transiently-stable, microfluidic-produced microbubbles were directly injected into the vasculature through a tail vein and successfully imaged with ultrasound. No acute harm was observed despite the large diameter of the microbubbles and co-administration of excess shell material with the microbubbles. Analysis of a one-dimensional model of microbubble oscillations suggests off-resonance insonation of large diameter microbubbles (> 10 μm) produces a larger acoustic response than insonation of small diameter microbubbles at resonance. In addition, analysis of a model of microbubble dissolution suggests lifetimes in vivo of less than 10 seconds, which was consistent with observed results. These observations provide additional support for a future paradigm whereby large transiently-stable microbubbles are fabricated in a catheter inside a vessel to provide enhanced image contrast.
Notes
Peak to peak pressure was calculated from the simulated echoes as the difference between the maximum and minimum pressure and normalized to the maximum value amongst all frequencies and diameters.
The energy of the simulated echo was calculated as the integral of the square of the absolute value of the signal. Each echo was normalized to the energy of their respective insonation pulse. All energy values were then normalized to the maximum energy amongst all diameters and frequencies.
References
D.R. Absolom, C.J. Van Oss, W. Zingg, A.W. Neumann, Determination of surface tensions of proteins II. surface tension of serum albumin, altered at the protein-air interface. Biochimica et Biophysica Acta (BBA) - Protein Structure. 670(1), 74–78 (1981). doi:10.1016/0005-2795(81)90050-7
B.D. Butler, B.A. Hills, The lung as a filter for microbubbles. J. Appl. Physiol. 47(3), 537–543 (1979)
E. Castro-Hernández, W. van Hoeve, D. Lohse, J.M. Gordillo, Microbubble generation in a co-flow device operated in a new regime. Lab. Chip. 11(12), 2023–2029 (2011). doi:10.1039/c0lc00731e
J.C. Chappell, J. Song, C.W. Burke, A.L. Klibanov, R.J. Price, Targeted delivery of nanoparticles bearing fibroblast growth factor-2 by ultrasonic microbubble destruction for therapeutic arteriogenesis. Small. 4(10), 1769–1777 (2008). doi:10.1002/smll.200800806
J.L. Chen, A.H. Dhanaliwala, A.J. Dixon, A.L. Klibanov, J.A. Hossack, Synthesis and characterization of transiently stable albumin-coated microbubbles via a flow-focusing microfluidic device. Ultrasound Med. Biol. 40(2), 400–409 (2014). doi:10.1016/j.ultrasmedbio.2013.09.024
J.J. Choi, J.A. Feshitan, B. Baseri, S. Wang, Y.S. Tung, M.A. Borden, E.E. Konofagou, Microbubble-size dependence of focused ultrasound-induced blood-brain barrier opening in mice in vivo. IEEE Trans. Biomed. Eng. 57(1), 145–154 (2010). doi:10.1109/TBME.2009.2034533
F. Conversano, R. Franchini, A. Lay-Ekuakille, S. Casciaro, In vitro evaluation and theoretical modeling of the dissolution behavior of a microbubble contrast agent for ultrasound imaging. IEEE Sensors J. 12(3), 496–503 (2012). doi:10.1109/JSEN.2011.2109707
N. De Jong, A. Bouakaz, P. Frinking, Basic acoustic properties of microbubbles. Echocardiography. 19(3), 229–240 (2002). doi:10.1046/j.1540-8175.2002.00229.x
A.H. Dhanaliwala, J.L. Chen, S. Wang, J.A. Hossack, Liquid flooded flow-focusing microfluidic device for in situ generation of monodisperse microbubbles. Microfluid Nanofluidics. 14(3–4), 457–467 (2013). doi:10.1007/s10404-012-1064-x
A.J. Dixon, A.H. Dhanaliwala, J.L. Chen, J.A. Hossack, Enhanced intracellular delivery of a model drug using microbubbles produced by a microfluidic device. Ultrasound Med. Biol. 39(7), 1267–1276 (2013). doi:10.1016/j.ultrasmedbio.2013.01.023
A. Doinikov, A. Bouakaz, Review of shell models for contrast agent microbubbles. IEEE Trans. Ultrason. Ferroelectr. Freq. Control. 58(5), 981–993 (2011). doi:10.1109/TUFFC.2011.1899
P.B. Duncan, D. Needham, Test of the EpsteinPlesset model for gas microparticle dissolution in aqueous media: effect of surface tension and gas undersaturation in solution. Langmuir. 20(7), 2567–2578 (2004). doi:10.1021/la034930i
P.S. Epstein, M.S. Plesset, On the stability of gas bubbles in liquid-gas solutions. J. Chem. Phys. 18(11), 1505 (1950). doi:10.1063/1.1747520
S.B. Feinstein, P.M. Shah, R.J. Bing, S. Meerbaum, E. Corday, B.L. Chang, G. Santillan, Y. Fujibayashi, Microbubble dynamics visualized in the intact capillary circulation. J. Am. Coll. Cardiol. 4(3), 595–600 (1984). doi:10.1016/S0735-1097(84)80107-2
K. Ferrara, R. Pollard, M. Borden, Ultrasound microbubble contrast agents: fundamentals and application to gene and drug delivery. Annu. Rev. Biomed. Eng. 9, 415–447 (2007). doi:10.1146/annurev.bioeng.8.061505.095852
R.T. Ferrell, D.M. Himmelblau, Diffusion coefficients of nitrogen and oxygen in water. J. Chem. Eng. Data. 12(1), 111–115 (1967). doi:10.1021/je60032a036
J.A. Feshitan, C.C. Chen, J.J. Kwan, M.A. Borden, Microbubble size isolation by differential centrifugation. J. Colloid Interface Sci. 329(2), 316–324 (2009). doi:10.1016/j.jcis.2008.09.066
A.M. Gañán-Calvo, Perfectly monodisperse microbubbling by capillary flow focusing: an alternate physical description and universal scaling. Phys. Rev. E Stat. Nonlin. Soft. Matter Phys. 69(2 Pt 2), 027,301 (2004)
A.M. Gañán-Calvo, J.M. Gordillo, Perfectly monodisperse microbubbling by capillary flow focusing. Phys. Rev. Lett. 87(27 Pt 1), 274,501 (2001)
D.F. Gaitan, L.A. Crum, C.C. Church, R.A. Roy, Sonoluminescence and bubble dynamics for a single, stable, cavitation bubble. J. Acoust. Soc. Am. 91(6), 3166–3183 (1992). doi:10.1121/1.402855
P. Garstecki, I. Gitlin, W. DiLuzio, G.M. Whitesides, E. Kumacheva, H.A. Stone, Formation of monodisperse bubbles in a microfluidic flow-focusing device. Appl. Phys. Lett. 85(13), 2649–2651 (2004). doi:10.1063/1.1796526
P. Garstecki, H.A. Stone, G.M. Whitesides, Mechanism for flow-rate controlled breakup in confined geometries: a route to monodisperse emulsions. Phys. Rev. Lett. 94(16), 164,501 (2005)
P. Garstecki, M.J. Fuerstman, H.A. Stone, G.M. Whitesides, Formation of droplets and bubbles in a microfluidic t-junctionscaling and mechanism of break-up. Lab Chip. 6(3), 437–446 (2006). doi:10.1039/B510841A
J.M. Gorce, M. Arditi, M. Schneider, Influence of bubble size distribution on the echogenicity of ultrasound contrast agents: a study of SonoVue. Invest. Radiol. 35(11), 661–671 (2000)
J.M. Gordillo, Z. Cheng, A.M. Gañán-Calvo, M. Marquez, D.A. Weitz, A new device for the generation of microbubbles. Phys. Fluids. 16(8), 2828 (2004). doi:10.1063/1.1737739
S. Hernot, A.L. Klibanov, Microbubbles in ultrasound-triggered drug and gene delivery. Adv. Drug. Deliv. Rev. 60(10), 1153–1166 (2008). doi:10.1016/j.addr.2008.03.005
K. Hettiarachchi, E. Talu, M.L. Longo, P.A. Dayton, A.P. Lee, On-chip generation of microbubbles as a practical technology for manufacturing contrast agents for ultrasonic imaging. Lab Chip. 7(4), 463–468 (2007). doi:10.1039/b701481n
A. Kabalnov, D. Klein, T. Pelura, E. Schutt, J. Weers, Dissolution of multicomponent microbubbles in the bloodstream: 1. theory. Ultrasound Med. Biol. 24(5), 739–749 (1998)
M. Kaya, S. Feingold, K. Hettiarachchi, A.P. Lee, P.A. Dayton, Acoustic responses of monodisperse lipid-encapsulated microbubble contrast agents produced by flow focusing. Bubble Sci. Eng. Technol. 2(2), 33–40 (2010)
M.W. Keller, S.B. Feinstein, D.D. Watson, Successful left ventricular opacification following peripheral venous injection of sonicated contrast agent: an experimental evaluation. Am. Heart J. 114(3), 570–575 (1987). doi:10.1016/0002-8703(87)90754-X
A. Klibanov, in Ultrasound contrast agents: Development of the field and current status, ed. by W. Krause. Contrast Agents II, Topics in Current Chemistry, vol. 222 (Springer, Berlin / Heidelberg, 2002), pp. 73–106
A. Kobayashi, M. Yamada, M. Seki. Bubble-based continuous separation system in microfluidic device. In: 14th International Conference on Miniaturized Systems for Chemistry and Life Sciences (Groningen, The Netherlands, 2010), pp. 1364–1366
T.G. Leighton. Cavitation inception and fluid dynamics. In: The Acoustic Bubble (Elsevier, 1994), pp. 100–106
A.K.P. Lim, N. Patel, R.J. Eckersley, S.D. Taylor-Robinson, D.O. Cosgrove, M.J.K. Blomley, Evidence for spleen-specific uptake of a microbubble contrast agent: A quantitative study in healthy volunteers. Radiology. 231(3), 785–788 (2004). doi:10.1148/radiol.2313030544
R.S. Meltzer, E. Glen Tickner, R.L. Popp, Why do the lungs clear ultrasonic contrast? Ultrasound Med. Biol. 6(3), 263–269 (1980). doi:10.1016/0301-5629(80)90022-8
L.D. Palma, M. Bertolotto, Introduction to ultrasound contrast agents: physics overview. Eur. Radiol. 9(3), S338–S342 (1999). doi:10.1007/PL00014069
K. Pancholi, U. Farook, R. Moaleji, E. Stride, M. Edirisinghe, Novel methods for preparing phospholipid coated microbubbles. Eur. Biophys. J. 37(4), 515–520 (2008). doi:10.1007/s00249-007-0211-x
L.C. Phillips, A.H. Dhanaliwala, A.L. Klibanov, J.A. Hossack, B.R. Wamhoff, Focused ultrasound-mediated drug delivery from microbubbles reduces drug dose necessary for therapeutic effect on neointima formation–brief report. Arterioscler. Thromb. Vasc. Biol. 31(12), 2853–2855 (2011). doi:10.1161/ATVBAHA.111.238170
K. Sarkar, A. Katiyar, P. Jain, Growth and dissolution of an encapsulated contrast microbubble: Effects of encapsulation permeability. Ultrasound Med. Biol. 35(8), 1385–1396 (2009). doi:10.1016/j.ultrasmedbio.2009.04.010
R. Shih, D. Bardin, T.D. Martz, P.S. Sheeran, P.A. Dayton, A.P. Lee, Flow-focusing regimes for accelerated production of monodisperse drug-loadable microbubbles toward clinical-scale applications. Lab Chip. 13(24), 4816–4826 (2013). doi:10.1039/C3LC51016F
K. Soetanto, M. Chan, Fundamental studies on contrast images from different-sized microbubbles: analytical and experimental studies. Ultrasound Med. Biol. 26(1), 81–91 (2000). doi:10.1016/S0301-5629(99)00107-6
E. Stride, M. Edirisinghe, Novel preparation techniques for controlling microbubble uniformity: a comparison. Med. Biol. Eng. Comput. 47(8), 883–892 (2009). doi:10.1007/s11517-009-0490-8
E. Talu, M.M. Lozano, R.L. Powell, P.A. Dayton, M.L. Longo, Long-term stability by lipid coating monodisperse microbubbles formed by a flow-focusing device. Langmuir. 22(23), 9487–9490 (2006). doi:10.1021/la062095+
E. Talu, R.L. Powell, M.L. Longo, P.A. Dayton, Needle size and injection rate impact microbubble contrast agent population. Ultrasound Med. Biol. 34(7), 1182–1185 (2008). doi:10.1016/j.ultrasmedbio.2007.12.018
Y.C. Tan, V. Cristini, A.P. Lee, Monodispersed microfluidic droplet generation by shear focusing microfluidic device. Sensors Actuators B Chem. 114(1), 350–356 (2006). doi:10.1016/j.snb.2005.06.008
T.M. Tran, F. Lan, C.S. Thompson, A.R. Abate, From tubes to drops: droplet-based microfluidics for ultrahigh-throughput biology. J. Phys. Appl. Phys. 114(11), 004 (2013). doi:10.1088/0022-3727/46/11/114004
E. Tumarkin, E. Kumacheva, Microfluidic generation of microgels from synthetic and natural polymers. Chem. Soc. Rev. 38(8), 2161 (2009). doi:10.1039/b809915b
Y.S. Tung, F. Vlachos, J.A. Feshitan, M.A. Borden, E.E. Konofagou, The mechanism of interaction between focused ultrasound and microbubbles in blood-brain barrier opening in mice. J. Acoust. Soc. Am. 130(5), 3059–3067 (2011). doi:10.1121/1.3646905
S. Wang, A.H. Dhanaliwala, J.L. Chen, J.A. Hossack, Production rate and diameter analysis of spherical monodisperse microbubbles from two-dimensional, expanding-nozzle flow-focusing microfluidic devices. Biomicrofluidics. 7(1), 014,103–014,103–12 (2013). doi:10.1063/1.4774069
J.H. Wible, J.K. Wojdyla, G.L. Bales, W.N. McMullen, E.A. Geiser, D.D. Buss, Inhaled gases affect the ultrasound contrast produced by albunex in anesthetized dogs. J. Am. Soc. Echocardiogr. 9(4), 442–451 (1996)
E. Wilhelm, R. Battino, R.J. Wilcock, Low-pressure solubility of gases in liquid water. Chem. Rev. 77(2), 219–262 (1977). doi:10.1021/cr60306a003
J.H. Xu, S.W. Li, Y.J. Wang, G.S. Luo, Controllable gas-liquid phase flow patterns and monodisperse microbubbles in a microfluidic t-junction device. Appl. Phys. Lett. 88(13), 133,506 (2006). doi:10.1063/1.2189570
M. Yamada, M. Seki, Hydrodynamic filtration for on-chip particle concentration and classification utilizing microfluidics. Lab Chip. 5(11), 1233–1239 (2005). doi:10.1039/B509386D
W. Yan, G.Y. Zhao, G.J. Chen, T.M. Guo, Interfacial tension of (methane + nitrogen) + water and (carbon dioxide + nitrogen) + water systems. J. Chem. Eng. Data. 46(6), 1544–1548 (2001). doi:10.1021/je0101505
Acknowledgements
This work is funded in part by National Institutes of Health grant NHLBI R01 HL090700 to ALK, JAH. The high speed camera was funded by a shared instrumentation grant S10 RR025594 to JAH. The high frequency small animal scanner was funded by a shared instrumentation grant S10 RR027333 to JAH. AJD is supported by a National Science Foundation Graduate Research Fellowship and AHD is supported by an American Heart Association Graduate Fellowship and the Virginia Space Grant Consortium. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH, NSF, VSGC, or AHA.
Author information
Authors and Affiliations
Corresponding author
Appendices
Appendix A: Microbubble oscillation model
Since the scattering cross section of microbubbles when insonated above their resonance frequency increases with the square of the diameter (Palma LD and Bertolotto M 1999), increasing microbubble size should increase acoustic contrast. This regime is relevant as the predicted resonance frequency of a 10 μm diameter microbubble is 0.6 MHz (De Jong et al. 2002), well below the imaging frequency in this study. However, insonating a microbubble at resonance is also known to increase its scattering cross section. To highlight the differences between insonating a large microbubble off-resonance compared to insonating a small microbubble at resonance, a 1-D spherically symmetric numerical model of microbubble oscillation (Keller Miksis-polytropic model (Gaitan et al. 1992)) was analyzed:
where R is the microbubble radius, \(\dot {R}\) is the microbubble wall velocity, \(\ddot {R}\) is the microbubble wall acceleration, ρ L is the density of surrounding liquid, c is the speed of sound in surrounding liquid, p d r i v e is the time delayed external driving acoustic pulse, p a m b is the ambient liquid pressure, and p i n t is the pressure inside the gas microbubble:
where σ is the gas-liquid surface tension, η L is the viscosity of the surrounding liquid, and κ is the gas polytropic exponent. While there are many 1-D numerical microbubble models (see Doinikov and Bouakaz (2011) for a complete review), a model of an unencapsulated gas microbubble was chosen as the transient stability of the microfluidic-microbubbles suggests a weak shell, which would have a minimal effect on oscillations. In addition, we sought only to analyze trends so a specific shell parameter was unnecessary.
Radius-time curves were solved using the ODE45 numerical solver and a tolerance of 1 × 10−5 in MATLAB (Mathworks, Natick, MA). Gaussian-ramped, 100 kPa, 4 cycle insonation pulses were used to insonate the microbubbles. Microbubbles with initial radii between 0.5 and 10 μm were simulated as this represents the range of microbubbles found in clinical microbubble formulations as well as those used in this study. Frequencies between 4 and 15 MHz were simulated as this covers the majority of clinical ultrasound frequencies. The properties of nitrogen were used for the internal gas and the properties of water were used for the liquid. The viscosity of water was doubled to account for thermal damping (Doinikov and Bouakaz 2011). Additional simulation parameters are listed in Table 1.
Finally, a relationship between pressure and velocity potential (Leighton 1994):
was used to convert the calculated radius-time curves to a predicted pressure wave, where P is pressure amplitude at a distance r from the microbubble.
Amongst all diameters, a resonance peak was only observed at an insonation frequency of 4 MHz, as the diameter of microbubbles in resonance at frequencies greater than 4 MHz were less than 1 μm (Fig. 8). Despite minimal radial excursions, large microbubbles produced increasingly higher acoustic pressures in the simulated echo. Analysis of peak to peak pressureFootnote 1 suggests that in certain specific cases, imaging of smaller microbubbles at resonance will produce greater contrast than the imaging of larger microbubbles – a 2.2 μm diameter microbubble imaged at 4 MHz results in an equivalent peak to peak pressure of a 13 μm diameter microbubble.

a Except when insonated at resonance, microbubbles with increasing diameters produced echos with increasing pressures. b The signal improvement due to resonance is abrogated when comparing energy versus pressure. c Normalized frequency spectra of microbubbles insonated with a 12.5 MHz pulse. The magnitude of the fundamental frequency decreased with decreasing microbubble diameter. For a 1 μm diameter microbubble harmonics were generated
When using energy as a metricFootnote 2, however, the effect of resonance is abrogated as compared to the improvements achieved with larger microbubbles. Energy of the simulated acoustic signal is perhaps a better representation of the relationship between microbubble size, insonation frequency, and acoustic contrast since it takes into account the effect of the microbubble on the entire insonation pulse. In addition, tissue attenuation and multiple-scattering effects from imaging a population of microbubbles will likely further reduce resonance enhancement.
Finally, an analysis of the frequency spectrum of the simulated echoes from microbubbles of various diameters when insonated at 12.5 MHz shows that the magnitude of the fundamental frequency decreases with microbubble diameter. The smallest microbubble had the lowest energy in the fundamental frequency; however, as its resonance frequency is near 12.5 MHz, it had the largest superharmonic magnitude. Of note, these simulation trends are in agreement with experimental trends obtained by Kaya et al. (2010).
Appendix B: Microbubble dissolution model
Given the difficulty in directly measuring microbubble lifetime in vivo, a model of microbubble dissolution was used to estimate microbubble lifetime. Kabalnov et al. (1998) modified the original Epstein-Plesset model of gas dissolution (Epstein and Plesset 1950) to incorporate the effect of blood pressure on microbubble lifetime:
where D is the diffusion coefficient of nitrogen gas in water, L is the Ostwald coefficient of nitrogen gas in water, and p b l o o d is the overpressure due to systemic blood pressure. In the model, the medium is considered fully gas saturated, the gas properties of nitrogen in blood are considered similar to the gas properties in water, and oxygen influx and non-steady state effects are neglected. Since the microbubbles have a non-crosslinked protein shell, the effect of the shell on gas diffusion (Sarkar et al. 2009; Conversano et al. 2012) is also assumed negligible. MATLAB was used to numerically solve the equation using the ODE45 solver with a tolerance of 1 × 10−8. Complete dissolution was set as the time when R=0. Given the assumptions and simplifications, the model only predicts dissolution times to one order of magnitude precision. Simulation parameters are listed in Table 2.
Surface tension is the major contributor to microbubble dissolution since small microbubbles produce large Laplace pressures. The surface tension of a pure nitrogen-water interface is 71.4 mN/m (Yan et al. 2001). The addition of albumin can reduce surface tension to 50 mN/m (Absolom et al. 1981), while the addition of lipid surfactants can reduce surface tension to as low as 25 mN/m (Duncan and Needham 2004). With a solid shell, such as lipid below its glass transition temperature, surface tension can be reduced to zero, resulting in microbubble lifetimes in excess of six hours (Duncan and Needham 2004). If the microfluidic-produced microbubbles are considered to be partially coated with albumin (50 <σ< 72 mN/m), the simulations predict lifetimes of approximately 10 seconds for microbubbles less than 20 μm when placed in water at 25 ∘C and 1 atm (Fig. 9a). These values may explain our unsuccessful attempts at measuring microbubble distribution using a Coulter counter as the measurement requires 30 seconds to acquire a measurement. Previous work using an acoustic metric of microbubble half-life also suggests lifetimes on the order of 10 seconds (Chen et al. 2014). It should be noted that although microbubbles with no surfactant are predicted to have lifetimes upward of 8 seconds, microfluidic-produced microbubbles produced without a surfactant coalesce immediately upon formation within the microfluidic device.

Simulated microbubble lifetime in (a) water at 25 ∘C and (b) blood at 40 ∘C. Isolines represent lifetime in seconds. Larger diameters and lower surface tensions increased lifetime. Simulated lifetime decreased in blood compared to water
When simulating microbubble lifetime in the blood, 20 μm diameter microbubbles were predicted to last 7 seconds at 40 ∘C and an overpressure of 4.3 kPa (Fig. 9b). These results are consistent within an order of magnitude of the lifetimes observed in vivo with the microfluidic-microbubbles, which lasted approximately 10 seconds after the end of the infusion. For comparison, a microbubble dissolution model for microbubbles composed of fluorocarbons (Kabalnov et al. 1998) predicts a lifetime in the bloodstream of 9.6 seconds for a 2 μm diameter microbubble composed of octafluoropropane and stabilized with albumin. For a similar microbubble with a 14 μm diameter, the predicted lifetime is 23 minutes. It should be noted that microbubbles composed purely of fluorocarbons are predicted to double in size before dissolving, whereas microbubbles composed purely of nitrogen are predicted to only shrink. Finally, while both the oscillation and dissolution models provide general trends, the both assume a single microbubble in an infinite medium. Microfluidic production and administration of microbubbles, however, results in high concentration and the single microbubble assumption is unlikely to be valid.
Rights and permissions
About this article

Cite this article
Dhanaliwala, A.H., Dixon, A.J., Lin, D. et al. In vivo imaging of microfluidic-produced microbubbles. Biomed Microdevices 17, 23 (2015). https://doi.org/10.1007/s10544-014-9914-9
Published:
DOI: https://doi.org/10.1007/s10544-014-9914-9