
Abstract
Despite lead widespread environmental pollution, its effect on humans and livestock's respiratory systems remains inadequately investigated. Similarly, furan is industrially relevant with enormous environmental presence. Lead and furan can be ingested -via lead pipes contaminated water and heat-treated food respectively. Thus, humans are inadvertently exposed continuously. Lead toxicity is well studied, and furan have earned a position on the IARC’s list of carcinogens. Here, we evaluate the effect of co-exposure to lead and furan on rat lungs. Thirty Wistar rats were grouped randomly into six cohorts (n = 6) consisting of a control group, furan alone group, lead acetate (PbAc) alone group and three other groups co-exposure to graded PbAc (1, 10 & 100 µg/L) alongside a constant furan (8 mg/kg) dose. After twenty-eight days, enzymatic and non-enzymatic antioxidant, oxidative stress and inflammatory biomarkers were biochemically evaluated. The ELISA-based technique was used to measure oxidative-DNA damage (8-OHG), tumour protein 53 (TP53) expressed and tumour necrotic factor-alpha (TNF-α) level. Dose-dependent increases (p < 0.05) in reactive oxygen and nitrogen species, malondialdehyde, nitric oxide, myeloperoxidase, TNF-α and TP53 level, with an associated decrease (p < 0.05) in enzymatic and non-enzymatic antioxidants were observed in the furan, PbAc and the co-treated rats relative to the control. In addition, PbAc and furan treatment impaired the histoarchitectural structures of rat lungs, exemplified by pro-inflammatory cell infiltration and trafficking into the bronchioles and alveolar spaces. Co-exposure to furan and PbAc may contribute to lung dysfunction via loss of redox balance, genomic damage/instability, inflammation and disrupted histoarchitectural features.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Humans and animals are constantly exposed to toxic chemicals due to a preponderant of industries, natural ores, explosives, gas flaring, combustibles, and other environmental anthropogenic activities (Pilsner et al. 2009; Sutton et al. 2012). Ecological toxicants, if left unmitigated, can build up in the body of animals and humans and cause severe local or systemic disorders (Wilks and Tsatsakis 2014). Furan is formed via thermally mediated reactions from different precursors, including carbohydrates, polyunsaturated fatty acids, amino acids, ascorbic acid and sugars (Bi et al. 2017). The presence of furan in food is a public health concern, and humans are exposed to furan primarily through heat-treated and canned foods and tobacco smoking (de Conti et al. 2017). Infants and young children risk furan toxicity following the detection of furan in infant formulas and cereals (Batool et al. 2021). The toxicity of furan is related to the activity of cytochrome P450 monooxygenase, especially CYP2E1. This enzyme mediates furan's metabolic activation to cis-but-2-ene-1,4-dialdehyde (BDA) (Varelis, Melton et al. 2018). BDA, a very reactive intermediate, is known to bind covalently to functional macromolecules, including proteins, lipids, and nucleic acids (Varelis, Melton et al. 2018). The CYP2E1-mediated ring-opening of furan triggers the formation of cis-2-but-ene-1,4-diol, a highly reactive metabolite that reacts with the exocyclic nitrogen of deoxyguanosine, deoxyadenosine and deoxycytidine to produce bicyclic derivatives (Batool et al. 2021). Experimental findings showed that furan increases cell proliferation and induces epigenetic changes, which result in mutations and carcinogenic changes (de Conti et al. 2016). In addition, sub-acute exposure to furan destabilised the delicate balance of antioxidants and pro-oxidants, thus mediating oxidative stress, inflammation and apoptosis (Owumi, Bello et al. 2021).
Lead is a heavy metal with important physicochemical properties which have ensured its continued use despite its toxicity (Wani et al. 2015). According to a recent report by the Agency for Toxic Substances and Disease Registry (ATSDR), lead is a frequently encountered hazardous heavy metal by both humans and livestock (ATSDR 2019), and the main route of ingestion of lead is via ingestion and inhalation (Andjelkovic, Buha Djordjevic et al. 2019a, b). Exposure to Pb and its compounds on humans and livestock occurs through various sources such as the combustion of leaded gasoline, industrial processes such as lead smelting and combustion, boat building, lead-based painting, lead-containing pipes, battery recycling, firearms and ammunition industry, dyes and pigments, and book printing (Wani et al. 2015). Lead (II) acetate Pb (CH3COO−)2 or sugar of lead is a derivative of lead used as an artificial sweetener. Nonetheless, the industrial application of lead has been heavily discouraged recently due to its apparent toxicities (Ibrahim et al. 2012). An avalanche of epidemiological and experimental studies correlates lead exposure to the onset of lung diseases, including pulmonary fibrosis, chronic obstructive pulmonary disease and asthma via oxidative stress mechanisms (Ibrahim et al. 2012; Han et al. 2019). Extensive research has shown that lead exerts its toxicity on almost all organs in the body, with the lungs highly predisposed to its toxicity (Wani et al. 2015). It exerts its noxious effects via modulation of the immune system by altering the production of cytokines and inflammatory agents (Zhang et al. 2020, Attafi, Bakheet et al. 2022), changing the redox balance (Balali-Mood, Naseri et al. 2021), and induction of pro-inflammatory mediators, including interleukin 1β (IL-1β) and tumour necrosis factor (TNF-α) (Boskabady et al. 2016). In another study, Ahamed et al., who studied the preventive effect of TiO2 nanoparticles on Pb-induced toxicity in human epithelial (A549) cells, observed that Pb alone decreased cell viability, damaged cell membrane, and perturbed antioxidant/pro-oxidant balance in A549 cells (Ahamed et al. 2019a, b).
Humans are exposed to mixtures of chemicals rather than individual chemicals. Therefore, it is essential to establish whether chemical mixtures produce a more pronounced effect than individual chemicals. In addition, people dwelling in Sub-Sahara Africa, where there is little or no attention to environmental health and food quality control assessments, are exposed to Pb from varied sources especially drinking water from public water supply systems. Further, they sometimes consume foods replete in furan due to thermal processing, particularly in homes where food hygiene is not prioritised. Therefore, the advertent drinking of Pb-contaminated water and consumption of furan-contaminated foods could induce long-term local or systemic toxicities in human or animal subjects. To this end, we hypothesised that co-exposure to Furan and PbAc might implicate possible synergy or antagonistic, additive, or novel effects that are not observed for a single exposure. To unravel this hypothesis, the present study was designed to investigate the impact of sub-acute co-exposure to furan and PbAc on the lungs of adult male Wistar Albino rats by explicitly evaluating the statuses of the redox, inflammatory, and genomic stability in the pulmonary system of rats as well as the histopathological changes of the lungs of experimental rats.
Materials and methods
Chemicals, reagents and kits
Furan, lead (II) acetate, thiobarbituric acid (TBA), 5′, 5′-dithiobis-2-nitrobenzoic acid (DTNB), 1-chloro-2,4-dinitrobenzene (CDNB), hydrogen peroxide (H2O2), epinephrine, and reduced glutathione (GSH) were purchased from Sigma Chemical Co. (St Louis, MO, USA). Enzyme-Linked Immunosorbent Assay (ELISA) kits for 8-hydroxyl-2′-deoxyguanosine (8-OHdG) (CAT No.: E-EL-0028), Tumour necrosis factor-α (TNF-α) (CAT No.: E-EL-R2856) and protein 53 (p53) (CAT No.: E-EL-0910) measurement were purchased from Elabscience Biotechnology Company (USA). All other reagents and chemicals used were obtained commercially and of analytical grade.
Animals, ethics, and experimental design
The study was designed in line with the 3R’s (replacement, reduction, and refinement) guidelines for the care and use of experimental animals (Abbassi et al. 2010; Gouveia and Hurst 2019) and the methodological approaches outlined as previously reported by Owumi et al., (Owumi, Otunla et al. 2021a, b). G* Power software version 3.1.9.4 was used for the sample size determination (Faul et al. 2007) to avoid type I and II errors. An effect size of 0.40 (Larger effect) at 0.05 alpha error of probability for one-way analysis of variance (ANOVA) (Cohen 1992) was utilised to obtain a sample size of 125 at 95% power. Out of 125, estimated experimental animals, 30 (consisting of n = five rats, i = 6) male Wistar rats with a mean weight of approximately 177 g body weight (b.w.) were bought at the Faculty of Veterinary Medicine, Experimental Animal Facility, the University of Ibadan, Nigeria. The animals were transported to the Animal House of the Department of Biochemistry, Faculty of Basic Medical Sciences, University of Ibadan, Nigeria, and maintained in a humane condition and a natural photoperiod of daily 12 h darkness/light cycle. Rats were fed with rat chows (Breedwell Feeds) and clean water ad libitum. The rats were acclimatised for seven days before the commencement of experimental treatments, consisting of six groups (n = 5 each). The different groups of rats were subjected to a 28-day of successive treatments. Stock solutions for dosing experimental rats, furan (8 mg/kg) and PbAc (0.1 mg/mL), were used daily. The doses of furan and PbAc utilised in the present study were established from previously available data (Owumi et al. 2020; Kataba et al. 2022). These studies showed that Pb and furan at the test doses triggered local and system toxicities even at a minimal concentration of 1.0 μg/L in the case of PbAc, alluding that there is no clear consensus for the permissible exposure limit (PEL) for Pb as many researchers have opined that even at a minimal dose below PEL of 10 μg/dL (Luo et al. 2019) or maximum permissible limit (MPL) of 12 μg/L as reviewed by (Kumar and Puri 2012)), Pb toxicity is inevitable. Briefly, furan stock solution (8 mg/kg) was prepared by dissolving 213 μL furan in corn oil to make a total volume of 20 mL and then administered to rats per os (po) according to their body weight (with an average volume of 0.6 mL). PbAc stock solution (0.1 mg/ml) was prepared by dissolving 50 mg PbAc in 50 mL of distilled water. From the stock of PbAc solution, 1.0, 10 and 100 μg/L were reconstituted by adding 0.02, 0.2 and 2 mL, respectively, of PbAc stock and were made up to 2 L each with distilled water. The volume of water was refilled daily to ensure a daily volume of 300 mL, and daily water intake was estimated after that. Rats in the group designated as the control received corn oil, Furan alone received 8 mg/kg b.w. of Furan p.o, PbAc alone received 100 μg/kg bw of PbAc p.o; Furan + PbAc1 received 8 mg/kg bw furan and 1 μg/kg b.w. PbAc, Furan + PbAc2 received 8 mg/kg b.w. furan and 10 μg/kg b.w. PbAc, and Furan + PbAc3 received 8 mg/kg b.w. furan and 100 μg/kg b.w. PbAc (Fig. 1).

Experimental protocol of Lead and Furan-induced pulmonary toxicity for 28 consecutive days. Furan alone: 8 mg/kg; PbAc alone: 100 µg/L; Furan + PbAc1: 8 mg/kg + 1 µg/L, Furan + PbAc2: 8 mg/kg + 10 µg/L, & Furan + PbAc3: 8 mg/kg + 100 µg/L. The scheme was modelled with Biorender.com by Arunsi U.O
Gravimetry
The body weight of all rats was recorded with a laboratory scale on day 1 of the experimental treatment and the day of sacrifice (Das et al. 2015). The difference in weight was calculated and reported as body weight gain.
Termination of the experiment and preparation of microsomal fraction
The experiment lasted for 28 days, and on day 29, 24 h after the last administration, the final weight of the rats was taken. The animals were sacrificed by cervical dislocation after carbon dioxide (CO2) asphyxiation (Avma 2001, Owumi, Bello et al. 2021). The rats were afterwards sacrificed, the lung was immediately removed, and the weight was measured using US Solid Digital Analytical Balance USS-DBS16 (Cleveland, OH, USA). The relative organ weight of the lungs was calculated according to the formula:
After the weight had been determined, the rat’s lungs were rinsed in 1.15% aqueous potassium chloride (4 °C) and processed for histological and biochemical assays. Using a glass-Teflon homogeniser, the samples used for biochemical assays were prepared by homogenising 2 g of the lung in 8 mL of homogenising buffer (0.1 M of phosphate buffer at pH 7.4). The homogenate was centrifuged for 15 min at 12,000 g in a cold centrifuge to obtain a clear supernatant (mitochondria fraction) which was collected and frozen in aliquots before biochemical analysis.
Evaluation of markers of oxidative stress, inflammation and genomic instability
The homogenate from the lungs of the control, PbAc, and furan-treated rats were subjected to biochemical analyses. The total protein concentration of the lungs was estimated according to the method of Lowry et al. (Lowry et al. 1951). Briefly, 7 μL, 23 μL and 150 μL of homogenate, distilled water and CuSO4 (aq), respectively, were added to a microplate and incubated at 25 °C for 10 min, after which 15 µL of Folin-C solution was added to the plate and further incubated for another 30 min. Absorbance was read against the reagent blank at 750 nm. Superoxide dismutase -SOD- activity was determined according to Misra and Fridovich’s method (Misra and Fridovich 1972). Succinctly, 50 μL of the sample was added to 2.5 mL of 0.05 M carbonate buffer (pH 10.2), and 0.3 ml of epinephrine in a cuvette, mixed by inversion and change in absorbance was monitored every 30 secs for 2 min at 480 nm. Catalase -CAT- activity was assayed according to Clairborne’s method (Clairborne 1995). Briefly, 2.95 mL phosphate buffered hydrogen peroxide was pipetted into a 1 cm quartz cuvette, after which 50 μL of tissue sample was added. The mixture was rapidly inverted to mix and placed in a spectrophotometer, and the change in absorbance was read at 240 nm every minute for 5 min. Glutathione S-transferase -GST- was assayed following Habig’s method as reported previously (monitored at 340 nm) (Habig et al. 1974; Owumi et al. 2022a, b, c). Accordingly, 10 μL of 1-chloro-2,4-dinitrobenzene (CDNB), 170 μL of the reaction mixture (20mls phosphate buffer, 0.5 ml GSH and 10.5mls distilled water) and 20 μL of the lung homogenate were mixed in a microplate and absorbance was monitored at 340 nm for 3 min every 30 s. To determine the activity of glutathione peroxidase -GPx-, 50 µL of phosphate buffer was kept in an Eppendorf tube, and to it was added 10 µl of sodium azide (NaN3), 20 µl of GSH, 10 µl of H2O2 and 50 µl of the sample (added last). The reaction mixture was incubated for 3 min at 37°C, after which 50 μL of trichloroacetic acid (TCA) was added, and the final mixture was centrifuged at 3000 g for 5 min. 50 μL of the supernatants, 100 μL of dipotassium hydrogen orthophosphate (K2HPO4.3H2O) and 50 μL of Ellman’s reagent were added, and the absorbance read against a reagent blank at 412 nm. This was done according to Rotruck et al. method (Rotruck et al. 1973). The total sulfhydryl group -TSH- was determined by the procedure described by Ellman (Ellman 1959). Briefly, 150 µL of the sample, 100 µL of phosphate buffer and 250 µL of distilled water were pipetted into an Eppendorf tube. The mixture was then allowed to stand, after which 15 µL of Ellman’s reagent was pipetted into a microplate along with 230 µL of the reaction mixture and allowed to stand for 2 min. The absorbance was then read at 412 nm. The level of GSH in lung homogenate was measured by adding equal volumes of sample and precipitating solution (80 µL), which was vortexed and centrifuged at 4000 g for 5 min. After that, 50 µl of the supernatant was added to 150 µL of Ellman’s reagent in a plate, and the absorbance of the reaction mixture was read at 412 nm according to the method described by Jollow et al. (Jollow et al. 1974). Also, Malondialdehyde -MDA- a lipid peroxidation biomarker, was quantified by the Okhawa method (Ohkawa 1979). An aliquot of 40 µL of the lungs was mixed with 160 µL of Tris-KCl buffer to which 50 µl of 30% TCA was added. Then 50 µL of 0.75% thiobarbituric acid (TBA) was added and incubated for 45 min at 80 °C. The mixture was cooled to 25 °C and centrifuged at 3000 g for 10 min. Finally, 200 µL of the clear supernatant was collected, and absorbance was measured against a reference blank of distilled water at 532 nm using a microplate reader. In addition, the level of reactive oxygen and nitrogen species were also quantified by preparing a reaction mixture consisting of 10 µL of the sample, 150 µL of 0.1 M potassium phosphate buffer (pH 7.4), 35 µL of distilled water and 5 µL of 2′, 7′-dichlorodihydrofluorescin diacetate (DCFH-DA). This was followed by an analysis of the fluorescence emission of DCF for 10 min at 488 nm excitation and 525 nm emission wavelengths. The biomarkers of inflammation, including nitric oxide -NO- were estimated by the protocols of Green et al., as previously reported (Green, Wagner et al. 1982, Owumi, Najophe et al. 2022). Briefly, equal volumes of sample and Griess reagent were incubated at 25 °C for 20 min, after which the optical density (OD) at 550 nm was determined spectrophotometrically. The MPO activity was assayed by adding 200 µL of O-dianisidine mixture and 50 µL of dilute H2O2 to 7 µL of tissue homogenate. Absorbance was read for 30 s intervals for 4 min at 460 nm following the described method of Granell et al. Field (Granell, Gironella et al. 2003). Xanthine oxidase was quantified by the method of Bergmeyer et al. (Buege and Aust 1978). Succinctly, 150 µL of phosphate buffer, 80 µL of xanthine solution and 8 µL of the sample were pipetted into a microplate. The mixture was mixed, and absorbance was taken every minute for 3 min at 290 nm. In addition, the levels of tumour necrosis factor-alpha -TNF-α-, 8-hydroxydeoxyguanosine (8-OHdG), and p53 were estimated using ELISA kits (Owumi et al. 2020; Owumi et al. 2022a, b, c). 100 µL of the standard working solution was added to seven wells of the microplate, and 100 µL of lung samples were added to the other wells. The plate was covered with the sealer provided in the kit and kept in a water bath for 90 min at 37 °C. After the incubation period, the liquid was removed from each well without washing, and 100 μL of biotinylated detection Ab working solution was immediately added. The plate was then covered with a plate sealer, gently mixed up and once again incubated for 1 h at 37 °C. Afterwards, the solution from each well was decanted, 350 μL of wash buffer was added, allowed to soak for 1–2 min, decanted from the wells, and patted dry against the clean absorbent paper. This wash step was repeated three times in total, after which 100 μL of horseradish peroxidase (HRP) conjugate working solution was added to each well and then incubated for 30 min at 37 °C. The solution was decanted from each well, and the wash process was repeated five times. After the wash process, 90 μL of substrate reagent was added to each well in the dark, covered with a plate sealer, and incubated for 15 min at 37 °C. Finally, the reaction was stopped by adding 50 μL of stop solution to each well, and the OD value was measured at once at 450 nm. The intensity of the colour developed is directly proportional to the concentration of TNF-α, 8-OHdG and p53 in the samples.
Lung histopathological assessment
Fixed in phosphate-buffered formalin (10%), the lungs were subjected to histopathological examination using haematoxylin and eosin (H&E) stains. Each section was examined under an optical microscope (Carl Zeiss Axio light microscope (Jena, Germany). Representative images were captured upon examination using a Zeiss Axiocam 512 camera (Jena, Germany) attached to the microscope by a pathologist with limited knowledge of the treatment protocol.
Statistical analysis
Paired student t-tests were performed to determine the significance level of rats' mean body weight before and after treatment. Also, a one-way analysis of variance (ANOVA) followed by a post-hoc test (Tukey) was used to determine the level of significance different across the different cohorts of rats using GraphPad Prism version 8.3.0 for Mac (www.graphpad.com; GraphPad, CA, USA,). The results are expressed as the mean ± SD of replicates, and statistically significant differences were set at p < 0.05.
Results
Co-exposure to furan and lead acetate alters rats’ mean weight change and organosomatic indices.
The result combined effect of PbAc and furan on weight change, and organosomatic indices of adult male rats are presented in Table 1. The results showed that furan and PbAc decreased the mean weight change of rats, and this effect was more significant (p < 0.05) in cohorts of rats treated with 10 and 100 μg/kg of PbAc at a constant amount of furan (8 mg/kg). Our result further revealed that treatment of rats with furan and PbAc slightly increased the lung weight and relative lung weight, indicating organomegaly. This result divulged that exposure to PbAc and furan altered the organosomatic indices of rats.
Co-exposure to furan and lead acetate alters redox balance in the pulmonary system of rats.
The effect of Furan and PbAc on antioxidant and oxidative biomarkers in the lungs of rats was investigated in this study, and the results are presented in Figs. 2–4. It was observed that co-exposure to PbAc and Furan altered the redox buffering system in the pulmonary system of the rats. Individual exposure to PbAc and Furan in experimental rats significantly waned (p < 0.05) the activities of SOD, CAT, and GPx. However, as the dosage of PbAc was raised at a constant amount of furan, the activities of these antioxidant enzymes decreased drastically (Fig. 2). Similarly, co-administration of furan and PbAc significantly (p < 0.05) downturned the activity of GST and levels of thiol groups, including GSH and TSH synergistically. The observed changes were substantial in cohorts of rats treated with 1.0, 10 and 100 μg/L of PbAc at a constant amount of furan (8 mg/kg) (Fig. 3). Co-treatment with PbAc and furan significantly increased the levels of MDA -a marker of lipid peroxidation (LPO) and RONS– a marker of oxidative and nitrosative stress in rats. As the doses of PbAc were increased (1.0, 10 and 100 μg/L) at a constant amount of furan (8 mg/kg), the concentrations of LPO and RONS were markedly raised (Fig. 4). The result indicates synergy in the alteration of the redox buffering capacity along the alveolar-arterial axis of rats exposed to PbAc and furan.

Effect of co-exposure of Furan and PbAc on SOD, CAT, and GPx activities in rat’s lungs for 28 days. Furan alone: 8 mg/kg; PbAc alone: 100 µg/L; Furan + PbAc1: 8 mg/kg + 1 µg/L, Furan + PbAc2: 8 mg/kg + 10 µg/L, & Furan + PbAc3: 8 mg/kg + 100 µg/L. Data analysis was performed by one-way analysis of variance (ANOVA) followed by a post-hoc test (Tukey). Data were expressed as mean ± SD of 5 rats, and significantly different values are indicated by *, **, *** or **** denoting p values at 0.05, 0.01, 0.001 and 0.0001, respectively. PbAc: lead acetate; SOD: superoxide dismutase; CAT: catalase; GPx: Glutathione peroxidase

Effect of co-exposure of Furan and PbAc on GST, GSH and TSH levels in rat’s lungs for 28 days. Furan alone: 8 mg/kg; PbAc alone: 100 µg/L; Furan + PbAc1: 8 mg/kg + 1 µg/L, Furan + PbAc2: 8 mg/kg + 10 µg/L, & Furan + PbAc3: 8 mg/kg + 100 µg/L. Data analysis was performed by one-way analysis of variance (ANOVA) followed by a post-hoc test (Tukey). Data were expressed as mean ± SD of 5 rats, and significantly different values are indicated by *, **, *** or **** denoting p values at 0.05, 0.01, 0.001 and 0.0001, respectively. PbAc: lead acetate; GST: Glutathione-S-transferase; GSH: reduced glutathione; TSH: total sulfhydryl groups

Effect of co-exposure of Furan and PbAc on LPO and RONS levels in rat’s lungs for 28 days. Furan alone: 8 mg/kg; PbAc alone: 100 µg/L; Furan + PbAc1: 8 mg/kg + 1 µg/L, Furan + PbAc2: 8 mg/kg + 10 µg/L, & Furan + PbAc3: 8 mg/kg + 100 µg/L. Data analysis was performed by one-way analysis of variance (ANOVA) followed by a post-hoc test (Tukey). Data were expressed as mean ± SD of 5 rats, and significantly different values are indicated by *, **, *** or **** denoting p values at 0.05, 0.01, 0.001 and 0.0001, respectively. PbAc: lead acetate; LPO: lipid peroxidation; RONS: reactive oxygen and nitrogen species
Co-exposure to furan and lead acetate triggers inflammation and genomic instability in the pulmonary system of rats.
Co-exposure to furan and PbAc can trigger inflammation and genomic instability in rats. To this end, we investigated the effect of the co-treatment of furan and PbAc on the pulmonary system of rats, and our results are presented in Figs. 5,6. The results revealed that cohorts of rats co-treated with PbAc and Furan increased (p < 0.05) pro-inflammatory mediator levels, including MPO, XO, and NO. The pro-inflammatory mediator levels were raised as the doses of PbAc increased at a constant amount of furan (Fig. 5). The results further divulged that co-administration of furan and PbAc significantly (p < 0.05) increased the concentrations of 8-OHdG, p53, and TNF-α. The orchestration of sub-acute inflammation, oxidative DNA damage and genomic instability manifoldly increased in cohorts of rats treated with 1.0, 10 and 100 μg/L of PbAc at a constant amount of furan (8 mg/kg) (Fig. 6). These results divulged that exposure to PbAc and furan abrogated genomic stability and induces inflammations in the pulmonary system of rats.

Effect of co-exposure of Furan and PbAc on MPO, XO, and NO levels in rat’s lungs for 28 days. Furan alone: 8 mg/kg; PbAc alone: 100 µg/L; Furan + PbAc1: 8 mg/kg + 1 µg/L, Furan + PbAc2: 8 mg/kg + 10 µg/L, & Furan + PbAc3: 8 mg/kg + 100 µg/L. Data analysis was performed by one-way analysis of variance (ANOVA) followed by a post-hoc test (Tukey). Data were expressed as mean ± SD of 5 rats, and significantly different values are indicated by *, **, *** or **** denoting p values at 0.05, 0.01, 0.001 and 0.0001, respectively. PbAc: lead acetate; XO: xanthine oxidase; MPO: myeloperoxidase; NO: nitric oxide;

Effect of co-exposure of Furan and PbAc on 8-OHdG, p53, and TNF-α levels in rat’s lungs for 28 days. Furan alone: 8 mg/kg; PbAc alone: 100 µg/L; Furan + PbAc1: 8 mg/kg + 1 µg/L, Furan + PbAc2: 8 mg/kg + 10 µg/L, & Furan + PbAc3: 8 mg/kg + 100 µg/L. Data analysis was performed by one-way analysis of variance (ANOVA) followed by a post-hoc test (Bonferroni). Data were expressed as mean ± SD of 5 rats, and significantly different values are indicated by *, **, *** or **** denoting p values at 0.05, 0.01, 0.001 and 0.0001, respectively. PbAc: lead acetate; 8-OHdG: 8-hydroxy-2′-deoxyguanosine; TNF-α: tumour necrotic factor-alpha
Co-exposure to furan and lead acetate alters the typical histoarchitectural features in the lungs of rats.
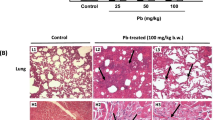
The effect of co-exposure to furan and PbAc on the histological features of rat lungs was also investigated. The result is presented in Fig. 7. Unlike the control, with normal alveolar epithelium, bronchioles, and the intra alveolar spaces mildly infiltrated by inflammatory cells, cohorts of rats treated with Furan alone and PbAc alone, revealed bronchioles with mild lymphocytes follicle, intra alveolar spaces infiltrated by inflammatory cells. However, as the doses of PbAc were raised at a constant amount of furan, the lungs of rats presented bronchioles with a gradual increase in lymphocytic follicular accumulation and alveolar spaces infiltrated by inflammatory cells (Fig. 8).

Photomicrograph of the sections of experimental rat lung stained with haematoxylin and eosin (H and E), at magnifications of × 100 top row; and × 400 bottom row. Control: the alveolar epithelium appears normal (double-headed arrow at × 100). The bronchiole and the intra-alveolar spaces are mildly infiltrated by inflammatory cells (black arrow at × 400). Furan alone: treated rat lung bronchiole shows mild lymphocyte follicle (black arrow). The intra-alveolar spaces infiltrated by inflammatory cells and the alveolar epithelium appear normal (red arrow). PbAc alone: rats show bronchiole with lymphocyte follicle (black arrow), and the alveolar epithelium appears normal (red arrow). Rats treated with Furan and graded doses of PbAc1—3 presented bronchiole with a gradual increase in lymphocytic follicular accumulation (thick black arrows) with areas of the lymphoid follicle. The intra-alveolar spaces are mildly infiltrated by inflammatory cells, and the alveolar epithelium appears normal (red arrow). (Color figure online)

Proposed mechanisms of inadvertent co-exposure of an experimental rat model to PbAc and furan in diet and water, respectively. Furan is converted to cis-but-ene-1,4-dialdehyde, which activates xanthine oxidase and myeloperoxidase to generate abundant ROS, including O2.−, H2O2, and HOCl, while Pb may trigger the Fenton and Haber–Weiss reaction to generate abundant OH−. These intermediate activations of iNOS mediate the generation of NO and other pro-inflammatory cytokines, such as TNF-α. Unresolved oxidative stress and inflammation will trigger oxidative DNA damage, which signals the activation of p53 to induce apoptosis in the pulmonary system of rats. The scheme was modelled with Biorender.com by Arunsi U.O
Discussion
Furan is a compound labelled as a potential human carcinogen (Batool et al. 2021). It is commonly produced in heat-processed foods such as breakfast cereals widely consumed by children, coffee, baby foods, meats or foods produced via heat treatment techniques such as jarring and canning Fields(FAO 2018). Both children and adults are at risk of furan toxicity. Pb exerts toxicity on all organs investigated; the lungs, made up of diverse cells, are mainly at risk of lead exposure and accumulation (Attafi, Bakheet et al. 2022). The current study showed additive effects in the co-exposed groups as the concentration of the toxicants increased. The key findings of the present work are that Furan and Pb, after oral administration for twenty-eight days individually, caused oxidative stress and induced inflammation. The treatment also resulted in DNA damage in the lungs of the exposed animals, while co-exposure to Pb (1, 10 & 100 μg/L) exacerbated the toxic effects of Furan.
Since experimental studies with rats have shown that exposure to toxicants may cause an alteration of body weight and organ-to-body weight ratios (Andjelkovic, Buha Djordjevic et al. 2019a, b), Furan and Pb’s effect on the mean weight change and lung weight was evaluated. Accordingly, a decrease in mean weight change and an increase in lung weight and relative lung weight is indicative of loss of appetite and proliferation of pro-inflammatory cells in the lungs of the animals, as observed in the study. This harmful effect of PbAc on the mean weight change and lung weight was elevated parallel with the increase of lead acetate doses, and the severe impact was found in the rats ingested and treated with 10 & 100 μg/L of lead acetate. The obtained result agrees with the study by Ibrahim et al., where they observed a similar trend in the body weight and organ weight of animals treated with Pb (Ibrahim et al. 2012). These results in body weight gain, which the toxic ions may cause, could be associated with several factors, one of which is the alteration in nutrient metabolism. Zinc is a potent activator of several enzymes involved in protein metabolisms, such as delta-aminolaevulinic acid dehydratase, carboxypeptidase A and alkaline phosphatase. Zinc has shown that lead acetate ingestion impairs zinc status in zinc-dependent enzymes necessary for many metabolic processes (McCall et al. 2000, Sherry M, Charlotte E et al. 2005, Ibrahim et al. 2012).
The activity of antioxidant enzymes can be viewed as a sensitive indicator of a cell's response to oxidative stress (Kurutas 2016; Ahamed et al. 2021). Changes in intracellular antioxidant enzyme activities, specifically lung SOD, CAT, GPx and GST activities, were assayed to learn more about the impacts of furan and Pb in rats. Crosschecked against the control group, the activities of these enzymatic antioxidants and thiol group levels decreased on exposure to Furan and Pb. The most significant decrease was observed in the rat cohort administered with Furan (8 mg/kg) and PbAc (100 μg/L), while the level of RONS and LPO were significantly (p < 0.05) increased. Decreased activities of SOD, CAT, GPx, GST, GSH and TSH, along with a concomitant increase in the levels of RONS & LPO in lead acetate and furan-treated rats, suggest an interaction between the accumulated free radicals and the active amino acids of these enzymes. Free radicals have been shown to inhibit the functional activity of enzymes by interacting with their structures (Das et al. 2015; Ahamed et al. 2019a, b). A variety of methods could cause Pb-induced oxidative damage. Pb interacts with negatively charged phospholipids in membranes, which may enhance the spread of lipid oxidation by inducing changes in membrane biophysical characteristics. Pb causes lateral phase separation, which impacts membrane-related functions such as membrane enzyme activity, endo- and exocytosis, solute transport across the bilayer, and signal transduction (Khalaf et al. 2012). Although the literature lacks documentation on the effects of furan on the lungs, there is ample evidence of its ability to elicit oxidative stress by decreasing the activity of endogenous antioxidant enzymes in organs, including the liver, kidney and reproductive tissues of rats (Owumi et al. 2020; Owumi et al. 2022a, b, c). According to a review of recent literature (Kaya et al. 2019; Yuan et al. 2021), furan caused oxidative stress in mice, which was associated with increased MDA levels and resulted in alterations in antioxidant enzyme activity.
Similarly, we discovered that the levels of MDA were raised in the groups given furan at 8 mg/kg body weight per day, indicating lipid peroxidation induction. The GSH and antioxidant enzyme activity levels were reduced in rats exposed to furan, confirming these findings. Apart from assessing the levels of MDA and GSH, we also checked the activity of CAT, GPx, GST, and SOD to see if furan administration may induce oxidative stress. The observed reductions in GSH levels and GST activity are most likely owing to increased GSH demand. Another mechanism for the observed decrease in GSH content in the present study could be the binding of the divalent metals of lead with –SH groups. The GSH depletion and elevated LPO & RONS levels, together with enhanced XO activity in the present study, indicate failure of the antioxidant defence mechanism, which resulted in the excess formation of free radicals, thus potentiating oxidative stress in the lungs of rats. The lung evolved as an organ capable of effectively promoting gas exchange throughout the body, but as a result, the lung is quite vulnerable to its surroundings. Both enzymatic and non-enzymatic reactions can convert oxygen into ROS and reactive nitrogen species (RNS), damaging DNA, proteins, and lipids. Antioxidants present intracellularly, and in the fluid surrounding, the lung's epithelial cells work to reduce ROS/RNS concentrations under normal circumstances. Oxidative stress occurs when the lung's antioxidant capacity is exceeded by accidental exposure to hazardous chemicals such as lead and furan.
A by-product of oxidative DNA damage is 8-hydroxy-2’-deoxyguanosine (8-OHdG), continually created in living cells. It is one of the most popular indicators for assessing oxidative stress (Lıu et al. 2019). In previous research, higher 8-OHdG levels were found in patients with diabetes, cancer, cardiovascular illnesses, and stable COPD (Kondo et al. 2000; Valavanidis et al. 2005, Tabur, Aksoy Ş et al. 2015). Previous studies have demonstrated that an imbalance between oxidants and antioxidants stimulates the inflammatory process and harms lung function (Lıu et al. 2019; Yang et al. 2022). The increased quantity of 8-OHdG adducts found in the current study after co-exposure to Furan and graded doses of PbAc (1, 10, 100 µg/L) is associated with lung function severity, suggesting that elevated oxidative stress hastens the loss of lung function. Asthma, chronic obstructive pulmonary disease (COPD), and other conditions directly associated with oxidative stress are developed due to these exposures and the following pulmonary reactions (Rogers and Cismowski 2018).
Pulmonary responses to oxidative stress include activation of oxidases, lipid peroxidation, increases in nitric oxide, and autophagy (Rogers and Cismowski 2018). Lung inflammation is known to be induced by environmental contaminants through stimulating inflammatory mediators such as cytokines and interleukins. In addition, the importance of toxicant-induced inflammation in lung damage is becoming increasingly evident (Attafi, Bakheet et al. 2022). Secondary injury from lead exposure may result via cell activation and a cascade of events involving numerous types of inflammatory and cytotoxic mediators (Das et al. 2015). When Attafi et al. exposed Wistar rats to varying doses of Pb for three days, they reported an increase in gene expression of inducible nitric oxide synthase (iNOS) and TNF-α, an observation associated with inflammation (Attafi, Bakheet et al. 2022). This result is further corroborated by Villa et al. They reported that induction of TNF-α and iNOS increases the risk of developing lung fibrosis and cancer via an inflammation-mediated pathway (Villa et al. 2017). In this study, the significant increase in NO and TNF-α levels and MPO activity in the lungs of animals co-exposed to Furan and alternate doses of PbAc is indicative of increased inflammation. In response to toxic substances, inflammatory cells, macrophages, epithelial cells and neutrophils of the lungs secrete chemokines, which promote local inflammation in the lungs. If unresolved, this inflammation could become chronic and damage the lung tissues (Moldoveanu et al. 2009).
Prolonged exposure to toxicants destroys cellular material, disrupts cellular genetics, and causes oxidative stress (Das et al. 2015; Owumi et al. 2022a, b, c). It is generally known that the p53 protein may control cell growth and induce apoptosis. When the p53 gene is mutated, it loses the ability to cause cell death, which causes unrestricted cell growth and promotes carcinogenesis (Huang et al. 2014), the level of p53 expression is thus used in both diagnostic and prognostic conditions. Akram et al. reported that lead exposure induced DNA damage (Akram et al. 2019), and El-Shetry suggested that the induction could be through activation of the p53 pathway (El-Shetry et al. 2021). Elevated p53 expression levels correlate with tumour progression (Huang et al. 2014). In this study, exposure of animals to furan or Pb led to increased expression of p53 protein, which was worsened by the co-exposure to these toxicants. The resultant elevation in p53 protein after co-exposure to these substances suggests changes in DNA structure and the inhibition of damaged DNA repair, which may lead to aberrant gene expression, thus potentiating the onset of cancer.
Interestingly, as the redox balance and the stability of the genome are lost, the pulmonary system of rats is subjected to inflammation, leading to the infiltration and trafficking of pro-inflammatory cells into the lungs. These cells interact with dangerous signals such as damaged DNA, proteins, and lipids, triggering the expressions of several gene signatures known to orchestrate pro-inflammation and programmed cell death (Li et al. 2022; Owumi et al. 2022a, b, c). The results showed that PbAc and furan co-treatment resulted in the infiltration and trafficking of pro-inflammatory cells into rats' bronchioles and alveolar spaces. Taken together, exposure to PbAc and furan synergistically abrogated the pulmonary redox balance, inflammation, genomic stability of rats, and histoarchitectural features of the lungs of rats.
Conclusions
This study aims to validate the possible impacts of co-exposure to dietary furan and drinking water containing PbAc in male Wistar Albino rats after 28 days using analytical, ELISA and histological measures. Our findings revealed that co-exposure to PbAc and furan abrogated pulmonary redox balance as evidenced by elevation in ROS levels (LPO and RONS) with a concurrent reduction in antioxidant systems (SOD, CAT, GPx, GST, GSH, TSH), triggered inflammatory responses by increasing the levels of MPO, XO, NO, and TNF-a, induced genomic instability by altering pulmonary levels of 8-OHdG and p53, and deranged the histological features of the pulmonary system of rats as depicted in our proposed mechanism of toxicity in Fig. 8. These findings draw attention to the possible dangers to public health posed by sub-acute or concurrent exposure to furan and PbAc. To further understand the potential toxicological mechanism of co-exposure to Furan and PbAc in rats, further studies on cytochrome P450 activity and carcinogenicity of furan and PbAc in the lungs are warranted.
Data availability
The dataset generated during the current study is available from the corresponding author upon reasonable request.
References
Abbassi R, Chamkhia N, Sakly M (2010) Chloroform-induced oxidative stress in rat liver: implication of metallothionein. Toxicol Ind Health 26(8):487–496
Ahamed M, Akhtar MJ, Alhadlaq HA (2019a) Preventive effect of TiO2 nanoparticles on heavy metal Pb-induced toxicity in human lung epithelial (A549) cells. Toxicol in Vitro 57:18–27
Ahamed M, Akhtar MJ, Khan MAM, Alrokayan SA, Alhadlaq HA (2019b) Oxidative stress mediated cytotoxicity and apoptosis response of bismuth oxide (Bi2O3) nanoparticles in human breast cancer (MCF-7) cells. Chemosphere 216:823–831
Ahamed M, Akhtar MJ, Khan MAM, Alhadlaq HA (2021) SnO2-Doped ZnO/Reduced graphene oxide nanocomposites: synthesis, characterization, and improved anticancer activity via oxidative stress pathway. Int J Nanomedicine 16:89–104
Akram Z, Riaz S, Kayani MA, Jahan S, Ahmad MW, Ullah MA, Wazir H, Mahjabeen I (2019) Lead induces DNA damage and alteration of ALAD and antioxidant genes mRNA expression in construction site workers. Arch Environ Occup Health 74(4):171–178
Andjelkovic M, Buha Djordjevic A, Antonijevic E, Antonijevic B, Stanic M, Kotur-Stevuljevic J, Spasojevic-Kalimanovska V, Jovanovic M, Boricic N, Wallace D, Bulat Z (2019) Toxic effect of acute cadmium and lead exposure in rat blood, liver, and kidney. Int J Environ Res Public Health 16(2):274
Andjelkovic M, Buha Djordjevic A, Antonijevic E, Antonijevic B, Stanic M, Kotur-Stevuljevic J, Spasojevic-Kalimanovska V, Jovanovic M, Boricic N, Wallace D, Bulat Z (2019b) Toxic effect of acute cadmium and lead exposure in rat blood, liver, and kidney. Int J Environ Res Public Health 16(2):274
ATSDR. (2019). “ATSDR (2019) The ATSDR 2019 Substance Priority List. Public Health Service; Agency for Toxic Substances and Disease Registry. .” Retrieved 3–05–2022, 2022, from https://www.atsdr.cdc.gov/spl/index.html.
Attafi IM, Bakheet SA, Ahmad SF, Belali OM, Alanazi FE, Aljarboa SA, Al-Alallah IA, Korashy HM (2022) Lead nitrate induces inflammation and apoptosis in rat lungs through the activation of NF-kappaB and AhR signaling pathways. Environ Sci Pollut Res Int. https://doi.org/10.1007/s11356-022-19980-8
Avma P, o. E. A. V. M. A. (2001) 2000 Report of the AVMA Panel on Euthanasia. J Am Vet Med Assoc 218(5):669–696
Balali-Mood M, Naseri K, Tahergorabi Z, Khazdair MR, Sadeghi M (2021) Toxic mechanisms of five heavy metals: mercury, lead, chromium, cadmium, and arsenic. Front Pharmacol. https://doi.org/10.3389/fphar.2021.643972
Batool Z, Xu D, Zhang X, Li X, Li Y, Chen Z, Li B, Li L (2021) A review on furan: Formation, analysis, occurrence, carcinogenicity, genotoxicity and reduction methods. Crit Rev Food Sci Nutr 61(3):395–406
Bi KH, Zhang L, Qiao XG, Xu ZX (2017) Tea polyphenols as inhibitors of furan formed in the maillard model system and canned coffee model. J Food Sci 82(5):1271–1277
Boskabady MH, Tabatabai SA, Farkhondeh T (2016) Inhaled lead affects lung pathology and inflammation in sensitized and control guinea pigs. Environ Toxicol 31(4):452–460
Buege JA, Aust SD (1978) Microsomal lipid peroxidation. Methods Enzymol 52:302–310
Clairborne A (1995) Catalase activity FL. CRC Press, Boca Raton
Cohen J (1992) A power primer. Psychol Bull 112(1):155–159
Das KK, Jargar JG, Saha S, Yendigeri SM, Singh SB (2015) α-tocopherol supplementation prevents lead acetate and hypoxia-induced hepatic dysfunction. Indian J Pharmacol 47(3):285–291
de Conti A, Tryndyak V, Doerge DR, Beland FA, Pogribny IP (2016) Irreversible down-regulation of miR-375 in the livers of Fischer 344 rats after chronic furan exposure. Food Chem Toxicol 98(Pt A):2–10
de Conti A, Beland FA, Pogribny IP (2017) The role of epigenomic alterations in furan-induced hepatobiliary pathologies. Food Chem Toxicol 109:677–682
Ellman GL (1959) Tissue sulfhydryl groups. Arch Biochem Biophys 82(1):70–77
El-Shetry ES, Mohamed AA-R, Khater SI, Metwally MMM, Nassan MA, Shalaby S, El-Mandrawy SAM, Bin Emran T, Abdel-Ghany HM (2021) Synergistically enhanced apoptotic and oxidative DNA damaging pathways in the rat brain with lead and/or aluminum metals toxicity: expression pattern of genes OGG1 and P53. J Trace Elem Med Biol 68:126860
FAO. (2018, 01/25/2018). "Questions and Answers on the Occurrence of Furan in Food." Chemical Contaminants in food. Retrieved 16–05–2022, 2022, from https://www.fda.gov/food/chemical-contaminants-food/questions-and-answers-occurrence-furan-food.
Faul F, Erdfelder E, Lang AG, Buchner A (2007) G*power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods 39(2):175–191
Gouveia K, Hurst JL (2019) Improving the practicality of using non-aversive handling methods to reduce background stress and anxiety in laboratory mice. Sci Rep 9(1):20305
Habig WH, Pabst MJ, Jakoby WB (1974) Glutathione S-transferases. the first enzymatic step in mercapturic acid formation. J Biol Chem 249(22):7130–7139
Han B, Li S, Lv Y, Yang D, Li J, Yang Q, Wu P, Lv Z, Zhang Z (2019) Dietary melatonin attenuates chromium-induced lung injury via activating the Sirt1/Pgc-1alpha/Nrf2 pathway. Food Funct 10(9):5555–5565
Huang K, Chen L, Zhang J, Wu Z, Lan L, Wang L, Lu B, Liu Y (2014) Elevated p53 expression levels correlate with tumor progression and poor prognosis in patients exhibiting esophageal squamous cell carcinoma. Oncol Lett 8(4):1441–1446
Ibrahim NM, Eweis EA, El-Beltagi HS, Abdel-Mobdy YE (2012) Effect of lead acetate toxicity on experimental male albino rat. Asian Pac J Trop Biomed 2(1):41–46
Jollow DJ, Mitchell JR, Zampaglione N, Gillette JR (1974) Bromobenzene-induced liver necrosis. Protective role of glutathione and evidence for 3,4-bromobenzene oxide as the hepatotoxic metabolite. Pharmacology 11(3):151–169
Kataba A, Botha TL, Nakayama SMM, Yohannes YB, Ikenaka Y, Wepener V, Ishizuka M (2022) Environmentally relevant lead (Pb) water concentration induce toxicity in zebrafish (Danio rerio) larvae. Comp Biochem Physiol C Toxicol Pharmacol 252:109215
Kaya E, Yilmaz S, Ceribasi S (2019) Protective role of propolis on low and high dose furan-induced hepatotoxicity and oxidative stress in rats. J Vet Res 63(3):423–431
Khalaf A, Moselhy WA, Abdel-Hamed MI (2012) The protective effect of green tea extract on lead induced oxidative and DNA damage on rat brain. Neurotoxicology 33(3):280–289
Kondo S, Toyokuni S, Tanaka T, Hiai H, Onodera H, Kasai H, Imamura M (2000) Overexpression of the hOGG1 gene and high 8-hydroxy-2’-deoxyguanosine (8-OHdG) lyase activity in human colorectal carcinoma: regulation mechanism of the 8-OHdG level in DNA. Clin Cancer Res 6(4):1394–1400
Kumar M, Puri A (2012) A review of permissible limits of drinking water. Indian J Occup Environ Med 16(1):40–44
Kurutas EB (2016) The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: current state. Nutr J 15(1):71–71
Li S, Wu P, Han B, Yang Q, Wang X, Li J, Deng N, Han B, Liao Y, Liu Y, Zhang Z (2022) Deltamethrin induces apoptosis in cerebrum neurons of quail via promoting endoplasmic reticulum stress and mitochondrial dysfunction. Environ Toxicol 37(8):2033–2043
Lıu X, Deng K, Chen S, Zhang Y, Yao J, Weng X, Zhang Y, Gao T, Feng G (2019) 8-Hydroxy-2’-deoxyguanosine as a biomarker of oxidative stress in acute exacerbation of chronic obstructive pulmonary disease. Turk J Med Sci 49(1):93–100
Lowry O, Rosebrough N, Farr AL, Randall R (1951) Protein measurement with the folin phenol reagent. J Biol Chem 193(1):265–275
Luo T, Shen M, Zhou J, Wang X, Xia J, Fu Z, Jin Y (2019) Chronic exposure to low doses of Pb induces hepatotoxicity at the physiological, biochemical, and transcriptomic levels of mice. Environ Toxicol 34(4):521–529
McCall KA, Huang C-C, Fierke CA (2000) Function and mechanism of zinc metalloenzymes. J Nutr 130(5):1437S-1446S
Misra HP, Fridovich I (1972) The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J Biol Chem 247(10):3170–3175
Moldoveanu B, Otmishi P, Jani P, Walker J, Sarmiento X, Guardiola J, Saad M, Yu J (2009) Inflammatory mechanisms in the lung. J Inflamm Res 2:1–11
Ohkawa HO, N., Yagi, K. (1979) Assay for lipid peroxidation in animal tissues by thiobarbituric acid reaction. Anal Biochem 95:351–358
Owumi SE, Adedara IA, Farombi EO, Oyelere AK (2020) Protocatechuic acid modulates reproductive dysfunction linked to furan exposure in rats. Toxicology 442:152556
Owumi SE, Bello SA, Idowu TB, Arunsi UO, Oyelere AK (2021) Protocatechuic acid protects against hepatorenal toxicities in rats exposed to Furan. Drug Chem Toxicol. https://doi.org/10.1080/01480545.2021.1890109
Owumi SE, Otunla MT, Arunsi UO, Najophe ES (2021b) 3-Indolepropionic acid upturned male reproductive function by reducing oxido-inflammatory responses and apoptosis along the hypothalamic-pituitary-gonadal axis of adult rats exposed to chlorpyrifos. Toxicology 463:152996
Owumi SE, Arunsi UO, Oyewumi OM, Altayyar A (2022a) Accidental lead in contaminated pipe-borne water and dietary furan intake perturbs rats’ hepatorenal function altering oxidative, inflammatory, and apoptotic balance. BMC Pharmacol Toxicol 23(1):76
Owumi SE, Bello SA, Idowu TB, Arunsi UO, Oyelere AK (2022b) Protocatechuic acid protects against hepatorenal toxicities in rats exposed to Furan. Drug Chem Toxicol 45(4):1840–1850
Owumi SE, Kazeem AI, Wu B, Ishokare LO, Arunsi UO, Oyelere AK (2022c) Apigeninidin-rich Sorghum bicolor (L. Moench) extracts suppress A549 cells proliferation and ameliorate toxicity of aflatoxin B1-mediated liver and kidney derangement in rats. Sci Rep 12(1):7438
Pilsner JR, Hu H, Ettinger A, Sánchez BN, Wright RO, Cantonwine D, Lazarus A, Lamadrid-Figueroa H, Mercado-García A, Téllez-Rojo MM, Hernández-Avila M (2009) Influence of prenatal lead exposure on genomic methylation of cord blood DNA. Environ Health Perspect 117(9):1466–1471
Rogers LK, Cismowski MJ (2018) Oxidative stress in the lung—the essential paradox. Curr Opin Toxicol 7:37–43
Rotruck JT, Pope AL, Ganther HE, Swanson AB, Hafeman DG, Hoekstra WG (1973) Selenium: biochemical role as a component of glutathione peroxidase. Science 179(4073):588–590
Sherry ML, Charlotte HE, Duane UE (2005) CHAPTER 13 - Nutrition and Nutritional Diseases. In: Sonia W-C (ed) The Laboratory Primate. Academic Press, London, pp 181–208
Sutton P, Woodruff TJ, Perron J, Stotland N, Conry JA, Miller MD, Giudice LC (2012) Toxic environmental chemicals: the role of reproductive health professionals in preventing harmful exposures. Am J Obstet Gynecol 207(3):164–173
Tabur S, Aksoy ŞN, Korkmaz H, Ozkaya M, Aksoy N, Akarsu E (2015) Investigation of the role of 8-OHdG and oxidative stress in papillary thyroid carcinoma. Tumour Biol 36(4):2667–2674
Valavanidis A, Vlahoyianni T, Fiotakis K (2005) Comparative study of the formation of oxidative damage marker 8-hydroxy-2’-deoxyguanosine (8-OHdG) adduct from the nucleoside 2’-deoxyguanosine by transition metals and suspensions of particulate matter in relation to metal content and redox reactivity. Free Radic Res 39(10):1071–1081
Varelis P, Melton L, Shahidi F (2018) Encyclopedia of Food Chemistry. Elsevier, Netherlands
Villa M, Gialitakis M, Tolaini M, Ahlfors H, Henderson CJ, Wolf CR, Brink R, Stockinger B (2017) Aryl hydrocarbon receptor is required for optimal B-cell proliferation. EMBO J 36(1):116–128
Wani AL, Ara A, Usmani JA (2015) Lead toxicity: a review. Interdiscip Toxicol 8(2):55–64
Wilks MF, Tsatsakis AM (2014) Environmental contaminants and target organ toxicities—new insights into old problems. Toxicol Lett 230(2):81–84
Yang X, Fang Y, Hou J, Wang X, Li J, Li S, Zheng X, Liu Y, Zhang Z (2022) The heart as a target for deltamethrin toxicity: Inhibition of Nrf2/HO-1 pathway induces oxidative stress and results in inflammation and apoptosis. Chemosphere 300:134479
Yuan Y, Wang Z, Nan B, Yang C, Wang M, Ye H, Xi C, Zhang Y, Yan H (2021) Salidroside alleviates liver inflammation in furan-induced mice by regulating oxidative stress and endoplasmic reticulum stress. Toxicology 461:152905
Zhang Z, Guo C, Jiang H, Han B, Wang X, Li S, Lv Y, Lv Z, Zhu Y (2020) Inflammation response after the cessation of chronic arsenic exposure and post-treatment of natural astaxanthin in liver: potential role of cytokine-mediated cell-cell interactions. Food Funct 11(10):9252–9262
Acknowledgements
Not applicable.
Funding
This research was done without a specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study’s conception and design. Conceptualisation: SO. Project administration, investigation, data curation, analysis: MO and UA. Supervision and Visualization: SO. Validation: UA. Writing, review, and editing: SO, MO and UA.
Corresponding author
Ethics declarations
Competing interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the University of Ibadan Animal Care and Use Research Ethics Committee (ACUREC), with approval number UI-ACUREC/032–0525/27.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Owumi, S.E., Otunla, M.T. & Arunsi, U.O. A biochemical and histology experimental approach to investigate the adverse effect of chronic lead acetate and dietary furan on rat lungs. Biometals 36, 201–216 (2023). https://doi.org/10.1007/s10534-022-00472-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10534-022-00472-8