
Abstract
Objectives
The aim of this work was to rapidly produce in plats two recombinant antigens (RBDw-Fc and RBDo-Fc) containing the receptor binding domain (RBD) of the spike (S) protein from SARS-CoV-2 variants Wuhan and Omicron as fusion proteins to the Fc portion of a murine IgG2a antibody constant region (Fc).
Results
The two recombinant antigens were expressed in Nicotiana benthamiana plants, engineered to avoid the addition of N-linked plant-typical sugars, through vacuum agroinfiltration and showed comparable purification yields (about 35 mg/kg leaf fresh weight).
Conclusions
Their Western blotting and Coomassie staining evidenced the occurrence of major in planta proteolysis in the region between the RBD and Fc, which was particularly evident in RBDw-Fc, the only antigen bearing the HRV 3C cysteine protease recognition site. The two RBD N-linked glycosylation sites showed very homogeneous profiles free from plant-typical sugars, with the most abundant glycoform represented by the complex sugar GlcNAc4Man3. Both antigens were specifically recognised in Western Blot analysis by the anti-SARS-CoV-2 human neutralizing monoclonal antibody J08-MUT and RBDw-Fc was successfully used in competitive ELISA experiments for binding to the angiotensin-converting enzyme 2 receptor to verify the neutralizing capacity of the serum from vaccinated patients. Both SARS-Cov-2 antigens fused to a murine Fc region were rapidly and functionally produced in plants with potential applications in diagnostics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The huge demand for diagnostic kits during this period of spread of the SARS-CoV-2 virus has highlighted not only the serious shortage of reagents (antigens and recombinant antibodies) but also of means to produce them (Lico et al. 2020). In this scenario, ‘Plant Molecular Pharming’ is a production strategy that has already demonstrated to be particularly successful for the production of SARS-CoV-1 virus antigens (Capell et al. 2020). In the last 3 years, during the pandemic period, there have been numerous the studies carried out for the production of recombinant SARS-CoV-2 antigens in plants, not only for diagnostic use but also for the production of vaccines.
Most immunological diagnostic tests for the detection of SARS-CoV-2 antibodies are based on the use of the N protein, or the whole spike (S) glycoprotein and its shorter receptor binding domain (RBD) and external S1 segment (Freeman et al. 2020; Klumpp-Thomas et al. 2021; Rosendal et al. 2020). Regarding the latter, a first study estimated that between 2 and 4 µg of RBD per g of leaf fresh weight could be produced using N. benthamiana plants (Diego-Martin et al. 2020). Similarly, Rattanapisit and colleagues produced the RBD domain of SARS-CoV-2 by transient expression in N. benthamiana with a maximum expression yield of 8 μg/g at 3 days post-agroinfiltration (Rattanapisit et al. 2020), obtaining an antigen that showed binding specificity for the angiotensin-converting enzyme 2 (ACE2) receptor and thus demonstrating its utility as a diagnostic reagent. To this purpose, a portion of the nucleoprotein (N) derived from SARS CoV-2 was also produced in N. benthamiana plants and used to develop an indirect enzyme-linked immunosorbent assay (ELISA) for the detection of antibodies against SARS-CoV-2 in human sera showing very satisfactory results, with a calculated diagnostic sensitivity of 96.41% (Williams et al. 2021). Further studies demonstrated that higher yields could be obtained in N. benthamiana plants reaching 10 µg/g (Shin et al. 2021) and 25 µg/g (Siriwattananon et al. 2021) of the RBD antigen. Yet, transient expression in N. benthamiana of different variants of the RBD of SARS-CoV-2 S protein (Shin et al. 2021) showed suitability of plant-based platforms for the production of serological reagents to address future pandemic outbreaks.
The production of recombinant SARS-CoV-2 antigens in plants can be considered an excellent strategy also to obtain vaccines in short times and at low costs to counteract the spread of the virus more efficiently. Most of the currently available COVID-19 vaccines use antigens based on the whole S protein. Vaccine approaches that use only the RBD region as an antigen have the advantage of being able to determine a more focused immune response; in fact, the RBD domain is specifically responsible for the binding and entry of the virus into the cells. Several RBD antigens produced as vaccine candidates have been successful used in preclinical studies and have advanced in clinical trials (Kleanthous et al. 2021).
Recently, it has been demonstrated the possibility to obtain valid vaccine candidates against SARS-CoV-2 with recombinant antigens produced in plants (Mamedov et al. 2021; Maharjan et al. 2021). Recombinant antigens consisting of the RBD domain of SARS-CoV-2 have been expressed in N. benthamiana plants obtaining purification yields that would be sufficient for commercialization, and able to induce a high antibody response with a potent neutralizing activity of the SARS-CoV-2 virus when administered to mice. The immunogenicity of RBD has also been demonstrated in primates by administering a recombinant antigen produced as a fusion protein between the RBD domain and the Fc portion of a human IgG1 cloned into a viral vector for expression in plants of N. benthamiana (Siriwattananon et al. 2021). These results provide a solid foundation for the further development of plant-expressed RBD antigens to be used as vaccines for the prevention of COVID-19, which ultimately are safe and cost-effective. In the study carried out by Ceballo and coworkers (Ceballo et al. 2022), it has been proven once again the efficacy of transient expression of the RBD of the S protein of the SARS-CoV-2 virus in N. benthamiana plants, obtaining the correct assembly of two of the four disulfide bonds. Furthermore, despite having used a low dosage, it was demonstrated that the use of specific anti-RBD antibodies inhibit the binding of RBD to the ACE2 receptor, confirming the immunogenicity of the antigen produced in plants. Therefore, this study suggests that transient expression in N. benthamiana allows to obtain correctly folded antigens that could be used as diagnostic or vaccines candidates against the SARS-CoV-2 virus (Ceballo et al. 2022).
In the present work, two recombinant antigens (RBDw-Fc and RBDo-Fc) containing the RBD of the S protein from SARS-CoV-2 variants Wuhan and Omicron, which were fused to the Fc portion of a murine IgG2a, were rapidly and functionally produced in glyco-edited N. benthamiana plants. Their two N-linked glycosylation sites within the RBD domain showed a very homogeneous profile free from plant-typical sugars. The RBD-Fc antigens produced in plants were specifically recognised by the anti-SARS-CoV-2 human neutralizing monoclonal antibody J08-MUT. The RBDw-Fc antigen was successfully used in competitive ELISA experiments for binding to the ACE2 receptor with the aim to verify the neutralizing capacity of the serum from vaccinated patients.
Materials and methods
RBD-Fc constructs
The RBDw-Fc gene (GenBank accession number, OR839188) encoding the SARS-CoV2 Wuhan RBD (319-541 aa SARS-COV-2, 2019-nCoV GenBank accession number NC_045512.2) fused to a murine IgG2a antibody Fc region; and the RBDo-Fc gene (GenBank accession number, OR839189) encoding the SARS-CoV2 Omicron RBD lineage B.1.1.529 also fused to a murine IgG2a antibody Fc region, were synthesized and supplied by GenScript Corporation USA in the pUC57 vector. Codon-usage optimization for the expression in N. benthamiana was carried using the OptimumGeneTM algorithm (GenScript, Piscataway NJ, USA). The sequences were cloned into the plant expression binary vector pBI-Ω (Marusic et al. 2007) using the BamHI/EcoRI restriction sites, yielding plasmids pBI-Ω-RBDo-Fc and pBI-Ω-RBDw-Fc. Both sequences encoded the PR1 signal peptide sequence from tobacco, UniProtKB—P08299 (PR1A_TOBAC). The pBI-Ω-RBDo-Fc and pBI-Ω-RBDw-Fc constructs were transformed in Agrobacterium tumefaciens (LBA 4404). The pBI-Ωp19 bearing the p19 silencing suppressor gene from artichoke mottled crinckle virus (AMCV) was also used.
Plant growth and agroinfiltration
N. benthamiana plants were hydroponically grown on rockwool (Cultilene 7.5 × 7.5 cm) (Cultilene Grodan, Rijen the Netherlands) with nutrient solution at 1:200 dilution in water (Hydro Grow type nutrient solution, Growth Technology Ltd, Taunton, UK), at 24 °C, under LED lamps (Valoya AP673L, Helsinki, Finland) with a 16 h light and 8 h dark cycle. Expression was performed in fucosyl and xylosyl transferase Knock-out (FX-KO) N. benthamiana plants optimized for the protein glycosylation profile by genome editing (Jansing et al. 2019). Transient expression was performed by vacuum agroinfiltration with LBA4404 A. tumefaciens suspensions harboring RBDo-Fc or RBDw-Fc constructs together with the P19 silencing suppressor protein. Bacteria were pelleted by centrifugation at 4000×g, resuspended in infiltration buffer (10 mM MES, 10 mM MgCl2, pH 5.8), and suspensions were mixed (RBDo-Fc and P19 or RBDw-Fc and P19 at a 1:1 ratio) reaching a final OD600 of 0.5 for each construct (Lombardi et al. 2009). Plants at the 6–7 leaves stage were infiltrated in parallel by completely submerging them in the different Agrobacterium-containing solutions inside a vacuum chamber. Plants were the grown for another 6 days post-infiltration in the same conditions used before (light intensity of 140μmol m2s−1, with a 16 h light and 8 h dark cycle, at 24 °C). For antibody purification, batches of agroinfiltrated leaves (40 g) were collected and stored at − 80 °C before use.
SDS-PAGE, western blot analysis and ELISA
To quantify the functional RBDw-Fc antigen present in the plant purified product, an indirect ELISA experiment was performed by carrying out a coating experiment with different concentrations of the plant RBDw-Fc, and comparing results with those obtained with the commercial RBDw-mFc fusion protein containing a mouse Fc portion and produced in HEK293 cells (SARS-CoV-2 Wuhan S Protein RBDw-mFc Tag Recombinant—RP-87700 Thermo Fischer Scientific). For the coating with commercial RBDw-mFc or RBDw-Fc, serial dilutions were made starting from the concentration of 0.25 or 1 μg/well of total protein, respectively. Wells were washed twice with PBS/0.1% w/v Tween (PBST), once with PBS, and then blocked with 300 μl/well of a solution containing 4% v/v skimmed milk in PBS, for 2 h, at room temperature. A hundred μl of a solution containing the mAbJ08 (1 μg/ml) antibody, at a 1:5000 dilution in a solution containing 2% v/v skimmed milk in PBS, were added to the wells and incubated for 2 h, at room temperature. After washing, 100 μl of a secondary anti-γ antibody, goat anti-human IgG γ-chain specific (SIGMA A8419), conjugated to horseradish peroxidase (HRP), at a 1:5000 dilution in PBS containing 2% skimmed milk, was added and incubated for 1 h, at room temperature. The development solution containing the substrate 2,2’-azino-di-3-ethylbenzthiazoline sulfonate (ABTS, KPL) was added to the wells, and the colorimetric reaction was monitored after 10 and 20 min by spectrophotometric readings at a wavelength of 405 nm, using an ELISA reader (TECAN-Sunrise, Groedig, Austria).
ELISA experiments were carried out to test the recognition of purified recombinant RBDw-Fc products by the serum of vaccinated human subjects or mAbJ08 plant-produced nAb. Wells of a polystyrene plate (NUNC Maxisorb) were coated with RBDw-Fc (200 ng/well) or the commercial RBDw-mFc fusion protein containing a mouse Fc portion and produced in HEK293 cells (SARS-CoV-2 S Protein RBDw-mFc Tag Recombinant- RP-87700 Thermo Fischer Scientific), at 4 °C, overnight; the latter protein was used as a positive control. Commercial Rituximab (mAbRTX) (hcd20-mab1, Invivogen, San Diego, CA, USA) was used as a negative control. Wells were washed with PBST, PBS, and finally blocked with 300 μl/well of a solution containing 5% v/v skimmed milk in PBS for 2 h, at room temperature. Plates were washed again and 100 μl of the sera diluted 1:100 in a solution containing 3% v/v skimmed milk in PBS or 100 μl of mAbJ08 (1 μg/ml) at different dilutions were added to the wells, and incubated for 1 h, at 37 °C. Then, 100 μl of goat anti-human IgG γ-chain specific antibody (SIGMA A8419), conjugated to horseradish peroxidase (HRP), diluted 1:5000 in PBS containing 3% v/v skimmed milk were added and incubated for 1 h, at 37 °C. ELISA readings were obtained with ABTS and performed as described above.
A commercial competitive ELISA (SARS-CoV-2 Neutralizing antibody ELISA kit, Thermo Fisher Scientific, Waltham, MA, USA) was used to test the functionality of the plant produced RBDw-Fc, evaluating as control the capacity of the serum from vaccinated individuals or of the mAb675 plant-produced nAb to compete for binding to ACE2 with RBD, following the manufacturer’s instructions. Briefly, control mAbJ08 (10, 25, 100, 1000 and 5000 ng/well) and serum sample (1:100 dilution) from a vaccinated individual (2 doses of Pfizer–BioNTech vaccine) and pre-vaccine serum from the same individual (C–) were incubated in RBDw-Fc (150 ng/well) and RBD kit coated wells (100 μl) for 30 min, at room temperature. The wells were washed 3 times and 100 μl of biotinylated ACE2 were added; resulting mixtures were incubated for 30 min, at room temperature. The plate was washed, and 100 μl of streptavidin-HRP conjugate was added and incubated for 15 min, at room temperature and washed again. Hundred μl of substrate solution were added and incubated for 15 min before stopping the enzymatic reaction. Absorbance was measured at 450 nm on a microtitre plate reader (TECAN-Sunrise, Mannedorf, Switzerland).
Protein extraction and protein-A affinity chromatography
Protein extraction from agroinfiltrated leaves was performed by grinding 100 mg of leaf tissue using a pestle and by adding 200 μl of extraction buffer made of PBS containing protease inhibitors (Complete Mini, Roche). The commercial RBDw-mFc fusion protein (∼ 51 kDa) containing a mouse Fc portion (25 ng) and produced in HEK293 cells (SARS-CoV-2 S Protein RBDw-mFc Tag Recombinant- RP-87700 Thermo Fischer Scientific) was used as a positive control. The samples were centrifuged at 20,000×g for 20 min, at 4 °C, and the supernatant containing total soluble proteins was recovered. Protein concentration in each sample was evaluated by Bradford colorimetric assay (Bio-Rad, Hercules, CA, USA). Plant extracts separated on 12% T SDS-PAGE acrylamide gels were analysed by Western blotting. Proteins were electrotransferred onto a PVDF membrane (Trans-Blot Turbo Mini 0.2 µm PVDF Transfer Packs, Bio-Rad, Hercules, CA, USA) using the Trans-Blot® Turbo™ Transfer System (1704150, Bio-Rad, Hercules, CA, USA). Membranes were blocked with PBS containing 4% v/v milk, overnight. For detection of RBDw-Fc and RBDo-Fc protein, the membrane was incubated with an anti-mouse-Fc HRP KPL (4741802) conjugated antibody, at a dilution of 1:5000 in PBS containing 2% v/v skimmed milk, for 1 h, room temperature, or with the anti-SARS antibody CoV-2 mAbJ08-MUT (1 μg/ml) (Frigerio et al. 2022), at a dilution of 1:5000 in PBS containing 2% v/v skimmed milk, for 2 h, room temperature. The membrane incubated with mAbJ08-MUT, was incubated again with the HRP-conjugated goat anti-human IgG γ-chain specific antibody (Sigma A8419), at a dilution of 1:5000 in PBS containing 2% v/v skimmed milk, for 1 h, room temperature. Proteins were detected by enhanced chemioluminescence (ECL™ Prime Western Blotting System, Merck, Darmstadt, Germany) with the Invitrogen iBright CL1500 imaging system (Thermo Fisher Scientific).
Plant tissues frozen in liquid nitrogen were ground into a powder by use of pestle and mortar adding 3.5 g of polyvinylpolypyrrolidone (PVPP) to 40 g of leaf tissue. Extraction buffer (2 ml/g of leaves) consisting of PBS containing protease inhibitor cocktail (CompleteTM Roche, Basel, Switzerland) and ascorbic acid (0.25 g in 80 ml) was added; resulting mixtures were homogenized with an Ultra-Turrax homogenizer T25 (IKA, Staufen, Germany). The slurry was filtered through Miracloth pore size 22–25 µm (Sigma-Aldrich, St. Louis, MO, USA), and clarified by a double centrifugation at 8000 × g for 20 min, at 4 °C. The supernatant was loaded onto a protein-A affinity column (1 ml HiTrapTM Protein-A FF, GE Healthcare, Chicago, IL, USA), at a flow rate of 1 ml/min. The column was washed with 10 vol of PBS, and each antibody was eluted with 200 mM Tris-HCl, 100 mM glycine, pH 3.0. Eluted fractions (0.5 ml each) were neutralized to about pH 7.0 with 100 μl of 1 M Tris-HCl pH 9.5. The eluted proteins were dialyzed/concentrated in PBS by ultrafiltration with Vivaspin® 5000 MWCO HY concentrator. Antibody concentration was finally determined by measuring the corresponding absorbance at 280 nm, and the corresponding purity was evaluated by SDS-PAGE followed by Coomassie blue staining.
Proteomic and N-linked glycosylation analysis
Purified proteins were separated on a non-reducing 10% T SDS-PAGE. The gel was stained with colloidal Coomassie, and the protein bands were excised, triturated, in-gel reduced and S-alkylated with iodoacetamide, and finally digested with trypsin (Roche, Basel, Switzerland) (Salzano et al. 2013). An aliquot of the peptide mixture was directly analysed by MALDI-TOF-MS on an Ultraflextreme instrument (Bruker Daltonics, Billerica, MA, USA) operating in reflectron mode (acquisition range m/z 400–6200, pulsed ion extraction 100 ns, laser frequency 1000 Hz). 2,5-Dihydroxy-benzoic acid (10 mg/ml in 50% v/v acetonitrile, 0.1% v/v trifluoroacetic acid) was used as matrix and mixed 1:1 to the sample before loading on the instrument target. Mass spectra were calibrated externally using Peptide Calibration Standard II (Bruker Daltonics, Billerica, MA, USA) and elaborated using the FlexAnalysis software (Bruker Daltonics).
Peptides were also extracted from the gel particles using 5% formic acid/acetonitrile (1:1 v/v), and desalted by using μZipTipC18 pipette tips (Millipore, Burlington, MA, USA). They were finally analyzed by nanoLC-ESI-Q-Orbitrap MS/MS using a LTQ XL Q-ExactivePlus mass spectrometer equipped with a Nanoflex ion source and connected to an UltiMate 3000 HPLC RSLC nano system-Dionex (Thermo Fisher Scientific, Waltham, MA, USA). Peptides were resolved on an Acclaim PepMap RSLC C18 column (150 mm-length × 75 μm-internal diameter, 2 μm-particle size, and 100-Å pore size) (Thermo Fischer Scientific, Waltham, MA, USA) as previously reported (Lonoce et al. 2019). Full mass spectra were acquired in the range m/z 375–1500, with nominal resolution 70,000 using a data dependent scanning procedure over the eight most abundant ions, using 20 s-dynamic exclusion. Mass isolation window and collision energy were set to m/z 1.2 and 28%, respectively.
MS/MS data were searched with Byonic™ software (v.2.6.46) (Protein Metrics, Cupertino, USA) against a database containing the sequence of the recombinant RBDw-Fc and RBDo-Fc and common contaminants. Searching parameters were trypsin as cleavage specificity, allowing also semi-specific cuts and 2 missed cleavages as maximum value, Cys carbamidomethylation as fixed modification, and Met oxidation, N-terminal Gln/Glu cyclization, Asn/Gln deamidation, and Asn N-glycosylation as variable modifications. Mass tolerance values for peptide matches were set to 10 ppm and 0.05 Da for precursor and fragment ions, respectively. A homemade glycan database containing common biantennary structures and typical plant N-linked glycoforms was used for identification of glycopeptides. Score thresholds for accepting peptide and glycopeptide identifications were Byonic™ score > 150. Glycopeptide identifications were manually validated to assign the glycoforms. The chromatographic area corresponding to glycopeptides EDYNSTLR, FPNITNLCPF, FPNITNL, GEVFNATR and DEVFNATR, and the matching unmodified species were calculated extracting the three most abundant ions observed in the corresponding spectra. The relative percentage of each glycoform was calculated as the ratio between the extracted ion area of the glycopeptide and the total extracted ion area of unmodified species and all corresponding glycopeptides. Two technical replicates were analysed for each antibody sample.
Statistical analysis
Statistical analysis was performed using GraphPad Prism for Windows (GraphPad Software, La Jolla, CA, USA, www.graphpad.com). A two-tailed Student’s t-test Welch’s correction for unpaired samples was used; α-level of 0.05 was used as threshold. P-values < 0.05 were considered statistically significant. F-test was used to compare variances.
Results
Cloning and expression of RBD-Fc recombinant antigens in N. benthamiana
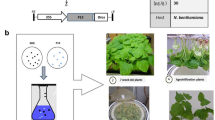
The sequences encoding for the RBD-Fc antigens were plant-codon-optimized and cloned under the control of the cauliflower mosaic virus 35S promoter (35S), translation enhancer sequence Ω and the terminator of nopaline synthase (nos t), and the resulting constructs pBI-RBDw-Fc and pBI-RBDo-Fc were used to transform A. tumefaciens (Fig. 1a). Schematic representation of RBD-Fc antigens is illustrated in Fig. 1b. In RBDw-Fc, the LEVLFQGP sequence of 8 amino acids, which is recognised by the human rhinovirus (HRV) 3C protease, was added between the RBD and the Fc hinge region (Fig. 1a); in RBDo-Fc, this sequence was omitted. N. benthamiana plants were vacuum infiltrated with a mix of A. tumefaciens strains carrying pBI-RBDw-Fc or pBI-RBDo-Fc and pBI-P19 constructs (Fig. 1b). As negative control, a group of plants was infiltrated only with Agrobacterium bearing pBI-P19.

Schematic representation of the constructs used for plant agroinfiltration. a pBI-RBDw-Fc: construct encoding the SARS-CoV2 Wuhan RBD (R319-F541 aa SARS-COV-2, 2019-nCoV GenBank accession number NC_045512.2) fused to a murine IgG2a antibody Fc region; pBI-RBDo-Fc: construct encoding the SARS-CoV2 Omicron RBD (R319-K529 aa SARS-CoV-2 S Omicron Spike B.1.1.529) fused to a murine IgG2a antibody Fc region; pBI-P19: construct encoding the P19 silencing suppressor protein from Artichoke Mottled Crinkle Virus (AMCV). The sequences are under the control of the cauliflower mosaic virus 35S promoter (35S), Ω translational enhancer sequence and nopaline synthase terminator (nos t). (L): signal peptide sequence from tobacco, PR1 UniProtKB—P08299 (PR1A_TOBAC). HRV3C: LEVLFQGP Human rhinovirus protease recognition site. b Schematic, structural representation of the RBD-Fc antigen fusions.
Expression and characterisation of RBD-Fc antigens
Leaves were typically sampled 5 days after agroinfiltration, and extraction of total soluble proteins (TSPs) was performed. The extracts were normalized for the total amount of TSPs and used to evaluate RBDo-Fc and RBDw-Fc antigens expression (Fig. 2a). Western blot analysis performed under non-reducing conditions, using an anti-mouse Fc, allowed to detect the presence of a band at about 110 kDa, which corresponds to the RBDo-Fc protein in dimeric form, also visible in the commercial RBDw-mFc protein used as positive control (C +), and three other bands at lower molecular weights probably due to unassembled monomeric antigen (~ 60 kDa) and to degradation fragments (Fig. 2a, left panel). In the case of RBDw-Fc (Fig. 2a, right panel), the band at about 110 kDa was less intense than the degradation bands, indicating a lower stability of the dimeric form and a higher proteolytic degradation propensity compared to the RBDo-Fc antigen.

Expression and purification of the RBD-Fc antigens. a 12% T SDS-PAGE analysis of RBDo-Fc and RBDw-Fc plant extracts under non-reducing conditions followed by Western Blotting using anti-mouse Fc conjugated to HRP (20 μl of extract normalised for TSPs). RBD-Fc-P: purified plant protein (1.32 μg). C + : commercial RBDw-mFc protein (125 ng). P19: extract of agro-infiltrated leaves with only the P19 construct used as a negative control (20 μl). M: molecular weight marker. b Analysis of RBDw-Fc and RBDo-Fc (15 µl ≈5 µg) purified from plant by 12% T SDS-Page under reducing (R) and non-reducing (NR) conditions, followed by staining with colloidal Coomassie
The purification of both RBD-Fc antigens was carried out by Protein A-affinity chromatography obtaining yields of 35.8 ± 5.8 and 31.6 ± 4 mg/kg for RBDw-Fc and RBDo-Fc, respectively. Purified RBD-Fc antigens were assayed by 12% T SDS-PAGE analysis under reducing and non-reducing conditions, which was followed by protein staining with colloidal Coomassie (Fig. 2b). In the reducing gel, two main bands were identified; the first one related to the intact antigen in its monomeric form (~ 60 kDa), which was less intense in RBDw-Fc, and a very intense second one at a molecular mass of ~ 30 kDa. This result indicated a significant degradation of the RBD-Fc purified antigens (Fig. 2b, left panel). On the other hand, a major band was identified in the non-reducing gel (indicated by an asterisk), which was related either to a proteolytic product or to the intact antigen in its monomeric form (~ 60 kDa). Another significant band also occurred in the non-reducing gel at a molecular mass around 110 kDa (indicated with an arrow), which was associated with the intact antigen in its dimeric form; the latter band appeared more intense in the case of RBDo-Fc. Other two less intense bands were also visible at a molecular mass of almost 30 and 20 kDa, which were probably due to additional degradation fragments (Fig. 2b, right panel). In the RBDo-Fc sample, a band at around 90 kDa was also visible.
Protein bands corresponding to the intact antigens (with a molecular mass of 110 kDa) or the corresponding degradation fragments (with a molecular mass of 20 kDa) were excised from the electrophoretic gel run under non-reducing conditions and directly subjected to trypsin digestion. The corresponding extracted peptides were directly analyzed by MALDI-TOF–MS, and the resulting spectral signals were interpreted based on the theoretical mass values of the expected tryptic peptides. For both antigens, the analysis of the molecular species with a mass of 110 kDa showed a full coverage of the entire protein sequence, indicating their correspondence to the intact dimeric forms, and also confirming the correct processing of their signal peptides (Supplementary Fig. 1). Instead, for the lowest molecular mass fragments with a mass of about 20 kDa, the coverage of the sequence for both antigens started downstream of the CPPCKC segment within the hinge region that allows Fc dimerization, thus indicating their correspondence to the corresponding C-terminal fragments in monomeric form (Supplementary Fig. 1).
Quantification of the intact RBDw portion in the plant-purified RBDw-Fc antigen was performed by ELISA using the RBD specific neutralising mAbJ08 against the Wuhan variant. The commercial RBDw-mFc fusion protein produced in HEK293 cells was used as a standard. Results showed that in 1 μg of plant purified antigen, 110 ng were represented by the RBDw portion specifically recognised by mAbJ08 (Supplementary Fig. 2).
Analysis of N-linked glycosylation in RBDw-Fc and RBDo-Fc
For both antigens, glycosylation analysis was performed on the bands corresponding to the intact protein species (~ 110 kDa). The glycoforms reported in Fig. 3 refer to the glycosylation at the N residue in the Fc domain as well as at N13 and N25 within the RBD domain of RBDw-Fc and RBDo-Fc. For simplicity, this numbering was given referring to the first amino acid residue (R319) of the RBD domain of both antigens. The glycopeptides EDYNSTLR in the Fc of both antigens, FPN13ITNLCPF and GEVFN25ATR in RBDw-Fc, FPN13ITNL and DEVFN25ATR in RBDo-Fc were identified in the tryptic digests of the corresponding bands after nLC-ESI-Q-Orbitrap MS/MS analysis and Byonic search of the resulting data. In the Fc domain of both antigens, no forms of plant-typical sugars (containing β1,2-xylose and α1,3-fucose) were detected, and the most abundant glycoform was represented by the complex sugar GlcNAc(4)Man(3). The same glycoform was the most representative glycoform present at N13 and N25 within the RBD of both antigens (red box in Fig. 3), having a relative percentage abundance of about 76.63% and 75.54% in RBDw-Fc, and of 84.95% and 89.62% in RBDo-Fc, respectively. In the RBD domain of RBDw-Fc, a α1,3-fucose-containing glycoform at N25 was selectively observed, with an abundance of 2.71%; the same amino acid also showed the presence of the high-mannose forms GlcNAc(4)Man(8) (12.43%) and GlcNAc(4)Man(9) (5.23%).

N-glycosylation analysis of the band corresponding to the intact RBD-Fc antigens. a Peptide EDYNSTLR in the mouse Fc region. b Peptides FPN13ITNLCPF and FPN13ITNL in the RBD domain of RBDw-Fc and RBDo-Fc, respectively. c Peptide GEVFN25ATR and DEVFN25ATR in the RBD domain of RBDw-Fc and RBDo-Fc, respectively
Western blot analysis of RBDw-Fc and RBDo-Fc with a potent SARS-CoV-2 neutralising antibody
The ability of the purified antigens to be recognized by the anti-SARS-CoV-2 monoclonal antibody mAbJ08- MUT (Andreano et al. 2021), which is specific for the receptor binding domain (RBD) of S protein of different SARS-CoV-2 strains, was then evaluated by Western blot analysis (Fig. 4). When corresponding electrophoresis was performed under non-reducing conditions, a band was detected in both samples at about 110 kDa, corresponding to the proteins in their dimeric form. A more intense signal was observed in RBDo-Fc, where an additional band at around 90 kDa was also visible. A major band migrating at around 60 kDa was also present in both RBDw-Fc and RBDo-Fc. Under reducing conditions, the presence of a band related to the intact monomeric antigen was present in both samples (~ 60 kDa), together with a more intense band around 30 kDa attributable to its degradation product. Commercial RBDw-mFc protein produced in HEK cells (C+) was used as positive control.

Western blot analysis under non-reducing (NR) and reducing (R) conditions using the anti-SARS-CoV-2 mAbJ08. RBDw-Fc Receptor Binding Domain of the Spike protein of the SARS-CoV2 Wuhan variant purified from plants (500 ng), RBDo-Fc Receptor Binding Domain of the Spike protein of the SARS-CoV2 Omicron variant purified from plants (500 ng). Commercial RBDw-mFc protein (200 ng) was used as a positive control
Recognition of RBDw-Fc by the serum from vaccinated human individuals
The RBDw-Fc antigen was first tested for its recognition by the mAbJ08 antibody through ELISA experiments. The antigen was coated on ELISA plate (200 ng/well) and then incubated with mAbJ08 (stock solution 1 mg/ml) at 3 different dilutions (1:2000, 1:4000 and 1:8000), using mAb RTX antibody (1 mg/ml) at the same dilutions or PBS as negative controls. Detection occurred after incubation with a secondary HRP-conjugated anti-human IgG antibody (Fig. 5a). Results showed a dilution-dependent specific binding by mAbJ08.

Indirect ELISA testing the recognition of RBDw-Fc by the anti-SARS-CoV-2 neutralising antibody mAbJ08 and by the serum of vaccinated human individuals. a The coating of the plate was obtained with RBDw-Fc at a concentration of 200 ng/well. The mAbJ08 (1 mg/ml) was then added as a primary antibody at 3 different dilutions (1:2000, 1:4000, 1:8000) and an anti-human IgG antibody conjugated to HRP as a secondary antibody. A plant-produced antitumor antibody mAb RTX (1 mg/ml) at 3 different dilutions (1:2000, 1:4000, 1:8000) and PBS were used as negative controls. b The coating of the plate was done with RBDw-Fc and with commercial RBD-mFc fusion protein (∼ 51 kDa) containing a mouse Fc portion and produced in HEK293 cells, both at a concentration of 200 ng/well. Sera were then added (1:100 dilution), comparing them with mAbJ08 (1 mg/ml) diluted 1:2500 used as a positive control and with the serum from unvaccinated subjects used as a negative control (C-Serum 1 and C-Serum 2). Finally, an anti-human IgG antibody conjugated to HRP (1:7500) was added. Values are the mean ± standard deviation (SD) (n = 3). Unpaired two-tailed Student’s t-test. *p < 0.05 for negative control serum from unvaccinated individuals compared to all the sera from vaccinated subjects and mAbJ08 for both RBDw-Fc and RBDw-mFc
The RBDw-Fc antigen was also tested for its recognition by antibodies present in the serum of vaccinated subjects (Fig. 5b). RBDw-Fc and commercial RBDw-mFc were used to coat ELISA plates (200 ng/well) and incubated with 1:100 diluted sera from human individuals vaccinated with 2 doses of Pfizer–BioNTech vaccine. Pre-vaccine serum from two of the same donors were used as negative controls. All the sera from vaccinated humans specifically recognized the RBDw-Fc antigen, with serum C showing slightly reduced ELISA signals. All the sera from vaccinated humans showed a statistically significant difference compared to the pre-vaccine serum 1 and 2 used as a control. Moreover, no significant difference was found in the recognition of RBDw-Fc and the commercial RBDw-mFc by the serum from vaccinated subjects or mAbJ08 used as control (Fig. 5b).
Neutralization assay based on RBD-binding competition with ACE2
The neutralizing capacity of antibodies present in the serum of vaccinated subjects and of the mAbJ08 antibody at different concentrations was also tested based on RBD-binding competition with ACE2. To this aim, an ELISA was performed using in parallel a coated plate with RBD provided by a commercial kit (SARS-CoV-2 Neutralizing antibody ELISA kit, ThermoFisher Scientific) and a plate coated with plant-purified RBDw-Fc. Samples with sera and mAbJ08 were incubated with excess amounts of biotinylated ACE2, and any ACE2 bound to the RBD was assayed with Streptavidin-HRP conjugate antibody. The results showed that the serum form the vaccinated individuals and the mAbJ08 dilutions (100 ng, 1000 ng and 5000 ng) had a statistically significant signal reduction compared to the C− Serum and C− kit controls (Fig. 6), indicating a strong binding competition to RBD with ACE2. No significant difference was noted between the plant-produced RBDw-Fc and the antigen provided by the kit. Furthermore, signal reduction with 100 ng of mAbJ08 was comparable to that observed for the vaccinated serum A and D at 1:100 dilution (Fig. 6).

Competitive ELISA using the plant derived RBDw-Fc was performed to assay the neutralization capacity of serum from vaccinated individuals using an anti-SARS-CoV-2 mAbJ08 as control. ELISA experiments were performed using in parallel a plate on which the RBD produced in mammalian cells provided by the kit had been immobilized (SARS-CoV-2 Neutralizing antibody ELISA kit, ThermoFisher Scientific) and a plate coated with RBDw-Fc (150 ng/well). The serum from vaccinated subjects (Vacc. Serum A, C and D), and the serum from a non-vaccinated subject (C-Serum) were added to the plates as a negative control (1:100), and mAbJ08 at different concentrations was added as a control (from 5 μg to 10 ng/well). Values are the mean ± standard deviation (SD) (n = 2). Unpaired two-tailed Student’s t-test, p < 0.05 for Vacc. Serum A, C, D, mAbJ08-MUT 100 ng, mAbJ08-MUT 1 μg and mAbJ08-MUT 5 μg versus C-serum for both RBDw-Fc and RBD kit
Discussion
The aim of this work was the production in plants of recombinant antigens containing the receptor binding domain (RBD) of the S protein from SARS-CoV-2. In the recent literature, several examples are reported on the production of the RBD domain of the Wuhan variant in N. benthamiana plants using different transient expression systems, many of which generally showing low protein yields are. For example, Rattanapisit et al. (2021) expressed a long version of the RBD (F318-C617) obtaining yields of 8 mg/g fresh weight, while slightly higher levels (10–20 mg/kg) were obtained for a shorter RBD variant (R319-S591) obtained using a virus-based transient expression vector (Mamedov et al. 2021). Even a shorter version of the RBD (R319-F541) was produced in plants (Diego-Meartin et al. 2020; Shin et al. 2021); however, this recombinant product showed even lower expression yields due to homodimer formation and protein aggregation (2–4 mg/kg fresh weight). Best results were obtained when expressing a truncated version of RBD (R319-L533) lacking C538; this recombinant product accumulated at levels of about 100 mg/kg fresh weight, showing that the yields of soluble antigen in plants may be enhanced by hindering the formation of RBD multimers (Shin et al. 2021).
With the aim of facilitating antigen purification and stabilization, in the present study we chose to fuse the RBD domain to the constant region of a murine immunoglobulin. Indeed, full-length immunoglobulins as well as recombinant antibody fragments are among the most stable and efficiently expressed proteins in plants (Donini and Marusic 2019). The choice of a murine IgG rather than a human IgG was necessary to avoid a certain background in diagnostic assays involving the use of an anti-human IgGs as a secondary antibody. The fusion with the constant moiety of an immunoglobulin generally allows for higher yields of plant-produced antigens, promotes the correct folding of the recombinant protein and simplifies its purification, as demonstrated by our group (Rage et al. 2019). For example, a recombinant chicken infectious bursal disease virus (IBDV) antigen was efficiently produced in N. benthamiana plants by fusing the immunodominant projection domain (PD) of the viral structural protein VP2 with the constant region of an avian IgY (FcY).
Western blot analysis of plant extracts expressing the two RBD-Fc antigens, performed using the plant-produced mAbJ08 as primary antibody, specific for the RBD domain, showed that the recombinant proteins were accumulated in plant cells and were able to form dimers with a molecular mass of about 110 kDa, which derived from the association of two Fc portions. An intense band at 60 kDa was also present for both antigens, indicating possible formation of unassembled monomers and/or degradation products. In the case of RBDw-Fc, a very intense band at 30 kDa was also observed, indicating a significant proteolytic degradation that was tentatively associated with the presence in this construct of the LEVLFQGP recognition site for the HRV 3C protease; this segment is located between the RBD and the Fc, just upstream of the hinge region. This sequence was inserted into the construct with the aim to obtain the RBD domain alone by eliminating the Fc portion, after the treatment of the purified antigen with the recombinant HRV 3C protease. Based on obtained results, it is possible to hypothesize that this HRV 3C protease recognition site can be specifically recognized also by plant cysteine proteases, also explaining the selective proteolysis observed for RBDw-Fc compared to RBDo-Fc. This result was also in perfect agreement with previous studies demonstrating that this sequence is strongly and specifically proteolyzed in plants (Jutras et al. 2018). It is also well known from previous studies that the hinge region of antibodies itself is particularly susceptible to proteolysis in plants (Donini et al. 2015), explaining the formation of several degradation products in both antigens. A previous work demonstrated that the expression of the SlCYS8 Cys protease inhibitor had a strong protective effect in specific regions of the heavy chain domains of antibodies (Jutras et al. 2016). In the future, the same approach could be assayed to increase the yield and quality of RBD-Fc fusions. Specific in planta proteolytic degradation was confirmed also by analysing the purified recombinant products, in which a major band with a molecular mass of about 20 kDa was present under both reducing and non-reducing conditions. Proteomic analysis allowed to characterise the nature of this component, which corresponded to a C-terminal fragment starting downstream of the CPPCKC sequence of the mouse Fc, confirming the susceptibility of the hinge region to plant proteases. Antigen purification carried out using protein A-chromatography showed similar yields for both RBDw-Fc and RBDo-Fc, with levels of 35 mg/kg of total purified protein. In the case of RBDw-Fc, using a RBD specific antibody, we quantified the fraction of intact and functional RBD that was present in the purified recombinant product; this percentage part corresponded to about 11% of the total purified protein. In the whole, the final yield of about 3.5 mg/kg of functional RBD was comparable to those reported in literature for other recombinant antigens, which contained the RBD alone or in fusion to a human Fc (Shin et al. 2021; Siriwattananon et al. 2021; Rattanapisit et al. 2021).
Genome edited plants were used for producing both antigens (Jansing et al. 2019), in which the genes coding for the enzymes responsible for the addition of α1,3-fucose and β1,2-xylose (typical plant sugars) were inactivated. This was done with the aim of obtain recombinant proteins with a glycosylation profile very similar to those eventually obtained in mammalian cells. Even if the two N-linked glycosylation sites present in the RBD are not part of the receptor-binding motif and are not involved in receptor binding (Walls et al. 2020; Yang et al. 2020), their post-translational modifications play an essential role in the S protein since it was demonstrated influencing either the correct folding of the above-reported domain and its successful expression in plants (Shin et al. 2021). Moreover, it was recently shown that a plant produced glycoengineered RBD with a human-type complex N-glycan profile with core α1,6-fucose was more reactive towards the neutralizing antibody S309 (Ruocco et al. 2023). Glycosylation analysis of the plant-derived RBDw-Fc and RBDo-Fc showed that the glycoforms present at N13 and N25 within the corresponding RBD domain had a similar and homogeneous modification profile. Both were substantially lacking plant-typical sugars and contained the complex sugar GlcNAc(4)Man(3) as the most abundant N-linked glycoform. A similar result in terms of the glycosylation profile of the RBD produced in N. benthamiana was previously obtained by Yun-Ji Shin and collegues (Shin et al. 2021), which also demonstrated the functionality of the antigen that successfully reacted with the serum of convalescent COVID-19 patients and bound to ACE2. Worth mentioning is also the fact that the RBD domain obtained in other mammalian expression systems, such as Chinese hamster ovary and human embryonic kindney cells, almost exclusively presented complex sugars N-linked at N13 and N25, as already shown by Tian and coworkers (Tian et al. 2021).
ELISA experiments demonstrated that recombinant RBDw-Fc antigen produced in plants was specifically recognized by the mAbJ08 antibody neutralizing the SARS-CoV-2 virus and by neutralizing antibodies present in the serum of subjects vaccinated with a double dose of Comirnaty vaccine (Pfizer/BioNTech). Results showed that the latter sera recognised RBDw-Fc at the same extent as the commercial RBD-mFc fusion protein produced in HEK293 cells, both at a concentration of 200 ng/well, thus indicating that the plant-produced antigen with homogeneous glycosylation is able to fold into its correct conformation and is fully functional. Similar results were obtained in a previous work in which a recombinant antigen, obtained by fusing the RBD to a mouse-Fc, was produced in CHO cells and used to set-up an indirect ELISA assay to test the serum from SARS-CoV-2-positive human subjects (Frumence et al. 2021).
In the present work, the neutralizing capacity of the mAbJ08 antibody and of the serum from vaccinated individuals was evaluated using a commercially available competition ELISA kit that assesses interference of antibodies on the binding of ACE2 to RBD domain-coated wells. This assay was also conducted in parallel by using ELISA plates coated with the plant-produced RBDw-Fc antigen. The results obtained with the latter antigen showed no substantial differences compared to those achieved with the commercial kit, confirming the possibility of using plant-derived RBDw-Fc both as a reagent in diagnostic assays for the detection of antibodies and in neutralization tests based on in vitro competition for ACE2 binding. Another study already compared the binding affinity of the conformation dependent anti-RBD antibody CR3022 and recombinant ACE2 to a plant-produced RBD versus a RBD produced in mammalian cells showing comparable affinity, thus indicating that the plant-derived antigen was correctly folded and fully functional (Shin et al. 2021).
The development of new strategies to rapidly produce antigens to be used in serological assays is becoming a high priority to face emerging epidemics. To this aim, we have reported here the production in plants of two antigens containing a murine Fc fused to the RBD domain of the SARS-CoV-2from Wuhan and Omicron variants. We have here demonstrated that these recombinant antigens can be rapidly produced with a mammalian-like homogeneous glycosylation profile and efficiently purified from plants, even if the specific proteolysis at the hinge region by plant proteases can reduce their overall yield. The Wuhan RBDw-Fc antigen performed well in recognition by a SARS-CoV-2 neutralising antibodies and was used to set-up an ELISA test to evaluate the neutralization efficiency of the serum from vaccinated subjects. In conclusion, the plant production of antigens to fused to murine Fc can represent a valid strategy to rapidly develop novel serological assays for diagnostic purposes.
Data availability
The data used to support the findings of this study are provided within this article. Further information can be provided by the corresponding author upon request.
References
Andreano E, Nicastri E, Paciello I, Pileri P, Manganaro N, Piccini G, Manenti A, Pantano E, Kabanova A, Troisi M, Vacca F, Cardamone D, De Santi C, Torres JL, Ozorowski G, Benincasa L, Jang H, Di Genova C, Depau L, Brunetti J, Agrati C, Capobianchi MR, Castilletti C, Emiliozzi A, Fabbiani M, Montagnani F, Bracci L, Sautto G, Ross TM, Montomoli E, Temperton N, Ward AB, Sala C, Ippolito G, Rappuoli R (2021) Extremely potent human monoclonal antibodies from COVID-19 convalescent patients. Cell 184:1821–1835. https://doi.org/10.1016/j.cell.2021.02.035
Capell T, Twyman RM, Armario-Najera V, Ma JK, Schillberg S, Christou P (2020) Potential applications of plant biotechnology against SARS-CoV-2. Trends Plant Sci 25:635–643. https://doi.org/10.1016/j.tplants.2020.04.009
Ceballo Y, López A, González CE, Ramos O, Andújar I, Martínez RU, Hernández A (2022) Transient production of receptor- binding domain of SARS-CoV-2 in Nicotiana benthamiana plants induces specific antibodies in immunized mice. Mol Biol Re 49:6113–6123. https://doi.org/10.1007/s11033-022-07402-4
Diego-Martin B, González B, Vazquez-Vilar M, Selma S, Mateos-Fernández R, Gianoglio S, Fernández-del-Carmen A, Orzáez D (2020) Pilot production of SARS-CoV-2 related proteins in plants: a proof of concept for rapid repurposing of indoor farms into biomanufacturing facilities. Front Plant Sci 11:612781. https://doi.org/10.3389/fpls.2020.612781
Donini M, Marusic C (2019) Current state-of-the-art in plant-based antibody production systems. Biotechnol Lett 41:335–346. https://doi.org/10.1007/s10529-019-02651-z
Donini M, Lombardi R, Lonoce C, Di Carli M, Marusic C, Morea V, Di Micco P (2015) Antibody proteolysis: a common picture emerging from plants. Bioengineered 6:299–302. https://doi.org/10.1080/21655979.2015.1067740
Freeman B, Lester S, Mills L, Rasheed MAU, Moye S, Abiona O, Hutchinson GB, Morales-Betoulle M, Krapinunaya I, Gibbons A, Chiang CF, Cannon D, Klena J, Johnson JA, Owen SM, Graham BS, Corbett KS, Thornburg NJ (2020) Validation of a SARS-CoV-2 spike protein ELISA for use in contact investigations and serosurveillance. bioRxiv. https://doi.org/10.1101/2020.04.24.057323
Frigerio R, Marusic C, Villani ME, Lico C, Capodicasa C, Andreano E, Paciello I, Rappuoli R, Salzano AM, Scaloni A, Baschieri S, Donini M (2022) Production of two SARS-CoV-2 neutralizing antibodies with different potencies in Nicotiana benthamiana. Front Plant Sci 13:956741. https://doi.org/10.3389/fpls.2022.956741
Frumence E, Lebeau G, Viranaicken W, Dobi A, Vagner D, Lalarizo-Rakoto M, Sandenon-Seteyen AL, Giry C, Septembre-Malaterre A, Raffray L, Gasque P (2021) Robust and low-cost ELISA based on IgG-Fc tagged recombinant proteins to screen for anti-SARS-CoV-2 antibodies. J Immunol Methods 495:113082. https://doi.org/10.1016/j.jim.2021.113082
Jansing J, Sack M, Augustine SM, Fischer R, Bortesi L (2019) CRISPR/Cas9-mediated knockout of six glycosyltransferase genes in Nicotiana benthamiana for the production of recombinant proteins lacking β-1,2-xylose and core α-1,3-fucose. Plant Biotechnol J 17:350–361. https://doi.org/10.1111/pbi.12981
Jutras PV, Marusic C, Lonoce C, Deflers C, Goulet MC, Benvenuto E, Michaud D, Donini M (2016) An accessory protease inhibitor to increase the yield and quality of a tumour-targeting mAb in Nicotiana benthamiana leaves. PLoS ONE 11:e0167086. https://doi.org/10.1371/journal.pone.0167086
Jutras PV, Goulet MC, Lavoie PO, D’Aoust MA, Sainsbury F, Michaud D (2018) Recombinant protein susceptibility to proteolysis in the plant cell secretory pathway is pH-dependent. Plant Biotechnol J 16:1928–1938. https://doi.org/10.1111/pbi.12928
Kleanthous H, Silverman JM, Makar KW, Yoon IK, Jackson N, Vaughn DW (2021) Scientific rationale for developing potent RBD-based vaccines targeting COVID-19. NPJ Vaccines 6:128. https://doi.org/10.1038/s41541-021-00393-6
Klumpp-Thomas C, Kalish H, Drew M, Hunsberger S, Snead K, Fay MP, Mehalko J, Shunmugavel A, Wall V, Frank P, Denson JP, Hong M, Gulten G, Messing S, Hicks J, Michael S, Gillette W, Hall MD, Memoli MJ, Esposito D, Sadtler K (2021) Standardization of ELISA protocols for serosurveys of the SARS-CoV-2 pandemic using clinical and at-home blood sampling. Nat Commun 12:113. https://doi.org/10.1038/s41467-020-20383-x
Lico C, Santi L, Baschieri S, Noris E, Marusic C, Donini M, Pedrazzini E, Maga G, Franconi R, Di Bonito P, Avesani L (2020) Plant molecular farming as a strategy against COVID-19—the Italian perspective. Front Plant Sci 1:609910. https://doi.org/10.3389/fpls.2020.609910
Lombardi R, Circelli P, Villani ME, Buriani G, Nardi L, Coppola V, Bianco B, Benvenuto E, Donini M, Marusic C (2009) High-level HIV-1 Nef transient expression in Nicotiana benthamiana using the P19 gene silencing suppressor protein of artichoke mottled crinckle virus. BMC Biotechnol 9:96
Lonoce C, Marusic C, Morrocchi E, Salzano AM, Scaloni A, Novelli F, Pioli C, Feeney M, Frigerio L, Donini M (2019) Enhancing the secretion of a glyco-engineered anti-CD20 scFv-Fc antibody in hairy root cultures. Biotechnol J 14:e1800081. https://doi.org/10.1002/biot.201800081
Maharjan PM, Cheon J, Jung J, Kim H, Lee J, Song M, Jeong GU, Kwon Y, Shim B, Choe S (2021) Plant-expressed receptor binding domain of the SARS-CoV-2 spike protein elicits humoral immunity in mice. Vaccines 9:978. https://doi.org/10.3390/vaccines9090978
Mamedov T, Yuksel D, Ilgın M, Gurbuzaslan I, Gulec B, Yetiskin H, Uygut MA, Islam Pavel ST, Ozdarendeli A, Mammadova G, Say D, Hasanova G (2021) Plant-produced glycosylated and in vivo deglycosylated receptor binding domain proteins of SARS-CoV-2 induce potent neutralizing responses in mice. Viruses 13:1595. https://doi.org/10.3390/v13081595
Marusic C, Nuttall J, Buriani G, Lico C, Lombardi R, Baschieri S, Benvenuto E, Frigerio L (2007) Expression, intracellular targeting and purification of HIV Nef variants in tobacco cells. BMC Biotechnol 7:12
Rage E, Drissi-Touzani C, Marusic C, Lico C, Göbel T, Bortolami A, Bonfante F, Salzano AM, Scaloni A, Fellahi S, El Houadfi M, Donini M, Baschieri S (2019) Functional characterization of a plant-produced infectious bursal disease virus antigen fused to the constant region of avian IgY immunoglobulins. Appl Microbiol Biotechnol 103:7491–7504. https://doi.org/10.1007/s00253-019-09992-9
Rattanapisit K, Shanmugaraj B, Manopwisedjaroen S, Purwono PB, Siriwattananon K, Khorattanakulchai N, Hanittinan O, Boonyayothin W, Thitithanyanont A, Smith DR, Phoolcharoen W (2020) Rapid production of SARS-CoV-2 receptor binding domain (RBD) and spike specific monoclonal antibody CR3022 in Nicotiana benthamiana. Sci Rep 10:17698. https://doi.org/10.1038/s41598-020-74904-1
Rattanapisit K, Bulaon CJI, Khorattanakulchai N, Shanmugaraj B, Wangkanont K, Phoolcharoen W (2021) Plant-produced SARS-CoV-2 receptor binding domain (RBD) variants showed differential binding efficiency with anti-spike specific monoclonal antibodies. PLoS ONE 18:e0253574. https://doi.org/10.1371/journal.pone.0253574
Rosendal E, Wigren J, Groening R, Yong-Dae G, Nilsson E, Sharma A, Espaillat A, Hanke L, Thunberg T, McInerney G, Puhar A, Cava F, Karlsson-Hedestam GB, Monsen T, Elgh F, Blomkvist B, Marklund I, Ahlm C, Evander M, Normark J, Johansson A, Överby AK, Forsell MNE (2020) Detection of asymptomatic SARS-CoV-2 exposed individuals by a sensitive S-based ELISA. medRxiv. https://doi.org/10.1101/2020.06.02.20120477
Ruocco V, Vavra U, König-Beihammer J, Bolaños Martínez OC, Kallolimath S, Maresch D, Grünwald-Gruber C, Strasser R (2023) Impact of mutations on the plant-based production of recombinant SARS-CoV-2 RBDs. Front Plant Sci 14:1275228. https://doi.org/10.3389/fpls.2023.1275228
Salzano AM, Novi G, Arioli S, Corona S, Mora D, Scaloni A (2013) Mono-dimensional blue native-PAGE and bi-dimensional blue native/urea-PAGE or/SDS-PAGE combined with nLC-ESI-LIT-MS/MS unveil membrane protein heteromeric and homomeric complexes in Streptococcus thermophilus. J Proteomics 94:240–261. https://doi.org/10.1016/j.jprot.2013.09.007
Shin Y-J, König-Beihammer J, Vavra U, Schwestka J, Kienzl NF, Klausberger M, Laurent E, Grünwald-Gruber C, Vierlinger K, Hofner M, Margolin E, Weinhäusel A, Stöger E, Mach L, Strasser R (2021) N-Glycosylation of the SARS-CoV-2 receptor binding domain is important for functional expression in plants. Front Plant Sc 12:689104. https://doi.org/10.3389/fpls.2021.689104
Siriwattananon K, Manopwisedjaroen S, Shanmugaraj B, Rattanapisit K, Phumiamorn S, Sapsutthipas S, Trisiriwanich S, Prompetchara E, Ketloy C, Buranapraditkun S, Wijagkanalan W, Tharakhet K, Kaewpang P, Leetanasaksakul K, Kemthong T, Suttisan N, Malaivijitnond S, Ruxrungtham K, Thitithanyanont A, Phoolcharoen W (2021) Plant-produced receptor-binding domain of SARS-CoV-2 elicits potent neutralizing responses in mice and non-human primates. Front Plant Sci 12:682953. https://doi.org/10.3389/fpls.2021.682953
Tian Y, Parsons LM, Jankowska E, Cipollo JF (2021) Site-specific glycosylation patterns of the SARS-CoV-2 spike protein derived from recombinant protein and viral WA1 and D614G strains. Front Chem 9:767448. https://doi.org/10.3389/fchem.2021.767448
Walls AC, Park YJ, Tortorici A, Wall A, McGuire AT, Veesler D (2020) Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181:281–292. https://doi.org/10.1016/j.cell.2020.02.058
Williams L, Jurado S, Llorente F, Romualdo A, González S, Saconne A, Bronchalo I, Martínez-Cortes M, Pérez-Gómez B, Ponz F, Jiménez-Clavero MA, Lunello P (2021) The C-terminal half of SARS-CoV-2 nucleocapsid protein, industrially produced in plants, is valid as antigen in COVID-19 serological tests. Front Plant Sci 12:699665. https://doi.org/10.3389/fpls.2021.699665
Yang Y, Xiao Z, Ye K, He X, Sun B, Qin Z, Yu J, Yao J, Wu Q, Bao Z, Zhao W (2020) SARS-CoV-2: characteristics and current advances in research. Virol J 17:117. https://doi.org/10.1186/s12985-020-01369-z
Acknowledgements
We acknowledge Julia Jansing and Luisa Bortesi for kindly providing N. benthamiana FX-KO seeds from RWTH Aachen University and Emanuele Andreano, Ida Paciello and Rino Rappuoli for providing sequences of mAbJ08.
Supporting information
Supplementary Figure 1—Sequence coverage of the recombinant antigens RBDw-Fc and RBDo-Fc purified by non-reducing SDS-PAGE, corresponding to the bands at about 110 kDa.
Supplementary Figure 2—Quantification of functional RBDw-Fc antigen present in the plant purified product as performed by ELISA.
Funding
The work has been funded by: the Italian Ministry of Education, Universities and Research with the 5X1000-2001 fund to the project “Plants as biofactories of vaccines and antibodies for the COVID-19 emergency”; the National Recovery and Resilience Plan, mission 4, component 2, investment 1.3, MUR call n. 341/2022 funded by the European Union-Next Generation EU for the project “One Health Basic and Translational Research Actions addressing Unmet Needs on Emerging Infectious Diseases (IN-FACT)”, concession decree 1554/2022, PE00000007.
Author information
Authors and Affiliations
Contributions
RF and FF performed the experiments and data analysis. AMS and AS carried out proteomic experiments, performed data analyses and helped revising the manuscript. CM contributed to the conception of the study, helped performing experiments and contributed to write the manuscript. MD conceived, designed the study, performed the experiments and wrote the manuscript. All the authors contributed to manuscript revision, read, and approved the submitted version.
Corresponding authors
Ethics declarations
Conflict of interest
The authors state that they have no conflict of interest. The funders did not play a role in the study’s design, data collection, analysis or interpretation, manuscript writing, or the decision to publish the findings.
Ethical approval
For the studies involving human serum samples, informed consent was obtained from the subjects involved in the study and written informed consent has been obtained from the subjects to publish this paper. The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the ENEA Local Ethics Committee.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Fagiani, F., Frigerio, R., Salzano, A.M. et al. Plant production of recombinant antigens containing the receptor binding domain (RBD) of two SARS-CoV-2 variants. Biotechnol Lett (2024). https://doi.org/10.1007/s10529-024-03517-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10529-024-03517-9