
Abstract
The influence of epipsammic biofilm developed on riverbed sediment on the sorption, uptake, mobility and transformation of AsV was studied. Native biofilm was incubated on sediment samples at microcosm level. Once the biofilm had developed, 500 µg L−1 AsV was spiked in two systems designated BAS and BASP, without P and with equimolar As:P concentration ratio, respectively, and compared with identical control (sterilized) systems (CAS and CASP). The evolution and speciation of arsenic (As) concentrations in the overlying water were followed during two additional weeks. The biofilm enhanced removal of AsV from the water up to 91 % of its initial concentration, while only ~70 % removal was attained in CAS. Presence of equimolar P concentration enhanced the amount of As removal up to 97 % in BASP, but had no effect in CASP. In the systems with biofilm, As was mostly (~97 %) in AsV form, whilst AsIII only accounted for ~1 % of total aqueous As. The organic species, monomethylarsonic acid (MMAV) and dimethylarsinic acid (DMAV), represented 0.6 and 0.7 % of total As, respectively. In contrast, in the systems devoid of biofilm, AsIII accounted for up to 39 % of aqueous As, whereas methylated aqueous species were negligible. The distribution of As in the biofilm showed that ~71 % of the retained As was extracellular, most (>99.5 %) in the form of AsV. Volatile As forms were only detected in the systems incorporating biofilm. It is concluded that biofilm covering sediments increases As retention, inhibits reduction of AsV to AsIII and methylates inorganic As, thus playing a key role in the biogeochemistry of As in river environments.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Arsenic (As) is a highly toxic metalloid which is widespread in the environment and causes severe and numerous health problems worldwide. Presence of As in soils, sediments and water is attributed to natural sources, such as weathering of minerals with high As content, and to human activities (use of arsenical fertilizers and pesticides, as well as industrial and mining activities) (Smedley and Kinniburgh 2002). The mobility and toxicity of As strongly depend on its chemical form (Oremland and Stolz 2003). AsIII, which is the predominant form in reduced environments, is more mobile and is considered more toxic than AsV, which is the predominant form in oxic environments. In turn, inorganic As (iAs) is generally recognized as more toxic than the organic forms (Sharma and Sohn 2009), with the exception of the methylated AsIII species (Petrick et al. 2000; Styblo et al. 2000).
In aquatic environments, As may undergo transformations in its chemical form as a consequence of changes in environmental physico-chemical conditions, and interaction with mineral surfaces (oxidation/reduction, surface complexation), but also through biologically mediated reactions (bio-transformation), which can strongly affect its fate, mobility, bioavailability and toxicity. Arsenic speciation and bioavailability in water and sediments are strongly influenced by biological activities (Oremland and Stolz 2005). Studying the effect of algae on As speciation, Hellweger and Lall (2004) proposed a model in which algae absorb AsV (mistaking it for phosphate). Then, inside the cell, an AsV detoxification mechanism takes place, which consists in reducing AsV to AsIII by methylating it to monomethylarsonic acid (MMAV), then MMAV to dimethylarsinic acid (DMAV). Finally, As is excreted as AsIII and/or DMAV, depending on the algal growth rate and the phosphate conditions (Hellweger and Lall 2004). Besides MMAV and DMAV, the products of methylation include trimethylarsine oxide (TMAO) (Duker et al. 2005) and the final product of the methylation pathway, the volatile trimethylarsine (TMAsIII) (Yin et al. 2011a).
At water–sediment interfaces, biofilms consisting of microorganisms (bacteria, fungi, cyanobacteria, algae and protozoa) embedded in extracellular polymeric substances (EPS) mainly composed of polysaccharides are commonly found (Costerton 2007). Consequently, biofilms are the first component to interact with dissolved substances such as nutrients, organic matter, metals and metalloids, as well as other toxicants in aqueous systems (Sabater et al. 2007).
It has been demonstrated that biofilms play a key role in retention of metals and metalloids from overlying water (Nelson et al. 1996, 1999; Friese et al. 1997; Headley et al. 1998; Decho 2000; Dong et al. 2000, 2007; Haack and Warren 2003; Morris et al. 2005; Serra et al. 2009; Beck et al. 2011; Drahota et al. 2014). Pollutants are removed by the biofilm through a variety of mechanisms, including (bio-)sorption, precipitation as sulphides or phosphates and microbial reductive precipitation (van Hullebusch et al. 2003). Biosorption consists of several mechanisms, including ion exchange, chelation, adsorption and diffusion through cell walls and membranes (van Hullebusch et al. 2003).
The interaction between As and biofilms in aquatic systems can be studied from two points of view: As behaviour or its effects on biofilms. Firstly, As biogeochemistry may be modified by the presence of biofilms. The potential for As enrichment in biofilms has been reported by Drewniak et al. (2008) with concentrations of up to 60 mg kg−1 in rock biofilm. Yang et al. (2011) demonstrated that multi-species biofilms, inoculated from a source receiving coal mining effluent, could both sequester and detoxify Se and As. Drahota et al. (2014), studying As adsorption from natural As sources onto natural surface coatings growing on glass slides, confirmed that dissolved As had been sorbed and retained by the biofilm. Tuulaikhuu et al. (2015), who investigated the fate and toxicity of As on periphytic and epipsammic biofilms using a simplified fluvial system including fish, biofilms and sediment, reported that periphytic biofilms also accumulated As, although it was predominantly retained by sediment. Regarding studies into the retention of As by epipsammic biofilms, Prieto et al. (2013) observed that epipsammic biofilms increased AsV sorption with respect to biofilm-devoid river sediments, with a more noticeable effect in the presence of phosphate. Secondly, presence of As in aquatic systems could affect periphyton communities. Thus, arsenate is responsible for inhibiting algal growth and photosynthetic capacity, as well as for decreasing total biofilm biomass, changing community composition (selecting tolerant species, reducing species richness and making biofilms more heterotrophic) and reducing diatom cell sizes and the ability of the community to use phosphorus (Blanck and Wangberg 1988, 1991; Wangberg et al. 1991; Rodríguez Castro et al. 2015; Tuulaikhuu et al. 2015; Barral-Fraga et al. 2016).
Despite this evidence, literature on (bio-)sorption, speciation and detoxification of As by epipsammic biofilms is scarce. Hence, the novelty of the present work is its contribution to understanding the role played by epipsammic biofilms in As biogeochemistry. The main objective is to study the influence of biofilms developed on riverbed sediments on the (bio-)adsorption and/or (bio-)uptake, mobility, (bio-)transformation and detoxification of AsV at microcosm scale using specifically designed systems to control As concentration and speciation over time. The kinetics of the removal process is also evaluated, along with changes in As aqueous speciation, As volatilization, the distribution of As species within biofilms and remobilization of previously retained As. The effect of phosphate as a potential competitor for As sorption and bio-uptake and as a nutrient for biofilm growth is also explored.
Materials and methods
Sediment characterization
Sediment was sampled from Anllóns River (NW Spain), where As contamination is a problem in some sections and where gold mining activities carried out during the Roman Empire, and in the early 20th century, brought about remobilization of associated arsenic and its accumulation in sediments (Devesa-Rey et al. 2008, 2011; Costas et al. 2011). It has been shown that As mobilization in Anllóns riverbed sediments is enhanced under conditions of high salinity, extreme pH or high P concentration, as well as during high-flow resuspension events (Rubinos et al. 2010, 2011).
For this study, sediment was sampled in an uncontaminated area upstream of the Au–As mineralized area known as Ponte de Eguas (43°13′24.46″N 8°45′44.61″W), which is located 8 km downstream from the Town of Carballo (population over 25,000). A complex sample was collected using a small plastic shovel from the top 5 cm at various points at this site and taken to the laboratory in hermetic plastic containers filled with river water to prevent oxidation. Eh and pH in the sediment were measured in situ using a portable device (Hanna Instruments, HI 9025 microcomputer). Eh values obtained with Pt–Ag/AgCl electrodes were corrected to refer them to the standard hydrogen electrode (SHE) by adding 245 mV.
Once in the laboratory, solid sediment samples were freeze-dried and sieved (<2 mm) for characterization. Only some organic debris was eliminated by sieving, so the <2 mm fraction used for the experiment practically represented bulk sediment. The grain size distribution was determined using wet sieving and the pipette method as described by Guitián and Carballas (1976). Total phosphorus (PT) was determined by means of acid digestion (HF:H2SO4:HCl 10:1:10) followed by colorimetric determination (Murphy and Riley 1962). Total organic carbon, nitrogen and sulphur content were analysed using a LECO TruSpec CHNS analyser. Major and trace constituents were determined by X-ray fluorescence spectrometry. The accuracy of the XRF measurements was checked by using certified reference materials. For instance, in the specific case of As, the As concentration (mg/kg) obtained for the reference material BCRCRM-277b was 45.4 ± 4.1 (certified value 47.3 ± 1.6). X-ray powder diffraction was used for semiquantitative mineralogical analysis of the mineral phases present in the sediment.
River water characterization
River water was collected and filtered using 0.45-μm cellulose nitrate membrane filters NCS 045 47 BC (ALBET LabScience, Dassel, Germany) to be used as a biofilm growth medium in the laboratory in order to mimic natural conditions for biofilm development. pH, Eh and electrical conductivity (EC) were measured in situ using portable electrodes (Hanna Instruments, HI 9025 and HI 9033, respectively). Soluble P was measured by means of acid digestion with H2SO4 followed by colorimetric determination with ammonium molybdate (APHA 2005). Total N was determined by segmented flow analysis and colorimetry with a Futura console (AMS Alliance) after filtration through a 0.45-µm membrane Millex-HM (Millipore). Nitrate was measured by ion chromatograph (model Metrohm 850 Professional IC), and ammonia was determined by ion-selective electrode (ISE; Thermo Scientific 9512BNWP). Alkalinity was measured by colorimetric determination using an AquaKem 250 analyser (Thermo Scientific, Waltham). Dissolved organic carbon (DOC) was measured using a total organic carbon analyser (model TOC-5000, Shimadzu, Kyoto). With this equipment, the DOC concentration is obtained by subtracting the inorganic carbon (IC) concentration from the total carbon (TC) concentration. TC was determined by the 680 °C combustion catalytic oxidation method, whereas IC was determined by acidification and sparging. The carbon dioxide generated in both determinations was detected using a non-dispersive infrared (NDIR) gas analyser. Na and K concentrations were measured by atomic emission spectrometry, and Ca and Mg by atomic absorption spectrometry (Spectra AA 220 FS, Varian Inc., Palo Alto). Total Fe, Mn and As concentration was measured by inductively coupled plasma mass spectrometry (ICP-MS, 820MS Varian Inc., Palo Alto), whereas As species were analysed using HPLC-ICP-MS as described below in “Arsenic analyses” section.
Native biofilm growth
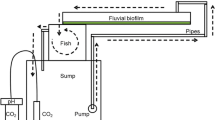
The sediment sample (500 g, 31 % water content) was incubated in bioreactors filled with 2 L of filtered As-free water, equipped with systems for air supply, sample collection and volatilized arsenic trapping (Fig. 1). All materials were previously sterilized by autoclaving at 121 °C for 30 min. The flasks were maintained for 3 weeks in an incubation chamber under optimal controlled conditions of light (day/night cycles, 12 h of light, intensity ca. 40 µmol photons m−2 s−1), temperature (20 °C) and air supply (ca. 1 L min−1). The water was replaced weekly to provide the necessary nutrients for biofilm growth. After 3 weeks, an appreciable biofilm layer was clearly perceived on the sediment surface. Epipsammic biofilms, covering sediments in the Anllóns River, are mainly constituted by Bacillariophyceae, which represent >86 % of the total abundance in superficial sediments (Martiñá Prieto et al. 2016). At the Eguas sampling site, Cocconeis, Navicula and Karayevia were identified as the predominant genera. Epipsammic biofilm inocula from this river have been satisfactorily incubated at laboratory scale in experimental fluvial channels, reaching maximum growth at 2–3 weeks, as indicated by chlorophyll-a and soluble carbohydrates contents (Prieto et al. 2016).

Scheme of experimental setup
Arsenic retention and speciation
Stock solution of 1000 mg L−1 AsV was prepared by dissolving Na2HAsO4·7H2O (Panreac, Spain) in Milli-Q water. Once the biofilm had developed, an aliquot of AsV solution was spiked, resulting in an initial 500 μg L−1 (6.67 µmol L−1) As exposure concentration. This As concentration, which exceeds the USEPA’s aquatic life criteria maximum concentration (CMC) (acute exposure) for As in freshwater, set at 340 µg L−1 (USEPA 2014), was selected because it was below the effective AsV concentration that produces a 20 % decrease (EC20) in the bioluminiscence of Aliivibrio fischeri model bacterium, which was set at 2.0 ± 0.6 mg L−1 by Rubinos et al. (2014) using the Microtox® acute toxicity screening bioassay and constitutes a measurable threshold of As toxicity, while still enabling detection of quantifiable changes in the As species concentration in the water column. The effect of phosphate on AsV retention and speciation was also studied in identical systems, using an equimolar As:P concentration ratio. P was added to the systems from stock solution of 1000 mg L−1 P, prepared by dissolving Na2HPO4·7H2O (Panreac, Spain) in Milli-Q water. The systems incorporating biofilm were labelled BAS and BASP, for the systems with and without added phosphate, respectively. In parallel, identical systems without biofilm (sediment and river water sterilized by three cycles of autoclaving at 120 °C for 30 min) were developed as reference systems (controls) for experiments, with and without phosphate (named CAS and CASP, respectively). The systems were maintained under the conditions indicated above for two additional weeks, during which aliquots (5 mL) of the water column were sampled daily, using single-use sterile polypropylene (PP) syringes (Braun Inkjet, B Braun AG, Melsungen). The samples were immediately filtered (sterile 0.45-μm Whatman Puradisc 25AS™ syringe filters, GE Healthcare Europe GmbH, Barcelona) and stored frozen (−80 °C), until analysis of total As and its species [AsIII, AsV, MMAV, DMAV, arsenobetaine (AsB)] by ICP-MS and HPLC-ICP-MS, respectively. Relative standard deviations for total As analysis by ICP-MS were <3 %. Additionally, dissolved P, Fe and Mn concentrations were measured (determined by ICP-MS) as well as dissolved organic carbon (DOC) (as previously determined by TOC analyser). At the end of the experiment, the As speciation in the sediment interstitial waters of the systems was also determined. Samples (10 mL) of interstitial water were filtered, frozen at −80 °C and analysed for As species as described above. The pH, Eh and sulphate concentration were also measured in the overlying water of the different systems at the end of the experiment, as described in “Sediment and river water characterization” section. All determinations were carried out in triplicate to ensure the quality of the values obtained.
Arsenic analyses
Analysis of dissolved As species was carried out using high-performance liquid chromatography together with inductively coupled plasma spectrometry (HPLC-ICP-MS). A Varian Prostar 230 HPLC, equipped with a guard column and a Hamilton PRP-X100 anion exchange column (4.1 × 250 mm and 10 µm), was used to separate five primary As species (AsV, AsIII, MMAV, DMAV and AsB) using a 13 min gradient LC method with 12.5 and 30 mM (pH 9) (NH4)2CO3 as mobile phase, flow rate of 1 mL min−1 and injection volume of 50 µL. For quantification, a Varian 820-MS ICP-MS, equipped with collision reaction interface (CRI) technology to reduce polyatomic interferences, was used. Relative standard deviations for total As analysis by ICP-MS were <3 %. The detection limits under the experimental conditions were 2.8, 4.1, 2.9, 4.6 and 2.5 ng L−1 for AsV, AsIII, MMAV, DMAV and AsB, respectively.
Volatilized arsenic
To quantify volatilized As, arsines were trapped using the AgNO3-based chemo-trapping approach described by Mestrot et al. (2009) and Yin et al. (2011a). In this method, arsine (AsH3), monomethylarsine (MeAsH2), dimethylarsine (Me2AsH) and trimethylarsine (TMAs or Me3As) react with AgNO3 and are preserved by oxidation to their pentavalent oxy-species (AsV, MMAV, DMAV and TMAO) (Mestrot et al. 2009).
To prepare the traps, silica gel (2.5–5 mm) was submersed overnight in 5 % HNO3 (w/v) solution and washed with Milli-Q water (18.2 MΩ cm−1 resistivity), then impregnated with 10 % AgNO3 (w/v) solution and placed overnight in an oven at 70 °C (covered with aluminium foil to avoid photodecomposition of AgNO3) (Yin et al. 2011a). Subsequently, the silica gel (~1 g) was loaded in the trap tubes (10-mL sterilized syringe) and held in place by a small quantity of glass wool washed QP (Panreac, Barcelona) at both ends. Trap tubes were again covered with aluminium foil to avoid photodecomposition of AgNO3 and coupled to the flask systems.
At the end of the experiment, 5 mL of 1 % (v/v) hot boiling HNO3 was used to elute the collected As in the trap tubes (Yin et al. 2011b). Eluates were filtered (0.45 µm) and frozen (−80 °C), and total As was measured by ICP-MS.
Remobilization and bioavailability of arsenic
Immediately after the retention, the overlying water was removed and the potential remobilization of As species was assessed by washing the As-loaded sediment–biofilm or sediment, with As-free filtered (0.45 µM) Anllóns River water (1:10 solid:liquid ratio). The aqueous extracts were filtered (0.45 µm), frozen (−80 °C) and analysed for As by HPLC-ICP-MS.
To evaluate As bioavailability, the diffusion gradient in thin films (DGT) technique was used. DGT devices, purchased from DGT Research Ltd. (Lancaster), accumulate metals on a binding agent after they have passed through a well-defined diffusive layer (Davison and Zhang 1994; Zhang and Davison 1995; Zhang et al. 1995). DGT incorporating Fe-oxide gels have been specifically developed for assessing As bioavailability (Stockdale et al. 2008, 2010; Luo et al. 2010).
Arsenic-specific DGT devices were placed onto the sediment surface of all the studied systems for 24 h to afford an operationally defined measure of the “bioavailable” fraction of As. Once removed from the systems, the devices were rinsed with Milli-Q water and opened for removal of the resin gels, which were then eluted with 1 mL of 7.2 M HNO3 for at least 24 h to allow complete extraction of As from the resin. An aliquot from the sample tube was pipetted and diluted 6 times with Milli-Q water prior to analysis by ICP-MS. The mass of As in the resin gel (M), the time-averaged DGT concentrations (CDGT) and the flux (F) of As measured by DGT were calculated according to Zhang and Davison (1995) and DGT® technical documentation.
To determine time-averaged DGT concentrations, the diffusion coefficient of total As was calculated from the diffusion coefficient of individual species using Eq. 1:
where x i and D i are the fraction of individual As species over the total As and the diffusion coefficient of the individual As species, respectively.
Values for the diffusion coefficient of AsIII and AsV have been reported in the ranges of 5.9–10.1 and 4.9–6.8 × 10−6 cm2 s−1, respectively (Fitz et al. 2003; Panther et al. 2008; Österlund et al. 2010, 2012; Bennett et al. 2010; Luo et al. 2010; Moreno-Jiménez et al. 2013), whereas diffusion coefficient values of 6.10 and 6.30 × 10−6 cm2 s−1 have been reported for MMAV and DMAV, respectively (Österlund et al. 2012).
Extracellular and intracellular arsenic
Extracellular As in the biofilms was determined using the extraction procedure described by Levy et al. (2005) consisting in phosphate extraction, followed by extraction of intracellular As by the method described by Miyashita et al. (2009), consisting of extraction of As in methanol–water from lysed cells. To this end, after exposure to AsV, biofilm samples (ca. 1 g) were gently harvested and rinsed with 10 mL of filtered Anllóns River water (1:10 solid:liquid ratio). Then, the solid phases were submitted to two washing cycles with 10 mL of 0.1 M KH2PO4/K2HPO4 buffer solution (pH 5.95) to extract any extracellular As. The suspensions were shaken for 30 s and allowed to stand for 20 min, then they were centrifuged (3000 rpm, 15 min), the supernatant was filtered (0.45-µm syringe filters), and the extraction cycle was repeated. The eluates of two washes were combined, frozen (−80 °C) and analysed for arsenic species (HPLC-ICP-MS). The remaining solid phases were gently washed with Milli-Q water (18.2 MΩ cm−1 resistivity), centrifuged (3000 rpm, 15 min) and frozen (−20 °C). For determination of intracellular As, solid phases were thawed and intracellular As was extracted with 10 mL of methanol/H2O (1:1, v/v) solution. After standing for 10 min, the suspensions were sonicated for 10 min and centrifuged at 3000 rpm for 15 min. Extraction was repeated twice with 5 mL of methanol/H2O (1:1, v/v) solution. The extracts were combined and evaporated under vacuum to dryness using a rotavapor (Büchi Rotavapor R-200, BÜCHI Labortechnik GmbH, Essen). The dried extracts were redissolved with Milli-Q water (18.2 MΩ cm−1 resistivity), filtered (0.45 µm), frozen (−80 °C) and analysed for As species (HPLC-ICP-MS). At the end of the sequential extractions, solid phases were dried at 105 °C to constant weight to determine the dry weight of the analysed samples. All determinations were carried out in triplicate.
Results and discussion
Sediment and river water characterization
The main physico-chemical properties of the bed sediments and river water are presented in Table 1. The sediment had slightly acidic pH (6.3) and Eh of 235 mV, which is indicative of a suboxic state (ranging between 100 and 400 mV at around neutral pH) of the surface layer, and showed a predominance of the sandy fraction. Semiquantitative mineralogical analysis of the mineral phases showed quartz as the predominant mineral, with about half of the total abundance, followed by microcline and hornblende. The presence of this highly weatherable mineral can be explained because, at this sampling site, the Anllóns River runs over basic rocks, namely peridotite, pyroxenite, amphibolite and serpentinite, and is indicative of a short transport distance of the sediments. The total organic matter content was 1.5 %, whereas P and N concentrations were 370 and 253 mg kg−1, respectively. The C/N ratio was 35.4, which is indicative of organic matter (OM) rich in lignin and cellulose as well as poor in N, attributable to terrestrial origin (Lamb et al. 2006). The total P concentration was lower than the lowest effect level (LEL) established by the Ontario sediment quality guidelines (Persaud et al. 1993), set at 600 mg kg−1. This level of pollution is expected to have no effect on the majority of sediment-dwelling organisms, and the sediment is considered clean to marginally polluted. The total As concentration of the sediment was 15 mg kg−1, which is lower than the values detected in the sampling campaign performed by Devesa-Rey et al. (2008), falling within the range of 33–264 mg kg−1. The As concentration slightly exceeded the “Effects Range-Low” (ERL) fixed by Long et al. (1995) at 8.2 mg kg−1, which represents the upper end of a range of concentrations at which effects would rarely be observed.
River water exhibited neutral pH (7.0), low alkalinity (23.3 mg L−1) and low concentrations of P (0.05 mg L−1), nitrate (1.59 mg L−1 as NO −3 -N) and ammonia (0.09 mg L−1 as NH +4 -N). These parameter values classify the river water as having high ecological status because they are below the limits for this status for Spanish rivers in Atlantic and Cantabrian watersheds, fixed at 0.07, 2.26 and 0.16 mg L−1 for P, nitrate (as NO −3 -N) and ammonia (as NH +4 -N), respectively (BOE 2015). The As concentration in the river water was 3.6 µg L−1, mainly in the form of AsV (92 %) with the remainder as AsIII. This concentration is in the range of those previously detected in Anllóns River fresh water by Costas et al. (2011) (0.16–3.96 µg L−1) and lower than the recommended permissible As level in drinking water, set at 10 µg L−1 by the World Health Organization (WHO 1993).
Arsenic retention
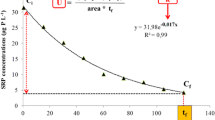
The As concentration in the overlying water decreased during the time course of the experiment down to a nearly constant final value. The removal of As was higher in the presence of biofilm (Fig. 2). After 14 days of exposure, the As retention in the BAS system was 91 % ({As}max = 32.5 μmol kg−1) in comparison with the initial concentration in water (500 µg L−1), but it only reached 70 % in CAS ({As}max = 27.8 μmol kg−1). Data were satisfactorily fitted (R 2 > 0.94) to the empirical exponential equation given by Eq. 2.
where [As] is the As concentration (µg L−1) at time t (days) and a, b and c are adjustment parameters. The equilibrium aqueous As concentration in BAS, defined by the a parameter of the fit, was 46.71 µg L−1, 3.6 times lower than in the system without biofilm (CAS) (167.4 µg L−1). These results are in agreement with those obtained by Prieto et al. (2013), who studied As removal by biofilm in batch experiments and reported an 18 % increase in average As retention for systems incorporating biofilm in comparison with sediments without biofilm.

Evolution of As concentration in overlying water throughout the experiment for BAS and CAS systems
In the environmental conditions of this study, a significant reduction of As concentration was achieved in the presence of biofilm, revealing its importance in natural aquatic systems and potential for application in biotechnological systems for water purification. Thus, the final concentrations in the BAS and CAS systems were lower than the USEPA’s aquatic life criteria maximum concentration (CMC), fixed at 340 µg L−1 in surface waters (USEPA 2014), but only the BAS value was lower than the criteria continuous concentration (CCC), fixed at 150 µg L−1. CMC and CCC are the highest concentrations in surface waters to which an aquatic community can be exposed briefly or indefinitely, respectively, without resulting in an unacceptable effect. The BAS final concentration was also lower than the permissible limit for irrigation water (100 µg L−1) (FAO 1985), although slightly higher than the environmental quality standards (EQS) for As in inland surface waters, set at 25 µg L−1 by the Priority Substances Directive in Surface Waters [S.I. No. 272/2009-European Communities Environmental Objectives (Surface Waters) Regulations 2009]. This EQS represents a threshold for annual average concentration of As in surface waters to ensure protection against long-term exposure to pollutants in an aquatic environment.
The epipsammic biofilm not only enhanced the retained As amount but also the retention rate. Thus, BAS exhibited higher initial retention rates (3.84 µmol As kg−1 day−1 at 7 days) than CAS (2.97 µmol As kg−1 day−1 at 7 days) up to 7 days, after which both values were similar.
The presence of the equimolar P concentration in the BASP system slightly enhanced the amount and rate of As removal, reaching 97 % of its initial concentration ({As}max = 36.3 μmol kg−1). The effect was lower in the absence of biofilm (CASP), where As removal reached only 69 % ({As}max = 26.1 μmol kg−1) (Fig. 3). In terms of water quality, the equilibrium As concentration in BASP determined by the exponential equation was 18.85 µg L−1, which is lower than the CMC and CCC values and than the FAO permissible limit for irrigation water but still higher than the EQS. The equilibrium As concentration in CASP was exactly the same as the value mentioned above for CAS (167.4 µg L−1). With respect to retention rates, BASP also exhibited higher values (4.92 µmol As kg−1 day−1) than CASP (3.28 µmol As kg−1 day−1) up to 7 days, after which the values for both systems were similar.

Evolution of As concentration in overlying water throughout the experiment for BASP and CASP systems
The soluble P decreased from 50 to 12 µg L−1 in BAS, while in BASP (to which P was added), it decreased from 220 to 23 µg L−1 (Fig. 4). The reduction of the P concentration was attributed to its consumption by the microorganisms composing the biofilm. In CAS, the soluble P concentration remained fairly constant, between 100 and 150 µg L−1, throughout the experiment, whereas in CASP it decreased in the first 3 days from an initial concentration of 312 to 100–150 µg L−1, which can be considered the equilibrium concentration in both systems.

Evolution of P concentration in overlying water throughout the experiment for BAS, CAS, BASP and CASP systems
Arsenic speciation
Five soluble As species were detected in the overlying water in the systems incorporating biofilm exposed to As-enriched solution. During the 2 weeks of the experiment, their concentrations followed the order: AsV ≫ AsIII > MMAV ≈ DMAV > AsB. For BAS, aqueous As was mostly (~98 %) in AsV form, whereas AsIII reached only 1.2 % of total As, and MMAV and DMAV represented only 0.6 and 0.7 % of total As, respectively, indicating slight (bio-)methylation by the epipsammic biofilm during the incubation period. In the absence of the biofilm, the speciation of As in the water column changed significantly. In this system, AsIII accounted for up to 39 % (80 μg/L) of the final aqueous As in CAS (Fig. 5a), with the remainder in the form of AsV, with no methylated forms present. The concentration of AsIII for the systems incorporating phosphate (BASP and CASP) coincided with the results observed for the systems without phosphate (Fig. 5b), while methylated aqueous species were again only detected in the presence of biofilm (Fig. 6a, b). The detection of methylated As species in the BAS and BASP systems suggests some occurrence of bio-methylation in the presence of biofilm, albeit insignificant, during the short running time of our study. Similar concentrations of AsIII were found in CASP and in CAS. In both control systems, AsIII was detected in the overlying water after 48 h of exposure. To investigate this fact, physico-chemical water conditions were investigated, as well as the behaviour of compounds susceptible to promote AsV reduction. The pH and Eh values in the overlying water at the end of the experiment showed that both sediment–biofilm and control systems were under near-neutral and oxic conditions (Table 2). There was no evidence of sulphur oxidation, as sulphate concentrations were similar at the end of the experiment for all the systems and similar to the initial concentrations in the river water. Also, Fe concentrations were similar throughout the experiment in all systems (Fig. 7a, b). On the other hand, in the systems without biofilm, Mn concentrations increased up to day 4 and then remained constant at values much higher (up to 1500-fold) than those observed in the systems with biofilm, in which Mn concentrations decreased over time (Fig. 7c, d). Similarly, higher DOC concentrations were measured for CAS (mean value 55.3 ± 4 mg L−1) in comparison with BAS (mean value of 14.0 ± 3.8 mg L−1). These results suggest that oxidation–reduction processes are occurring in the systems without biofilm, as explained in “General discussion” section. Biofilms seem to inhibit reduction of added AsV, by covering the mineral surfaces of the sediments and thus hindering their interaction with aqueous As and maintaining an oxygenated biofilm–sediment interface.

AsIII concentration in overlying water throughout the experiment for BAS and CAS systems (a) and for BASP and CASP systems (b)

DMAV concentration in overlying water throughout the experiment for BAS and CAS systems (a) and for BASP and CASP systems (b)

Fe and Mn total concentrations in overlying water throughout the experiment for BAS and BASP (a and c, respectively) and for CAS and CASP (b and d, respectively)
As in interstitial water
Similarly to what occurred in the overlying water, As concentrations in interstitial water were notably higher (at least 9 times) in the CAS and CASP systems, with a higher percentage of AsIII, than in the BAS and BASP systems, where detectable concentrations of DMAV were also found (Table 3).
Volatilized arsenic
Volatile As forms were only detected in systems incorporating biofilm. This fact, together with the detection of methylated As species, may indicate the occurrence of detoxification processes. It has been reported that some microorganisms (bacteria, fungi and algae) form arsine in order to decrease intracellular As, as a (bio-)transformation pathway to cope with As toxicity (Wang et al. 2014). The mean amount of volatilized As after 14 days of exposure was 17.0 ng As, and the bio-volatilization rate was 2.4 ng of volatilized As per mg of added AsV and day. This value is higher than those calculated from reported data (0.28–0.54 ng mg−1 day−1) by Yin et al. (2011a) for Microcystis, Nostoc and Synechocystis cyanobacteria, treated with either 7.5 or 30 mg L−1 AsV for 6 weeks, but falls within the range of those reported (1.3–9.3 ng mg−1 day−1) by Yin et al. (2011b) for As biotransformation by the protozoan Tetrahymena thermophila after 48 and 72 h of exposure to 150–3000 µg L−1 AsV concentrations. With respect to the initial As concentration, the maximum percentage of volatilized As was only 0.002 %. This value is slightly lower than the percentage (<0.1 %) calculated by Yin et al. (2011a) and than the percentages (0.006–1 %) reported by Yin et al. (2011b). In summary, these results may suggest that the epipsammic biofilm carried out detoxification processes with production of volatilized As, although analysing the global As balance, its contribution was of minor relevance.
Remobilization and bioavailability of arsenic
Regarding As remobilization, the release to water of previously retained As was low and varied between 113.0 ± 9.4, 166.8 ± 10.0, 181.1 ± 9.1 and 176.5 ± 7.9 µg kg−1 for BAS, CAS, BASP and CASP, respectively, representing 4.7, 6.1, 8.7 and 9.0 % of the retained As, respectively. In the presence of biofilm, 94 % of the released As was AsV, with only 3 % being AsIII. In contrast, AsIII accounted for 21 % of the released As in the systems without biofilm, where the concentration of remobilized AsIII was 10 times higher than in the systems with biofilm. MMAV was only detected (3.3 % of total As released) in the extracts from the systems with biofilm, while DMAV was not found in any case.
To estimate As bioavailability, the concentrations of As species were determined in the eluates from the DGT devices and are presented in Table 4, jointly with the parameters calculated from these concentrations. Once again, the values obtained were similar for the different systems. It is worth highlighting that the speciation of the DGT extracts revealed that the percentage of AsV was higher than 77 % in all cases, and DMAV was only detected in the BAS samples.
Extracellular and intracellular arsenic
In biofilm-enriched samples obtained from the BAS and BASP systems, 71.1 ± 1.5 % of the retained As was extractable with 0.1 M KH2PO4/K2HPO4 phosphate buffer, being attributed to extracellular As (Levy et al. 2005), most of it (>99.6 %) in the form of AsV. This fraction may represent an estimation of easily leachable As (Gleyzes et al. 2002). DMAV was identified in phosphate extracts, revealing once again the occurrence of a (bio-)methylation process by the epipsammic biofilm. Regarding intracellular As, extractable with methanol/H2O (1:1, v/v) solutions (Miyashita et al. 2009), the systems with biofilm revealed 28.9 ± 1.5 % of As inside the cells, almost exclusively (>99.8 %) in the form of AsV.
General discussion
In this study, the influence of epipsammic biofilm on the behaviour of AsV in freshwater environments was evaluated. The results reveal that biofilms increase the amount and rate of AsV retention by the sediment. Prieto et al. (2013) also observed, in batch experiments with short-term (24 h) As exposure, that epipsammic biofilm increased AsV sorption by sediments from the Anllóns River and that this effect was more noticeable in the presence of phosphate.
Sediments may retain As due to their content of Fe, Al and Mn (oxy)hydroxides (Oscarson et al. 1981; Jiang et al. 2005), clay mineral (Manning and Goldberg 1997) and organic matter (Thanabalasingam and Pickering 1986). Additionally, the favourable effect of biofilm on As removal may be attributed to the addition of two combined effects: (bio-)sorption and (bio-)accumulation. Firstly, As (bio-)sorption is improved because the biofilm may increase the specific surface area, and the number of sorption sites and consequently As sinks. It has been observed that sorption of metals by biofilms is governed by their interactions with the biofilm matrix, constituted by cells and extracellular polymeric substances (EPS). EPS are mainly constituted by polysaccharides and proteins, as well as nucleic acids, lipids or humic substances, and EPS molecules contain ionizable functional groups which can sequester toxic compounds (van Hullebusch et al. 2003). Moreover, Fe and Mn oxides precipitated as biominerals in biofilms have been identified as responsible for retention of trace elements (Dong et al. 2000; Warren and Haack 2001). Drahota et al. (2014) reported enhancement of As retention by precipitation of poorly crystalline biogenic Mn oxides, in turn induced by growth and accumulation of biofilms. Secondly, AsV (bio-)accumulation takes place when AsV enters cells via phosphate transporters (Páez-Espino et al. 2009). In our study, the extracellular fraction or As biosorption represents the main compartment of the retained As in the systems with biofilm.
Addition of equimolar phosphate concentration had no effect on the As adsorption in the CASP control system. Although phosphate usually competes with arsenate for sorption sites in soils and sediments due to their chemical similarities (Manning and Goldberg 1996; Hongshao and Stanforth 2001), there was no competition between As and P in CASP; the reason may be that phosphate rapidly decreased in the solution (a reduction of 63 % in the first 3 days) while As retention continued up to day 10. The amount of As retained in the control systems was only slightly lower than the maximum adsorption capacity (2.0 µg g−1) determined using a Langmuir model by Prieto et al. (2013) for sediments from the same site (6.6 µg g−1), which could explain the high As concentration that remained in the overlying water of the CAS and CASP control systems.
The presence of phosphate had a slight positive effect on the retention of As by the epipsammic biofilm. This effect could be due to the stimulative effect of phosphate on the growth of the microorganisms, as proved by the almost complete absorption of phosphate by the biofilm, which is supposed to contribute to its growth. Other possible explanations are the increase in the efficiency and/or number of phosphate/arsenate cellular transporters or to the alleviative effect of phosphate against AsV toxicity. This latter effect was observed by Karadjova et al. (2008) for the green microalga Chlorella salina in sea water, by Wang et al. (2013) for two freshwater green algae, and by Rubinos et al. (2014) for A. fischeri bacterium.
The biofilm inhibited the reduction of added AsV, which may be due to the coverage of the mineral surfaces of the sediments, thus preventing their interaction with aqueous As, and/or to the maintenance of a more oxygenated interface due to photosynthesis. Several key factors are reported in literature to control As speciation, such as pH and Eh (Smedley and Kinniburgh 2002), presence of redox pairs such as Fe3+/Fe2+, Mn4+/Mn2+, SO 2−4 /HS− (Cherry et al. 1979), and the effect of natural organic matter (NOM) (Sharma and Sohn 2009). Among all the conditions evaluated, DOC and Mn concentrations were notably higher in the systems devoid of biofilm. Lower DOC concentrations in BAS and BASP could be attributed to DOC release from the sediment, which was inhibited in the presence of biofilm, or to DOC consumption by biofilm heterotrophs. This fact is relevant because an important role in the geochemical behaviour of As is attributed to NOM (Buschmann et al. 2006; Klitzke and Lang 2009). Thus, it has recently been demonstrated that addition of arsenic to DOM solutions results in arsenate reduction as well as arsenite oxidation (Redman et al. 2002). On the other hand, the release of Mn2+ is attributed to the reduction and dissolution of Mn oxyhydroxides of the sediment. Therefore, the results of this study suggest that dissolution and reductive processes are occurring in CAS and CASP, involving AsV reduction and Mn mobilization jointly with NOM, which were inhibited in both the BAS and BASP systems. This inhibition may come about because of the ability of autotrophic microorganisms, which make up the biofilm, to oxygenate the water–sediment interface and hence avoid reductive processes. The inhibition of AsV reduction by the biofilms has relevant geochemical and toxicological implications, since AsIII is usually considered more mobile and toxic than AsV (Sharma and Sohn 2009; Huang 2014).
Arsenic methylation has been demonstrated in different aerobic and anaerobic microorganisms (Kuehnelt and Goessler 2003). The presence of methylated As species in the overlying water, in the interstitial water and within the biofilm in BAS and BASP are indicative of (bio-)methylation processes taking place. Among the methylated As compounds, DMAV was the main species detected in the overlying and interstitial water. MMAV was detected at lower concentrations. This fact could be explained because MMAV is an intermediate in the As methylation process, with a rapid intracellular metabolism and ten times lower permeability to membranes than DMAV (Cullen et al. 1994a, b, c). Higher DMAV concentrations compared with MMAV have also been found in natural environments such as in a German forested catchment by Huang and Matzner (2007), in eutrophic and mesotrophic lakes by Hasegawa et al. (2009) and in marine sediments by Fauser et al. (2013).
In summary, epipsammic biofilms play a key role in the biogeochemistry of As in river environments, by enhancing removal of AsV from water and by strongly affecting As speciation. Biofilms inhibit the occurrence of aqueous AsIII in the water column, which is attributed to their potential ability to oxygenate the biofilm–sediment interface and to carry out detoxification processes inside the cells via production of methylated and volatilized species.
Conclusions
Epipsammic biofilms covering riverbed sediments enhance the removal and retention rate of AsV from the water and also strongly affect the speciation of As in the water column by inhibiting the occurrence of aqueous AsIII, due to their ability to oxygenate aquatic environments and to bring about DOC consumption by biofilm heterotrophs. This fact has noteworthy toxicological and geochemical relevance, for example, for remediation purposes, considering the frequent greater toxicity and mobility of AsIII species. The detoxification of As driven by epipsammic biofilms is supported by the presence of methylated arsenic species such as MMAV and DMAV in the overlying water and within the biofilm, as well by the detection of volatilized arsenic. The enhancement of the retention and toxicity of As in the presence of biofilms highlights their importance in natural aquatic systems and their potential for application in biotechnological water purification systems.
References
APHA (2005) Standard methods for the examination of water and wastewater. American Public Health Association. 21st ed. American Water Works Association, Water Environment Federation, Alexandria
Barral-Fraga L, Morin S, Rovira MDM, Urrea G, Magellan K, Guasch H (2016) Short-term arsenic exposure reduces diatom cell size in biofilm communities. Environ Sci Pollut Res. doi:10.1007/s11356-015-4894-8
Beck AJ, Janssen F, de Beer D (2011) The influence of phototrophic benthic biofilms on Cd, Cu, Ni, and Pb transport in permeable sediments. Biogeochemistry 102(1–3):167–181
Bennett WW, Teasdale PR, Panther JG, Welsh DT, Jolley DF (2010) New diffusive gradients in a thin film technique for measuring inorganic arsenic and selenium(IV) using a titanium dioxide based adsorbent. Anal Chem 82(17):7401–7407
Blanck H, Wangberg S-A (1988) Validity of an ecotoxicological test system: short-term and long-term effects of arsenate on marine periphyton communities in laboratory systems. Can J Fish Aquat Sci 45(10):1807–1815
Blanck H, Wangberg S-A (1991) Pattern of cotolerance in marine periphyton communities established under arsenate stress. Aquat Toxicol 21(1–2):1–14
BOE (2015) Real Decreto 817/2015 del 11 de septiembre, por el que se establecen los criterios de seguimiento y evaluación del estado de las aguas superficiales y las normas de calidad ambiental. https://www.boe.es/boe/dias/2015/09/12/pdfs/BOE-A-2015-9806.pdf
Buschmann J, Kappeler A, Lindauer U, Kistler D, Berg M, Sigg L (2006) Arsenate and arsenite binding to dissolve humic acids: influence of pH, type of humic acid, and aluminium. Environ Sci Technol 40:6015–6020
Cherry JA, Shaikh AU, Tallman DE, Nicholson RV (1979) Arsenic species as an indicator of redox conditions in groundwater. J Hydrol 43:373–392
Costas M, Prego R, Filgueiras AV, Bendicho C (2011) Land–ocean contributions of arsenic through a river–estuary–ria system (SW Europe) under the influence of arsenopyrite deposits in the fluvial basin. Sci Total Environ 412–413:304–314
Costerton JW (2007) The biofilm primer. Springer, Berlin
Cullen WR, Harrison LG, Li H, Hewitt G (1994a) Bioaccumulation and excretion of arsenic compounds by a marine unicellular alga, Polyphysa peniculus. Appl Organomet Chem 8(4):313–324
Cullen WR, Herring FG, Nelson JC (1994b) Employing permeability coefficients to understand the biomobility and bioaccumulation of compounds sensitive to the environment. Bull Environ Contam Toxicol 52:171–175
Cullen WR, Li H, Hewitt G, Reimer KJ, Zalunardo N (1994c) Identification of extracellular arsenical metabolites in the growth-medium of the microorganisms Apiotrichum humicola and Scopulariopsis brevicaulis. Appl Organomet Chem 8(4):303–311
Davison W, Zhang H (1994) In situ speciation measurements of trace components in natural waters using thin-film gels. Nature 367:546–548
Decho AW (2000) Microbial biofilms in intertidal systems: an overview. Cont Shelf Res 20:1257–1273
DGT® technical documentation. DGT-for measurements in waters, soils and sediments. http://www.dgtresearch.com/dgtresearch/dgtresearch.pdf
Devesa-Rey R, Paradelo R, Díaz-Fierros F, Barral MT (2008) Fractionation and bioavailability of arsenic in the bed sediments of the Anllóns river (NW Spain). Water Air Soil Pollut 195(1–4):189–199
Devesa-Rey R, Díaz-Fierros F, Barral MT (2011) Assessment of enrichment factors and grain size influence on the metal distribution in riverbed sediments (Anllóns river, NW Spain). Environ Monit Assess 179:371–388
Dong D, Nelson YM, Lion LW, Shuler ML, Ghiorse WC (2000) Adsorption of Pb and Cd onto metal oxides and organic material in natural surface coatings as determined by selective extractions: new evidence for the importance of Mn and Fe oxides. Water Res 34:427–436
Dong D, Liu L, Hua X, Lu Y (2007) Comparison of lead, cadmium, copper and cobalt adsorption onto metal oxides and organic materials in natural surface coating. Microchem J 85:270–275
Drahota P, Skaloud P, Nováková B, Mihaljevic M (2014) Comparison of Pb, Zn, Cd, As, Cr, Mo and Sb adsorption onto natural surface coatings in a stream draining natural As geochemical anomaly. Bull Environ Contam Toxicol 93:311–315
Drewniak L, Styczek A, Majder-Lopatka M, Sklodowska A (2008) Bacteria, hypertolerant to arsenic in the rocks of an ancient gold mine, and their potential role in dissemination of arsenic pollution. Environ Pollut 156(3):1069–1074
Duker AA, Carranza EJM, Hale M (2005) Arsenic geochemistry and health. Environ Int 31:631–641
FAO (1985) Water quality guidelines for maximum crop production. In: Wastewater quality guidelines for agricultural use. Food and Agriculture Organization of the United Nations. http://www.fao.org/docrep/T0551E/t0551e04.htm#2.4. Accessed 10 Jan 2016
Fauser P, Sanderson H, Hedegaard RV, Sloth JJ, Larsen MM, Krongaard T, Bossi R, Larsen JB (2013) Occurrence and sorption properties of arsenicals in marine sediments. Environ Monit Assess 185(6):4679–4691
Fitz WJ, Wenzel WW, Zhang H, Nurmi J, Štipek K, Fischerova Z, Schweiger P, Köllensperger G, Ma LQ, Stingeder G (2003) Rhizosphere characteristics of the arsenic hyperaccumulator Pteris vittata L. and monitoring of phytoremoval efficiency. Environ Sci Technol 37(21):5008–5014
Friese K, Mages M, Wendt-Potthoff K, Neu TR (1997) Determination of heavy metals in biofilms from the river Elbe by total-reflection X-ray fluorescence spectroscopy. Spectrochim Acta, Part B 52:1019–1025
Gleyzes C, Tellier S, Astruc M (2002) Chapter 4: Sequential extraction procedures for the characterisation of the fractionation of elements in industrially-contaminated soils. In: Quevauviller Ph (ed) Methodologies for soil and sediment fractionation studies. Royal Society of Chemistry, Cambridge, pp 66–104
Guitián F, Carballas T (eds) (1976) Técnicas de Análisis de Suelos, Pico Sacro, Santiago de Compostela
Haack EA, Warren LA (2003) Biofilm hydrous manganese oxyhydroxides and metal dynamics in acid rock drainage. Environ Sci Technol 37:4138–4147
Hasegawa H, Rahman MA, Matsuda T, Kitahara T, Maki T, Ueda K (2009) Effect of eutrophication on the distribution of arsenic species in eutrophic and mesotrophic lakes. Sci Total Environ 407(4):1418–1425
Headley JV, Gandrass J, Kuballa J, Peru KM, Gong Y (1998) Rates of sorption and partitioning of contaminants in river biofilms. Environ Sci Technol 32:3968–3973
Hellweger FL, Lall U (2004) Modeling the effect of algal dynamics on arsenic speciation in Lake Biwa. Environ Sci Technol 38:6716–6723
Hongshao Z, Stanforth R (2001) Competitive adsorption of phosphate and arsenate on goethite. Environ Sci Technol 35:4753–4757
Huang JH (2014) Impact of microorganisms on arsenic biogeochemistry: a review. Water Air Soil Pollut 225:1848
Huang JH, Matzner E (2007) Biogeochemistry of organic and inorganic arsenic species in a forested catchment in Germany. Environ Sci Technol 41(5):1564–1569
Jiang W, Zhang S, Shan X-Q, Feng M, Zhu Y-G, Mc Laren RG (2005) Adsorption of arsenate on soils. Part 2: modeling the relationship between adsorption capacity and soil physicochemical properties using 16 Chinese soils. Environ Pollut 138:285–289
Karadjova IB, Slaveykova VI, Tsalev DL (2008) The biouptake and toxicity of arsenic species on the green microalga Chlorella salina in seawater. Aquat Toxicol 87:264–271
Klitzke S, Lang F (2009) Mobilization of soluble and dispersible lead, arsenic, and antimony in a polluted, organic-rich soil—effects of pH increase and counterion valency. J Environ Qual 38:933–939
Kuehnelt D, Goessler W (2003) Organoarsenic compounds in the terrestrial environment. In: Craig PJ (ed) Organometallic compounds in the environment. Wiley, Heidelberg, pp 223–275
Lamb AL, Wilson GP, Leng MJ (2006) A review of coastal palaeoclimate and relative sea-level reconstructions using δ13C and C/N ratios in organic material. Earth-Sci Rev 75(1–4):29–57
Levy JL, Stauber JL, Adams MS, Maher WA, Kirby JK, Jolley DF (2005) Toxicity, biotransformation, and mode of action of arsenic in two freshwater microalgae (Chlorella sp. and Monoraphidium arcuatum). Environ Toxicol Chem 10:2630–2639
Long E, MacDonald D, Smith S, Calder F (1995) Incidence of adverse biological effects within ranges of chemical concentrations in marine and estuarine sediments. Environ Manag 19:81–97
Luo J, Zhang H, Santner J, Davison W (2010) Performance characteristics of diffusive gradients in thin films equipped with a binding gel layer containing precipitated ferrihydrite for measuring arsenic(V), selenium(VI), vanadium(V), and antimony(V). Anal Chem 82(8903):8909
Manning BA, Goldberg S (1996) Modeling competitive adsorption of arsenate with phosphate and molybdate on oxide minerals. Soil Sci Soc Am J 60:121–131
Manning BA, Goldberg S (1997) Adsorption and stability of arsenic(III) at the clay mineral-water interface. Environ Sci Technol 31:2005–2011
Martiñá Prieto D, Devesa-Rey R, Paradelo R, Díaz-Fierros F, Barral MT (2016) Monitoring benthic microflora in river bed sediments: a case study in the Anllóns river (Spain). J Soil Sediments 16:1825–1839
Mestrot A, Uroic K, Plantevin T, Islam MR, Krupp E, Feldmann J, Meharg AA (2009) Quantitative and qualitative trapping of arsines deployed to assess loss of volatile arsenic from paddy soil. Environ Sci Technol l43:8270–8275
Miyashita S, Shimoya M, Kamidate Y, Kuroiwa T, Shikino O, Fujiwara S, Francesconi KA, Kaise T (2009) Rapid determination of arsenic species in freshwater organisms from the arsenic-rich Hayakawa River in Japan using HPLC-ICP-MS. Chemosphere 75:1065–1073
Moreno-Jiménez E, Six L, Williams PN, Smolders E (2013) Inorganic species of arsenic in soil solution determined by microcartridges and ferrihydrite-based diffusive gradient in thin films (DGT). Talanta 140:83–89
Morris JM, Nimick DA, Farag AM, Meyer JS (2005) Does biofilm contribute to diel cycling of Zn in High Ore Creek, Montana? Biogeochemistry 76:233–259
Murphy J, Riley J (1962) A modified single solution method for the determination of phosphate in natural waters. Anal Chim Acta 27:31–36
Nelson YM, Lion LW, Shuler ML, Ghiorse WC (1996) Modelling oligotrophic biofilm formation and lead adsorption to biofilm components. Environ Sci Technol 30:2027–2035
Nelson YM, Lion LW, Shuler ML, Ghiorse WC (1999) Lead binding to metal oxide and organic phases of natural aquatic biofilms. Limnol Oceanogr 44:1715–1729
Oremland RS, Stolz JF (2003) The ecology of arsenic. Science 300:939–944
Oremland RS, Stolz JF (2005) Arsenic, microbes and contaminated aquifers. Trends Microbiol 13(2):45–49
Oscarson DW, Huang PM, Defosse C, Herbillon A (1981) Oxidative power of Mn(IV) and Fe(III) oxides with respect to As(III) in terrestrial and aquatic environments. Nature 291:50–51
Österlund H, Chlot S, Faarinen M, Widerlund A, Rodushkin I, Ingri J, Baxter DC (2010) Simultaneous measurements of As, Mo, Sb, V and W using a ferrihydrite diffusive gradients in thin films (DGT) device. Anal Chim Acta 682(1–2):59–65
Österlund H, Faarinen M, Ingri J, Baxter DC (2012) Contribution of organic arsenic species to total arsenic measurements using ferrihydrite-backed diffusive gradients in thin films (DGT). Environ Chem 9(1):55–62
Páez-Espino D, Tamames J, De Lorenzo V, Cánovas D (2009) Microbial responses to environmental arsenic. Biometals 22:117–130
Panther JG, Stillwell KP, Powell KJ, Downard AJ (2008) Perfluorosulfonated ionomer-modified diffusive gradients in thin films: tool for inorganic arsenic speciation analysis. Anal Chem 80(24):9806–9811
Persaud D, Jaagumagui R, Hayton A (1993) Guidelines for the protection and management on aquatic sediment quality in Ontario. Ontario Ministry of the Environment and Energy, Ontario
Petrick JS, Ayala-Fierro F, Cullen WR, Carter DE, Aposhian HV (2000) Monomethylarsonous acid (MMAIII) is more toxic than arsenite in Chang human hepatocytes. Toxicol Appl Pharmacol 163:203–207
Prieto DM, Devesa-Rey R, Rubinos DA, Díaz-Fierros F, Barral MT (2013) Arsenate retention by epipsammic biofilms developed on streambed sediments. Influence of phosphate. BioMed Research International. http://dx.doi.org/10.1155/2013/591634
Prieto DM, Devesa-Rey R, Rubinos DA, Díaz-Fierros F, Barral MT (2016) Biofilm formation on river sediments under different light intensities and nutrient inputs: a flume mesocosm study. Environ Eng Sci 33(4):250–260
Redman AD, Macalady DL, Ahmann D (2002) Natural organic matter affects arsenic speciation and sorption onto hematite. Environ Sci Technol 36:2889–2896
Rodríguez Castro MC, Urrea G, Guasch H (2015) Influence of the interaction between phosphate and arsenate on periphyton’s growth and its nutrient uptake capacity. Sci Total Environ 503–504:122–132
Rubinos D, Iglesias L, Devesa-Rey R, Díaz-Fierros F, Barral MT (2010) Arsenic release from river sediments in a gold-mining area (Anllóns river basin, Spain): effect of time, pH and phosphorous concentration. Eur J Mineral 22(5):665–678
Rubinos D, Iglesias L, Díaz-Fierros F, Barral MT (2011) Interacting effect of pH, phosphate and time on the release of arsenic from polluted river sediments (Anllóns river, Spain). Aqua Geochem 17:281–306
Rubinos D, Calvo V, Iglesias L, Barral MT (2014) Acute toxicity of arsenic to Aliivibrio fischeri (Microtox® bioassay) as influenced by potential competitive–protective agents. DOI, Environ Sci Pollut Res. doi:10.1007/s11356-014-2715-0
Sabater S, Guasch H, Ricart M, Romaní A, Vidal G, Klünder C, Schmitt-Jansen M (2007) Monitoring the effect of chemicals on biological communities. The biofilm as an interface. Anal Bioanal Chem 387(4):1425–1434
Serra A, Corcoll N, Guasch H (2009) Copper accumulation and toxicity in fluvial periphyton: the influence of exposure history. Chemosphere 74:633–641
Sharma VK, Sohn M (2009) Aquatic arsenic: toxicity, speciation, transformations and remediation. Environ Int 35:743–759
S.I. No. 272/2009-European Communities Environmental Objectives (surface waters) Regulations (2009)
Smedley PL, Kinniburgh DG (2002) A review of the source, behaviour and distribution of arsenic in natural waters. Appl Geochem 17(5):517–568
Stockdale A, Davison W, Hao Zhang (2008) High-resolution two-dimensional quantitative analysis of phosphorus, vanadium and arsenic, and qualitative analysis of sulfide, in a freshwater sediment. Environ Chem 5(2):143–149
Stockdale A, Davison W, Hao Zhang (2010) 2D simultaneous measurement of the oxyanions of P, V, As, Mo, Sb, W and U. J Environ Monit 12:981–984
Styblo M, Del Razo LM, Vega L, Germolec DR, LeCluyse EL, Hamilton GA, ReedW Wang C, CullenWR Thomas DJ (2000) Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in rat and human cells. Arch Toxicol 74:289–299
Thanabalasingam P, Pickering WF (1986) Arsenic sorption by humic acids. Environ Pollut 12:233–246
Tuulaikhuu B-A, Romaní AM, Guasch H (2015) Arsenic toxicity effects on microbial communities and nutrient cycling in indoor experimental channels mimicking a fluvial system. Aquat Toxicol 166:72–82
USEPA (2014) National recommended water quality criteria: aquatic life criteria. Environmental Protection Agency. http://water.epa.gov/scitech/swguidance/standards/criteria/aqlife/index.cfm
van Hullebusch ED, Zandvoort MH, Lens PNL (2003) Review metal immobilisation by biofilms: mechanisms and analytical tools. Environ Sci Bio/Technol 2:9–33
Wang N, Li Y, Deng X, Miao A, Ji R, Yang L (2013) Toxicity and bioaccumulation kinetics of arsenate in two freshwater green algae under different phosphate regimes. Water Res 47:2497–2506
Wang P, Sun G, Jia Y, Meharg AA, Zhu Y (2014) A review on completing arsenic biogeochemical cycle: microbial volatilization of arsines in environment. J Environ Sci 26:371–381
Wangberg SA, Heyman U, Blanck H (1991) Long-term and short-term arsenate toxicity to fresh-water phytoplankton and periphyton in limnocorrals. Can J Fish Aquat Sci 48(2):173–182
Warren LA, Haack EA (2001) Biogeochemical controls on metal behaviour in freshwater environments. Earth Sci Rev 54:261–320
WHO (1993) Guidelines for drinking water quality, 2nd edn. Switzerland, Geneva
Yang SI, Lawrence JR, Swerhone GDW, Pickering IJ (2011) Biotransformation of selenium and arsenic in multi-species biofilm. Environ Chem 8(6):543–551
Yin X, Chen J, Qin J, Sun G, Rosen BP, Zhu Y (2011a) Biotransformation and volatilization of arsenic by three photosynthetic cyanobacteria. Plant Physiol 156:1631–1638
Yin X, Zhang Y, Yang J, Zhu Y (2011b) Rapid biotransformation of arsenic by a model protozoan Tetrahymena thermophila. Environ Pollut 159:837–840
Zhang H, Davison W (1995) Performance characteristics of the technique of diffusion gradients in thin-films (DGT) for the measurement of trace metals in aqueous solution. Anal Chem 67:3391–3400
Zhang H, Davison W, Miller S, Tych W (1995) In situ high resolution measurements of fluxes of Ni, Cu, Fe and Mn and concentrations of Zn and Cd in porewaters by DGT. Geochim Cosmochim Acta 59:4181–4192
Acknowledgments
The authors wish to thank the Spanish Ministry of Economy and Competitiveness (MINECO-FEDER) for financial support (project ref. CGL2010-22059 and CGL2013-46003P). D.M.P. wishes to acknowledge the support of the Spanish Ministry of Economy and Competitiveness for his personal funding (FPI Fellowship, ref. BES-2011-044514). D.A.R. is also grateful for his personal funding from the Xunta de Galicia (Plan Galego de Investigación, Innovación e Crecemento—I2C, Consellería de Educación e Ordenación Universitaria) and from the European Social Fund.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Responsible Editor: Breck Bowden
Rights and permissions
About this article

Cite this article
Prieto, D.M., Rubinos, D.A., Piñeiro, V. et al. Influence of epipsammic biofilm on the biogeochemistry of arsenic in freshwater environments. Biogeochemistry 129, 291–306 (2016). https://doi.org/10.1007/s10533-016-0232-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10533-016-0232-6