
Abstract
Lytic polysaccharide monooxygenases (LPMOs) have emerged as key proteins for depolymerization of cellulose. These copper-containing enzymes oxidize C-1 and/or C-4 bonds in cellulose, promoting increased hydrolysis of the oxidized cellulose chains. The LPMO from Thermoascus aurantiacus, a thermophilic ascomycete fungus, has been extensively studied and has served as a model LPMO. A method was developed to purify the LPMO from culture filtrates of T. aurantiacus along with its native cellobiohydrolase and endoglucanase. The activity of the purified LPMO was measured with a colorimetric assay that established the Topt of the native LPMO at 60 °C. Purification of the components of the T. aurantiacus cellulase mixture also enabled quantification of the amounts of cellobiohydrolase, endoglucanase and LPMO present in the T. aurantiacus culture filtrate, establishing that the LPMO was the most abundant protein in the culture supernatants. The importance of the LPMO to activity of the mixture was demonstrated by saccharifications with Avicel and acid-pretreated corn stover.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The conversion of plant biomass to biofuels and biochemicals is a critical component in implementing the bioeconomy (Scarlat et al 2015). This conversion comprises chemical pretreatment of lignocellulose followed by enzymatic depolymerization of the polysaccharides to sugars for microbial conversion (Blanch et al 2011). Cellulose is the most abundant polysaccharide in plant biomass, and cellulases produced by filamentous fungi have been harnessed for cellulose depolymerization on an industrial scale (Kuhad et al 2016). Fungi produce a variety of hydrolytic enzymes (cellobiohydrolases, endoglucanases and beta-glucosidases) that hydrolyze glycosidic bonds in the cellulose chain and in cellobiose, the hydrolytic product of the cellobiohydrolases (Payne et al 2015). Recently, an enigmatic glycoside hydrolase family, GH61, which was comprised of low activity endoglucanases, was reclassified as lytic polysaccharide monooxygenases (LPMOs) in the auxiliary activity family 9 (AA9) (Forsberg et al 2019). LPMOs are monocopper-containing proteins that oxidize C-1 and/or C-4 bonds in cellulose, promoting increased hydrolysis of the oxidized cellulose chains and lowering the protein loading requirements for high levels of glucose release from cellulose (Harris et al 2010; Quinlan et al 2011). LPMOs require a reductant for activity and have been shown to use a variety of reductants, including: exogenously added compounds (ascorbate and gallate), aromatics derived from biomass, redox-active proteins (cellobiose dehydrogenase) and photochemistry (Kracher et al 2016). Initial studies identified O2 as the co-substrate for LPMOs; recent work has amassed evidence for H2O2, which LPMOs can produce from O2, being the primary co-substrate for cellulose oxidation (Bissaro et al 2017) (Müller et al 2018).
The LPMO identified in Thermoascus aurantiacus has been central to the understanding of the biochemistry of these oxidative enzymes. A boosting effect of the T. aurantiacus supernatants on the celluloytic activity of Cellulcast, a commercial cellulase mixture, was observed and subsequently attributed to the activity of T. aurantiacus LPMO by expressing it in a Trichoderma reesei strain that produces high levels of the hydrolytic enzymes (Rosgaard et al 2006) (Harris et al 2010). The structure of the T. aurantiacus LPMO defined its active site as having a unique N-methylated histidine residue that binds strongly binds Cu2+ and spectroscopy of the Cu active site indicated that the Cu(I) form of the active site adopts a three coordinate T-shaped structure (Quinlan et al 2011) (Kjaergaard et al 2014; Petrović et al 2018). Recent spectroscopic work on the T. aurantiacus LPMO has identified a Cu(III) oxidation state, possibly stabilized by a tyrosyl radical, that is produced by reaction with H2O2 and may be involved in the catalytic cycle (Singh et al 2020).
Previous studies on T. aurantiacus enzyme production have shown that the LPMO is produced at high levels when T. aurantiacus is grown on xylan, cellulose or concentrations of xylose that do not trigger carbon catabolite repression (McClendon et al 2012) (Schuerg et al 2017a, 2017b). Under these conditions T. aurantiacus produces the LPMO along with glycoside hydrolases (cellobiohydrolase, endoglucanase, xylanase, and beta-glucosidase). Purification of the T. aurantiacus LPMO along with the other cellulase enzyme will allow the the LPMO to be characterized and its abundance in comparison with with the other cellulases enzymes quantitively assessed. Here we describe purification and characterization of the native T. aurantiacus LPMO along with its native cellobiohydrolase and endoglucanase.
Materials and methods
Protein production by T. aurantiacus
Thermoascus aurantiacus ATCC 26,409 was grown on potato dextrose agar plates for 2 days at 50 °C followed by 4 days at 45 °C. Precultures (250 ml) containing McClendon’s salts with 2% sucrose and 0.8% soymeal peptone (Amersco, VWR, Radnor, PA, USA) were inoculated with agar plugs from the plates and incubated at 50 °C and 200 rpm for 2 days (McClendon et al 2012). The precultures were filtered under sterile conditions and the recovered mycelia were inoculated into a medium with McClendon’s salts, 0.8% soy meal peptone and 1% beechwood xylan (Megazyme, Bray, Ireland). After 3 days of incubation, the cultures were filtered through Miracloth (Millipore, Burlington, MA, USA) and the filtrate was centrifuged for 10 min at 4000 rpm and subsequently filtered through a 0.22 μm filter. After filtration, Bradford protein assays indicated a protein concentration of ~ 1 mg/mL. The filtrate was concentrated to 2 mg/mL at 4 °C in vacuo with a 10 kDa molecular weight cutoff membrane (Millipore, Burlington, MA, USA).
LPMO purification
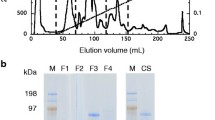
The culture filtrate (2 mg/mL, 10 mL) was concentrated to 2.5 mL and diluted to 5 mL with loading buffer (30 mM NaOAc pH 3.5). The diluted filtrate was loaded on to a PD-10 (GE Healthcare Life Sciences, Chicago, IL, USA) column and eluted with loading buffer. The eluted sample was concentrated to 1 mL and filtered through a 0.22 μm filter (Millipore, Burlington, MA, USA). The sample was loaded on to a 1 mL SP-Sepharose HP column (GE Healthcare Life Sciences, Chicago, IL, USA) that had been equilibrated with 1 mL of loading buffer and, after loading, was washed with 5 mL of loading buffer. The endoglucanase and cellobiohydrolase were eluted with a 7 mL NaCl gradient (0 to 0.5 M) in loading buffer; the endoglucanase and cellobiohydrolase eluted at 125 mM NaCl. The cation exchange column was re-equilibrated with 4 mL of loading buffer and the LPMO eluted at pH 6 with a 10 mL pH gradient, starting with loading buffer (30 mM NaOAc, pH 3.5) and continuously increased inflow of a potassium phosphate buffer (30 mM) with a finishing pH of 8. The xylanase co-eluted with the LPMO. The salt gradient fractions were rebuffered with elution buffer (potassium phosphate buffer, 30 mM, pH 6), concentrated to 0.5 mL and loaded on to a Superdex Increase 75 10/300 GL size exclusion column (24 mL) (GE Healthcare Life Sciences, Chicago, IL, USA), which was pre-equilbrated with 30 mM potassium phosphate buffer pH 6 containing 20 mM NaCl (elution buffer). The cellobiohydrolase and endoglucanase were separated by washing of the column with 1.5 volumes of elution buffer. In a separate purification step, the pH gradient sample was concentrated to 0.5 mL and loaded on to the Superdex Increase 75 10/300 GL column. The LPMO and xylanase were separated by elution with 1.5 column volumes. The LPMO was estimated to be > 90% pure by SDS-PAGE (Fig. 1d).

Purification of native cellobiohydrolase, endoglucanase and LPMO from T. aurantiacus. SDS-PAGE of a Cation exchange chromatography. Gel represents loaded culture filtrate (1), column wash fractions (2,3), NaCl-gradient (0–0.5 M) fraction containing CBH and EG, eluted at 125 mM NaCl (4), pH-gradient (pH 3.5–8) fractions containing LPMO and xylanase, eluted at pH 6 (5–8). b Size exclusion chromatography of CBH and EG. SDS-PAGE analysis of loaded sample (1) and elution fractions (2–9). CBH was predominantly found in fractions of the first elution peak (4–5), pure EG eluted within a second peak (8–9). c Size exclusion chromatography of xylanase and LPMO. Lane 1 represents loaded sample, LPMO eluted within the first peak (3–9) followed by co-elution of xylanase co-eluted (10–12). d Summary of stepwise protein purification. Comparison of initially loaded culture filtrate (1), NaCl-gradient purified CBH and EG (2), SEC-purified CBH (3) and EG (4), pH-gradient purified Xylanase and LPMO (5) and SEC-purified LPMO. 4–12% pre-cast gels were run for 45 min at 120 V in Tris/glycine SDS running buffer and stained with SimplyBlue™ SafeStain, ladder: Novex™ sharp pre-stained protein standards (3.5 to 260 kDa). Cation exchange and size exclusion chromatography were performed on an ÄKTA avant 150 chromatography system with a HiTrap™ SP HP and Superdex Increase 75 10/300 GL column, respectively (d)
Enzyme assays
Endoglucanase activity was measured by the DNS method using carboxymethylcellulose as the substrate (Gladden et al 2011). In brief, enzymes and substrates (5 μL and 75 μL, respectively) in NaOAc buffer (pH 5, 50 mM) were incubated at 65 °C for 60 min (CMC) followed by addition of 80 μL DNS and incubation at 90 °C for 5 min. Release of glucose was determined with optical density readings at λ = 540 nm. Cellobiohydrolase activity was measured using the p-nitrophenyl-β-d-cellobioside method (Park et al 2012). Enzyme samples and 10 mM pNPC (final concentration) in 100 mM NaOAc (pH 5.0) were incubated for 15 min at 50 °C. Spectrophotometric measurement of p-nitrophenol release was performed at λ = 415 nm after addition of 2 M Na2CO3 to the reaction mixtures.
LPMO activity was measured by the H2O2-dependent oxidation of 2,6-dimethoxyphenol to coerulignone as previously described (Breslmayr et al 2018). The temperature dependence of the LPMO activity was measured by pre-equilibrating the reaction mixture at the chosen temperature for 30 min and then adding the LPMO. One unit of CMCase, cellobiohydrolase or LPMO activity was defined as nmol of released glucose, p-nitrophenol or coerulignone per min, respectively. Protein concentration assays were performed using the Bradford method as previously described.
Protein quantification on SDS-PAGE gels
Quantification of cellobiohydrolase, endoglucanase and LPMO in the culture filtrate was performed with SDS-PAGE gel analysis using ImageJ (https://imagej.nih.gov/ij/features.html). Laemmli buffer (5 Ll) and 2-mercaptoethanol (1 µL) were added to a dilution series of purified proteins and culture filtrate. 15 µL samples and 5 µL of Novex™ sharp pre-stained protein standard molecular weight marker (Thermo Fisher Scientific, Waltham, MA, USA) were loaded a 4–12% precast protein gel (Bio-Rad) and run at 140 V for 45 min in Tris/glycine SDS running buffer (Bio-Rad, Hercules, CA, USA). Gels were stained with SimplyBlue™ SafeStain solution (Thermo Fisher Scientific, Waltham, MA, USA) for 1 h and destained according to the manufacturers protocol. The gel was pictured with a chemiDoc XRS + imaging system (Bio-Rad, Hercules, CA, USA) and uploaded to ImageJ. Rectangular areas of each band were selected and histograms indicating the band intensities were calculated by the software. Numerical intensity values of bands of each dilution series and applied amounts of protein were plotted for standard curve calculations. Protein amounts in the culture filtrate were measured by obtaining the band intensities as described above.
LPMO assays with enzymatic mixtures
Thermoascus aurantiacus culture filtrates produced by controlled feeding of xylose and cellobiose were used in this experiment (Schuerg et al 2017b). The filtrates were produced by preculturing T. aurantiacus in minimal medium with McClendon’s salts and 2% glucose/0.8% soymeal peptone as described above. The pre-cultures were incubated for 48 h at 50 °C in a rotary shaker and filtered in vacuo under sterile conditions. The mycelia (1.5 g) were weighed into baffled 250 ml Erlenmeyer culture flasks containing 50 ml of culture medium without a carbon substrate. Solutions of 1% xylose and cellobiose were autoclaved separately and added by a peristaltic pump (BT100-1L Multi-Channel Peristaltic Pump (Langer Instruments Corp., Boonton, NJ, USA)) according to a previously established protocol (Schuerg et al 2017b). The filtrates were were isolated as described above. Saccharifications were performed in 96-well plates with 2% w/v biomass (Avicel or acid-pretreated corn stover) suspended in 2 mL of 50 mM NaOAc pH 5.0 containing 0 or 1 mM sodium ascorbate. Aliquots of T. aurantiacus filtrates from xylose or cellobiose-grown cultures were added to provide reaction mixtures with 20 mg of enzymes/g of biomass. Cellic CTec2 (Novozymes A/S, Bagsværd, Denmark) was also added at 20 mg/g of biomass. The plates were incubated for 24 h at 50 °C and 180 rpm and the mixtures subsequently analyzed for glucose production using a YSI analyzer (YSI, Yellow Springs, Ohio, USA).
Results and discussion
Protein purification
A purification protocol was developed for the T. aurantiacus LPMO using culture supernatants obtained from mycelia grown on sucrose and shifted to induction medium with beechwood xylan as the carbon substrate. This T. aurantiacus cultivation method produces high levels of extracellular proteins (> 1 mg/mL), and visual inspection of the SDS-PAGE gels of the supernatant indicated that the cellobiohydrolase (58 kDa) and LPMO (23 kDa) were the dominant proteins in the fraction, with the endoglucanase (35 kDa) and xylanase (32 kDa) present at lower levels in an overlapping band (McClendon et al 2012). A purification protocol was devised that targeted the cellulases and would separate the LPMO from the hydrolytic proteins. Binding of the supernatant to cation exchange column and elution with a salt gradient separated the cellobiohydrolase and the endoglucanase from the LPMO and xylanase, which were eluted with a pH gradient (Fig. 1a). The cellobiohydrolase and endoglucanase were separated by gel filtration chromatography, with the cellobiohydrolase eluting first, as predicted by its higher molecular weight (Fig. 1b). In contrast, for gel filtration chromatography of the fraction containing the LPMO and xylanase, the lower molecular weight LPMO eluted first and the xylanase co-eluted with a second fraction of the LPMO (Fig. 1c). The specific activity of the endoglucanase and LPMO increased after the gel filtration step; however the specific activity of the cellobiohydrolase decreased (Table 1). Previously, purification of T. aurantiacus cellobiohydrolase and endoglucanase was reported from a strain isolated in India and grown with blotting paper as the carbon source (Khandke et al 1989). The reported molecular weights (52 kDa for the CBH and 32 kDa for the EG) are consistent with the molecular weights for the protein isolated here (Fig. 1d). The specific activity for the CBH from the Indian strain, which was measured by hydrolysis of Avicel, also decreased during the purification, suggesting that either other proteins may contribute to the cellobiohydrolase activity in the supernatant or the CBH is unstable during purification (Khandke et al 1989). Interestingly, SDS-PAGE of the supernatant from the Indian T. aurantiacus strain lacked a prominent band for an LPMO, indicating that strains of T. aurantiacus may respond differently to lignocellulosic substrates or not encode an LPMO.
Temperature dependence of LPMO assay
Previous studies of the T. aurantiacus LPMO measured its activity through its ability to enhance the hydrolysis of cellulose by enzymatic mixtures with glycoside hydrolases or by MALDI-TOF detection of cellodextrins upon incubation of phosphoric-acid swollen cellulose with the LPMO and beta-glucosidase (Harris et al 2010; Quinlan et al 2011). These studies were performed at 50 °C, which is generally the temperature which saccharifications with commercial enzymatic mixtures are performed. Recently, a colorimetric assay was developed to measure LPMO activity by taking advantage of the ability of the LPMO to act as a peroxidase, coupling oxidation of 2,6-dimethoxyphenol to coerulignone to reduction of H2O2 to water (Breslmayr et al 2018). This colorimetric assay was used to test the temperature dependence of LPMO activity (Fig. 2), which reached a maximum at 60 °C, which is in the same range (60–70 °C) as the Topt previously measured for the T. aurantiacus cellobiohydrolase and endoglucanase (Khandke et al 1989). The observation of a Topt of ≥ 60 °C for the three cellulases from T. aurantiacus is consistent with the maintenance of high saccharification rates for T. aurantiacus supernatant enzyme mixture at elevated temperatures compared with commercial enzymatic mixtures (Schuerg et al 2017b).

Temperature-dependent relative activities of LPMO in preincubated buffer. The LPMO was assayed with 1 mM 2,6-DMP and 100 μM H2O2 in 100 mM sodium acetate buffer pH 6. After preincubation of the reaction mixture for 30 min, the assay was started by adding the LPMO. Formation of coerulignone (ε469 = 53,200 M−1 cm−1) was followed over 10 min by spectrophotometric measurement of absorbance at λ = 469 nm. Error bars show the standard deviation of two independent protein preparations measured in triplicates. A relative activity of 100% was set at 60 °C based on the observed temperature maxima of the two experiments
Quantification of LPMO in T. aurantiaucs supernatant
Though the prominence of the LPMO in the T. aurantiacus culture supernatant was evident in SDS-PAGE, quantification of the LPMO in this mixture has not been reported. The simplicity of the enzyme mixture produced by T. aurantiacus and availability of purified LPMO, CBH and EG enabled the quantification of the amount of LPMO relative to the total protein in the mixture (Fig. 3). Band intensities of culture filtrate samples (lanes 1–3) were compared to dilution series of CBH, EG/xylanase and LPMO (lane 4–7, 8–10, 11–13, respectively). To determine relative quantities of each enzyme in the filtrate, lane 3 was quantitatively analyzed. In total, 2.5 µg of culture filtrate protein was separated and plotting the band intensities to those of the dilution series, 1.8 µg of target enzymes were found in the native culture filtrate. This analysis results in relative quantities of 34%, 31%, 7%, and 28% for LPMO, CBH, EG/Xylanase and other proteins, respectively. Previous studies adding the heterologously produced LPMO from T. aurnatiacus to commercial hydrolytic enzyme mixtures (Celluclast) demonstrated that at optimal boosting of activity was achieved with the LPMO set at 15% of the total enzymatic mixture. Therefore, the relative proportion of the LPMO in the native T. aurnatiacus mixture is more than double than the optimal loading in enzymatic mixtures (Müller et al 2015; Chylenski et al 2017).

Quantification of LPMO in T. aurantiacus culture filtrate using SDS-PAGE imaging. a SDS-PAGE gel electrophoresis of dilution series of whole culture filtrate (1–3) and purified cellobiohydrolase (4–7), endoglucanase/xylanase (8–10) and LPMO (11–13). Standard curves were obtained by plotting band intensities and concentration of proteins applied to the gel. b ImageJ protein quantification analysis. Graph represents relative quantities based on band intensities of lane 3 (2.5 μg total protein)
LPMO Activity in T. aurantiacus enzyme mixtures
The identification of the LPMO as the most abundant protein produced by T. aurantiacus suggested that the influence of the LPMO on the overall activity of the T. aurantiacus enzymatic mixture may be larger than in commercial enzymatic mixtures. To test this prediction, the effect of ascorbate on saccharification of Avicel and acid-pretreated corn stover by the T. aurantiacus enzyme mixture was performed and compared to CTec2. The saccharification was performed at 50 °C since this temperature is optimal for CTec2. Previous results with commercial enzymatic mixtures and purified proteins has demonstrated that a reducing agent such as ascorbate was required to activate the T. aurantiacus LPMO in Avicel saccharifications, but native phenolic compounds served as reductants for the LPMO in saccharifications with acid-pretreated corn stover (Quinlan et al 2011). T. aurantiacus supernatants from fed-batch incubations with xylose and cellobiose were tested (Schuerg et al 2017b). Saccharifications were performed in the presence or absence of 1 mM ascorbate and 20 mg/g biomass; CTec2 was used as a control. For Avicel saccharifications, CTec2 produced 5.8–8.1 × glucose in 24 h compared to the T. aurantiacus supernatants in the absence of ascorbate (Fig. 4a). In the presence of 1 mM ascorbate, glucose release from Avicel was increased 46% for the CTec2 saccharifications. However, glucose release from saccharifications with the T. aurantiacus enzymes was increased 91–151% when ascorbate was present, consistent with the higher level of LPMO in the T. aurantiacus supernatant. For acid-pretreated corn stover, CTec2 produced 2.0–2.5 × glucose compared to the T. auranaticus supernatants, and addition of 1 mM ascorbate had a negligible effect on glucose release (Fig. 4b). The comparative results for glucose release of CTec2 and the T. aurantiacus supernatants on acid-pretreated corn stover after 24 h are comparable to saccharifications reported previously for xylose-grown T. aurantiacus cultures (Schuerg et al 2017b). Of particular interest in these saccharifications is the divergence between glucose release on Avicel, which strongly favors CTec2, versus corn stover, where the glucose release is comparable. These results may reflect the differences in protein content of each enzymatic mixture. In the commercial cellulase mixtures, cellobiohydrolase is the most abundant protein (Taylor et al 2018), while in the T. aurantiacus mixture the LPMO is the most abundant protein. This observation suggests that the LPMO-dominated enzymatic mixture from T. aurantiacus is relatively poor at depolymerizing cellulose in purified preparations, but performs well on cellulose still embedded in the lignocellulose matrix. The abundance of the T. aurantiacus LPMO raises questions about the biological role of the LPMO and the other cellulases in biological biomass deconstruction by T. aurantiacus. In particular, how important is the LPMO in T. aurantiacus growth on cellulose and will eliminating the LPMO reduce or prevent growth? In addition, are there accessory enzymes that are required for the in vivo activity of the LPMO?

Saccharification of Avicel and acid-pretreated corn stover by CTec2 and culture filtrates obtained from xylose (Xyl) and cellobiose (Cell)-grown T. aurantiacus cultures with/without the reductant ascorbic acid (AscA) added. a Saccharification of 2% Avicel; b Saccharification of 2% acid-pretreated corn stover. Saccharification reactions were performed at 50 °C and 180 rpm for 24 h with an enzyme loading of 20 mg/g biomass. Error bars represent the standard deviation of 4 technical replicates, asterisks represent significant difference among conditions (p value < 0.05)
Conclusions
Purification of the native LPMO from T. aurantiacus has enabled a measurement of its temperature dependence and demonstrated that it is the most abundant protein produced by fungus in cultures with xylan as the inducing substrate. The importance of the LPMO to the overall activity of the T. auranaticus enzyme mixture was shown in up to 150% increases in glucose release from Avicel in the presence of ascorbate. The abundance of the LPMO in proteins produced by T. aurantiacus suggests that cellulolytic enzymatic mixtures may be designed with LPMOs as the dominant protein, as opposed to CBHI, which is the most abundant protein in most commercial enzymatic mixtures (Taylor et al 2018; Hamre et al 2019)..
References
Bissaro B, Røhr ÅK, Müller G et al (2017) Oxidative cleavage of polysaccharides by monocopper enzymes depends on H2O2. Nat Chem Biol 13:1123–1128. https://doi.org/10.1038/nchembio.2470
Blanch HW, Simmons BA, Klein-Marcuschamer D (2011) Biomass deconstruction to sugars. Biotechnol J 6:1086–1102. https://doi.org/10.1002/biot.201000180
Breslmayr E, Hanžek M, Hanrahan A et al (2018) A fast and sensitive activity assay for lytic polysaccharide monooxygenase. Biotechnol Biofuels 11:79. https://doi.org/10.1186/s13068-018-1063-6
Chylenski P, Petrović DM, Müller G et al (2017) Enzymatic degradation of sulfite-pulped softwoods and the role of LPMOs. Biotechnol Biofuels 10:177. https://doi.org/10.1186/s13068-017-0862-5
Forsberg Z, Sørlie M, Petrović D et al (2019) Polysaccharide degradation by lytic polysaccharide monooxygenases. Curr Opin Struct Biol 59:54–64. https://doi.org/10.1016/j.sbi.2019.02.015
Gladden JM, Allgaier M, Miller CS et al (2011) Glycoside hydrolase activities of thermophilic bacterial consortia adapted to switchgrass. Appl Environ Microbiol 77:5804–5812. https://doi.org/10.1128/AEM.00032-11
Hamre AG, Strømnes A-GS, Gustavsen D et al (2019) Treatment of recalcitrant crystalline polysaccharides with lytic polysaccharide monooxygenase relieves the need for glycoside hydrolase processivity. Carbohydr Res 473:66–71. https://doi.org/10.1016/j.carres.2019.01.001
Harris PV, Welner D, McFarland KC et al (2010) Stimulation of lignocellulosic biomass hydrolysis by proteins of glycoside hydrolase family 61: structure and function of a large, enigmatic family. Biochemistry 49:3305–3316. https://doi.org/10.1021/bi100009p
Khandke KM, Vithayathil PJ, Murthy SK (1989) Purification of xylanase, β-glucosidase, endocellulase, and exocellulase from a thermophilic fungus, Thermoascus aurantiacus. Arch Biochem Biophys 274:491–500
Kjaergaard CH, Qayyum MF, Wong SD et al (2014) Spectroscopic and computational insight into the activation of O2 by the mononuclear Cu center in polysaccharide monooxygenases. Proc Natl Acad Sci USA 111:8797–8802. https://doi.org/10.1073/pnas.1408115111
Kracher D, Scheiblbrandner S, Felice AKG et al (2016) Extracellular electron transfer systems fuel cellulose oxidative degradation. Science 352:1098–1101. https://doi.org/10.1126/science.aaf3165
Kuhad RC, Deswal D, Sharma S et al (2016) Revisiting cellulase production and redefining current strategies based on major challenges. Renew Sustain Energy Rev 55:249–272. https://doi.org/10.1016/j.rser.2015.10.132
McClendon SD, Batth T, Petzold CJ et al (2012) Thermoascus aurantiacus is a promising source of enzymes for biomass deconstruction under thermophilic conditions. Biotechnol Biofuels 5:54. https://doi.org/10.1186/1754-6834-5-54
Müller G, Chylenski P, Bissaro B et al (2018) The impact of hydrogen peroxide supply on LPMO activity and overall saccharification efficiency of a commercial cellulase cocktail. Biotechnol Biofuels 11:209. https://doi.org/10.1186/s13068-018-1199-4
Müller G, Várnai A, Johansen KS et al (2015) Harnessing the potential of LPMO-containing cellulase cocktails poses new demands on processing conditions. Biotechnol Biofuels 8:187. https://doi.org/10.1186/s13068-015-0376-y
Park JI, Steen EJ, Burd H et al (2012) A thermophilic ionic liquid-tolerant cellulase cocktail for the production of cellulosic biofuels. PLoS ONE 7:e37010. https://doi.org/10.1371/journal.pone.0037010
Payne CM, Knott BC, Mayes HB et al (2015) Fungal cellulases. Chem Rev 115:1308–1448
Petrović DM, Bissaro B, Chylenski P et al (2018) Methylation of the N-terminal histidine protects a lytic polysaccharide monooxygenase from auto-oxidative inactivation. Protein Sci 27:1636–1650. https://doi.org/10.1002/pro.3451
Quinlan RJ, Sweeney MD, Lo Leggio L et al (2011) Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc Natl Acad Sci USA 108:15079–15084. https://doi.org/10.1073/pnas.1105776108
Rosgaard L, Pedersen S, Cherry JR et al (2006) Efficiency of new fungal cellulase systems in boosting enzymatic degradation of barley straw lignocellulose. Biotechnol Prog 22:493–498. https://doi.org/10.1021/bp050361o
Scarlat N, Dallemand J-F, Monforti-Ferrario F, Nita V (2015) The role of biomass and bioenergy in a future bioeconomy: Policies and facts. Environ Dev 15:3–34. https://doi.org/10.1016/j.envdev.2015.03.006
Schuerg T, Gabriel R, Baecker N et al (2017a) Thermoascus aurantiacus is an intriguing host for the industrial production of cellulases. CBIOT 6:89–97. https://doi.org/10.2174/2211550105666160520123504
Schuerg T, Prahl J-P, Gabriel R et al (2017b) Xylose induces cellulase production in Thermoascus aurantiacus. Biotechnol Biofuels 10:271. https://doi.org/10.1186/s13068-017-0965-z
Singh RK, Blossom BM, Russo DA et al (2020) Detection and characterization of a novel copper-dependent intermediate in a lytic polysaccharide monooxygenase. Chem Eur J 26:454–463. https://doi.org/10.1002/chem.201903562
Taylor LE, Knott BC, Baker JO et al (2018) Engineering enhanced cellobiohydrolase activity. Nat Commun 9:1186. https://doi.org/10.1038/s41467-018-03501-8
Acknowledgements
The authors are grateful for funding support from the DOE Joint BioEnergy Institute (https://www.jbei.org) supported by the U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research, through contract DE-AC02-05CH11231 between Lawrence Berkeley National Laboratory and the U.S. Department of Energy. Novozymes is acknowledged for the generous gift of Cellic CTec2. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes. The authors thank Melvin Tucker of the National Renewable Energy Laboratory for providing acid-pretreated corn stover. We thank Andy DiGiovanni of the Joint BioEnergy Institute for assistance with protein purification.
Author information
Authors and Affiliations
Contributions
SWS and RG. designed research; SF, CH JG and RG performed research.
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Fritsche, S., Hopson, C., Gorman, J. et al. Purification and characterization of a native lytic polysaccharide monooxygenase from Thermoascus aurantiacus. Biotechnol Lett 42, 1897–1905 (2020). https://doi.org/10.1007/s10529-020-02942-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-020-02942-w