
Abstract
Objective
To survey genome-scale protease profiles regulated by the Aspergillus niger transcription factor PrtT and further controlled by carbon sources.
Results
The PrtT disruption mutant (delprtT) and overexpression (OEprtT) strains were successfully generated and further confirmed by phenotypic and protease activity analysis. RNA-seq analysis of WT and mutants identified 32 differentially expressed protease genes, which mostly belonged to serine-type peptidases, aspartic-type endopeptidases, aminopeptidases and carboxypeptidases. Furthermore, based on the MEME predicted motif analysis of the PrtT promoter, EMSA and phenotypic and qRT-PCR analyses confirmed that the carbon metabolism regulator AmyR directly regulated the protease genes and their regulatory factor PrtT.
Conclusion
Thirty-two PrtT-regulated protease genes were identified by RNA-seq, and the secondary carbon source regulator AmyR was found to have a negative regulatory effect on the expression of PrtT and its target protease genes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Proteases have long been recognized as one of the bottlenecks in the expression of target proteins in Aspergillus (Punt et al. 2008). However, Aspergillus proteases have also been developed for industrial production (Budak et al. 2014). A genome survey of A. niger based on the protein sequences of the verified proteases and Pfam domains revealed that 314 of the 11,162 genes are putative proteases (Budak et al. 2014).
Two sets of regulatory genes have been thought to be involved in the regulation of proteases to date: wide domain and pathway-specific regulators. The wide domain regulators, including CreA, AreA and PacC, are proposed to be involved in the overall regulation of protease expression in Aspergillus (Jarai and Buxton 1994). As the carbon catabolism repression regulator, CreA is considered the major wide domain regulator affecting proteases in protease production and is severely repressed in medium containing a preferred carbon source (Katz et al. 2000; Ruijter and Visser 1997). However, there is little literature on the regulation of protease production by carbon sources.
The transcription factor PrtT, identified as the pathway-specific regulator of extracellular proteases in Aspergilli, affects the expression of a broad spectrum of enzymes (Ballester et al. 2019; Bergmann et al. 2009; Chen et al. 2014; Hagag et al. 2012; Punt et al. 2008; Sharon et al. 2009; Shemesh et al. 2017). In addition, PrtT may compete with AmyR to inhibit five amylase genes, because the promoter region of these genes contains overlapping binding sites for AmyR and PrtT as shown for Penicillium oxalicum (Chen et al. 2014). Whether the regulation of carbon and nitrogen metabolism can be linked through AmyR and PrtT is worthy of further study, and it is also important to control the carbon source to manage protease expression in the process of A. niger fermentation. The reports on the function of prtT in A. niger are mainly about reducing protease secretion and increasing heterologous protein production, while the genome-wide protease expression profile regulated by PrtT and the relationship between secondary carbon sources and protease expression are not well studied.
To comprehensively evaluate the role of PrtT as the master regulator of the proteases in A. niger, we successfully generated a homokaryotic prtT disruption mutant (delprtT) and an overexpression mutant (OEprtT) and performed transcriptome analysis of the delprtT and OEprtT strains to more thoroughly characterize the effect of PrtT on protease regulation. Additionally, based on the analysis of transcriptome data, we further demonstrated that the secondary carbon metabolism regulator AmyR is involved in the transcriptional regulation of proteases and their regulatory factor.
Materials and methods
Strains and culture conditions
The Escherichia coli strain Mach 1T1 (Invitrogen, USA) was used for cloning and plasmid propagation purposes, while E. coli BL21 (DE3) (TaKaRa, Japan) served as the host for heterologous expression of AmyR. The A. niger strain HL-1 and its mutants were cultivated in minimal medium (Czapek–Dox (CD) medium) containing 2% (w/v) glucose, 0.3% (w/v) NaNO3, 0.1% (w/v) KH2PO4, 0.2% (w/v) KCl, 0.05% (w/v) MgSO4·7H2O, and 0.001% (w/v) FeSO4·7H2O (pH = 5.5); or in complete medium (DPY medium) containing 2% (w/v) glucose, 1% (w/v) peptone, 0.5% (w/v) yeast extract, 0.1% (w/v) KH2PO4, and 0.05% (w/v) MgSO4·7H2O. For the protease activity assay, transformants were further cultured in Mandel’s solution (Chen et al. 2014) supplemented with different carbon sources. For the testing of cell wall integrity, PDA medium added with 500 μg/ml CR or 100 μg/ml CFW was used.
Construction of plasmids for prtT deletion and overexpression
All primers used in this study are listed in Supplementary Table 1. To construct the prtT deletion plasmid, the prtT upstream (ATG start codon) (1 kb) and downstream (TAA stop codon) (1 kb) regions and the pyrG gene (1398 bp) were amplified by prtTup1kb-F/R, prtTdown1kb-F/R and pyrG-F/R, respectively. The three fragments obtained were cloned into the pMD20-T vector using an NEBuilder HiFi Assembly Kit (Cat: E2621L, NEB) to generate the plasmid pΔprtT::pyrG. The prtT deletion cassette fragment obtained from digestion of pΔprtT::pyrG with EcoRI was introduced into A. niger by PEG-mediated protoplast transformation.
To construct the plasmid for prtT overexpression, the primer sets PglaA-F/R, prtT-F/R, TglaA-F/R and pyrG-F/pyrG-pMD20-R were used to amplify the glucoamylase promoter (PglaA) (976 bp), prtT cDNA, the glucoamylase terminator (TglaA) (688 bp), and the pyrG gene, respectively. The four DNA fragments were cloned into the pMD20-T vector using the NEBuilder HiFi Assembly Kit (Cat: E2621L, NEB), yielding pOEprtT::pyrG. The overexpression cassette was sequenced, released from pOEprtT::pyrG via ApaI restriction digestion, and used for A. niger transformation.
Extracellular protease activity assay
An extracellular protease activity assay was performed according to a previously described method (Sharon et al. 2009). Fermentation supernatants were collected by centrifugation. A 400 µl volume of azocasein solution (5 mg/ml azocasein) was mixed with 100 µl of each supernatant and incubated for 2 h at 37 °C with shaking at 200 rpm. Reactions were stopped by adding 150 µl of 12% (w/v) trichloroacetic acid (TCA), and the reaction mixtures were allowed to stand at room temperature for 30 min. The tubes were then centrifuged for 5 min at 10,000*g, and 100 µl of each supernatant was added to 100 µl of 1 M NaOH. The absorbance at 436 nm of the released azo dye was determined with a Multiscan Spectrum (Tecan, Switzerland).
RNA isolation and quantitative real-time PCR (qRT-PCR) analysis
Total RNA was isolated using RNAisoPlus (TaKaRa) according to the manufacturer’s instructions. Reverse transcription was performed with a PrimeScript RT-PCR Kit (TaKaRa, Japan). The gene-specific primers used for expression analysis are listed in Supplementary Table 1. Quantitative real-time PCR (qRT-PCR) was performed using an ABI 7500 Fast Real-Time PCR System with gpdA (glyceraldehyde-3-phosphate dehydrogenase) as the reference gene. The qRT-PCR was carried out with three replicates per prepared cDNA sample, and the results were analyzed with the 2−ΔΔCT method.
RNA-seq analysis
The WT strain and mutant strains (delprtT and OEprtT) were cultivated in DPY medium at 30 °C with shaking at 200 rpm for 40 h (exponential phase). The hypha was frozen by liquid nitrogen and ground into powder with mortar and pestle. Then total RNA was extracted using the HiPure Fungal RNA kit (Magen, China) according to the manufacturer’s protocols. RNA quality was determined by the Agilent Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA) to confirm that all samples had an RNA integrity value above 7.0. Libraries were constructed for delprtT, WT, and OEprtT, with two biological replicates per sample (delprtT_1, 2; WT_1, 2; OEprtT_1, 2). The resulting libraries were sequenced at the Beijing Genomics Institute (BGI) with 50 bp single-end reads on a BGISEQ-500 platform, averagely generating 24,134,007 raw sequencing reads and then 23,940,410 reads after filtering low quality (Supplementary Table S2). After filtering, the clean reads were mapped to the A. niger genome and A. niger cDNA using HISAT and Bowtie2. The gene expression level was quantified by RSEM (Li and Dewey 2011). The NOISeq method (Tarazona et al. 2015) was used to screen differentially expressed genes between the two groups. The raw sequencing data were deposited in the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra/) of the National Center for Biotechnology Information under the accession number PRJNA542223.
Electrophoretic gel mobility shift assay (EMSA)
The amyR cDNA sequence was amplified with the primer set amyR-F/amyR-R and integrated into the expression vector pET-22b( +) (TaKaRa, Japan) to generate the plasmid pET-amyR. The AmyR-6 × His protein was purified from E. coli BL21 (DE3) cells. A double-stranded probe was prepared by PCR with the biotin-labeled primer sets prtT-5′biotin-F/prtT-5′biotin-R and pepA-5′biotin-F/pepA-5′biotin-R. A specific competitor was prepared by the same method as both non-labeled primers. The purified AmyR-6 × His protein and probes were then subjected to EMSA performed according to the manufacturer’s instructions (Beyotime, China). The protein-probe complex transferred to the nitrocellulose membrane was imaged by Image Station 4000MM PRO (CareStream Health, USA) after UV cross-linking, blocking, and antibody incubation.
Results
Generation and protease activity analysis of the delprtT and OEprtT strains
Deletion of the prtT gene was performed by inserting the pyrG cassette into the prtT ORF (Fig. 1a). The correct delprtT mutant was confirmed by diagnostic PCR using primer sets P1/P2 and P3/P4 to amplify 1.8 kb and 1.5 kb DNA fragments, respectively, in contrast with the expected absence of a fragment in the WT strain (Fig. 1a). The homozygous deletion strain was also confirmed by PCR using the primer set P5/P6 to amplify the inner sequence of prtT (Fig. 1a). To obtain the OEprtT strain, we integrated prtT cDNA, which was under the control of the strong A. niger glucoamylase promoter (PglaA), into the WT strain. The primer set P7/P8 was used to confirm the prtT overexpression strain (Fig. 1b). A 2.8 kb band was detected for the prtT overexpression strain, while, as expected, no band was observed for the wild-type strain (Fig. 1b). The expression level of prtT under the control of the glaA promoter was increased by 22.5-fold in complete medium compared to that in the WT strain, while no transcription was observed for the deletion strain (Fig. 1c), suggesting that the prtT deletion strain and the overexpression strain were successfully obtained.

Construction of the delprtT and OEprtT strains and phenotypic analysis. a Confirmation of prtT gene deletion by PCR analysis. The entire ORF of prtT was deleted and replaced with the pyrG marker. b Confirmation of the generated prtT gene overexpression strain by PCR analysis. c qRT-PCR analysis of prtT. d Phenotypic analysis. Colony morphology of the WT, delprtT and OEprtT strains grown on CD containing 0.05% Triton X-100 and 1.5% SM or 1% casein at 30 °C for 5 days. e Determination of the extracellular protease activity of the wild-type and mutant strains under shake flask fermentation conditions
Growth of the WT, delprtT, and OEprtT strains on CD medium containing skim milk (SM) or casein as a nitrogen source was assessed (Fig. 1d). The OEprtT strain showed the largest hydrolytic halo, while the delprtT strain lost its proteolytic capacity (Fig. 1d). The extracellular protease activities of the cultured wild-type strain (WT) and the mutants were further quantified under shake flask fermentation conditions (Fig. 1e). At different time points, the protease activity consistently exhibited a gradually increasing tendency from delprtT to OEprtT (Fig. 1e). Even though the delprtT mutant had no hydrolytic halo, it still exhibited weak protease activity during the fermentation process (Fig. 1e). The above results confirmed that PrtT is indeed a key regulator of protease activity in A. niger.
Determine the regulatory function of PrtT by transcriptome analysis and GO classification
To characterize the effect of prtT on gene expression at the transcriptional level, we performed transcriptome analysis. A total of 1281 genes were differentially expressed between WT and delprtT (Supplementary Table 3 p1), comprising 598 upregulated (46.7%) and 683 downregulated (52.3%) genes (Fig. 2a). A total of 1000 significant DEGs between the delprtT and OEprtT samples were identified (Supplementary Table 3 p3), comprising 467 upregulated (46.7%) and 533 downregulated (53.3%) genes (Fig. 2a). Only 706 DEGs were identified between WT and OEprtT (Supplementary Table 3 p2), with 304 upregulated (43.1%) and 402 downregulated (56.9%) genes (Fig. 2a). A Venn diagram of the distribution of the DEGs showed that the union set of differentially transcribed genes contained 1888 DEGs (Fig. 2b).

Analysis of differentially expressed genes in the WT, delprtT and OEprtT strains. a Overview of the number of differentially expressed genes via pairwise comparisons of the WT vs delprtT, WT vs OEprtT, and delprtT vs OEprtT strains. b Venn diagrams of the number of overlapping and nonoverlapping upregulated or downregulated genes via pairwise comparisons of the WT vs delprtT, WT vs OEprtT, and delprtT vs OEprtT strains. c ClueGO analysis of differentially up-regulated or down-regulated genes in the delprtT strain. Representation of the main significantly up-regulated and down-regulated GOTerm (p < 0.05) in the delprtT mutant strain compared with the WT strain. d ClueGO analysis of differentially up-regulated or down-regulated genes in the OEprtT strain. Representation of the main significantly up-regulated and down-regulated GOTerm (p < 0.05) in the OEprtT mutant strain compared with the WT strain
To gain insight into the functional categories of the DEGs, the 1281 DEGs in WT-vs-delprtT group and 706 DEGs in WT-vs-OEprtT group were categorized into 42 and 36 GO terms, respectively (Fig. 2c and d; Supplementary Table 4). Figure 2c showed that deletion of prtT resulted in down-regulated of protease genes, cation transporters, and genes related to nitrogen metabolism, while genes involved in secondary metabolic process, oxidoreductase activity, transporters and binding activity were up-regulated. Meanwhile, Overexpression of prtT in wild type strain can up-regulate genes involved in proteolysis, carbohydrate metabolism and secondary metabolic processes (Fig. 2d). Conversely, the down-regulated genes in the OEprtT mutant were enriched in metabolism of nitrogen source, response to extracellular stimulation and oxidoreductase activity.
Thirty-two putative protease genes are regulated by PrtT
Through analysis of the union set of differentially transcribed genes, the transcriptional level of 32 genes annotated as putative proteases was found to change significantly between the host strain and the mutants (delprtT and OEprtT) (Table 1), including the mainly acid proteases pepA, pepB and pepF. However, pepD which was shown no significant expression in prtT deficient strain by Northern blot (Punt et al. 2008) did not belong to the DEGs in our RNA-seq data for its FPKM value < 0.5 (Supplementary Table S3 p4). Furthermore, the PCR results revealed pepD was scarcely transcribed in 5 different A. niger strains (CBS513.88, SH2, FGSC A1279, AS3.350 and HL-1) (Fig. 3a). Signal peptide analysis of these protein degradation genes using SignalP 4.0 revealed that 24 of the proteases contained a signal peptide and the other 8 contained no signal peptide, indicating that PrtT is not only involved in the regulation of extracellular proteases but is also involved in the regulation of intracellular proteases. Surprisingly, three protease genes (An12g08560, An15g06280, and An16g06560) were shown to be upregulated in delprtT, two of which are intracellular proteases. Functional classification of the 32 proteolytic enzymes via FungiFun (https://sbi.hki-jena.de/fungifun/fungifun.php) (Supplementary Table 5) showed that 31 of the proteases had proteolytic activity, including 7 serine-type peptidases (GO:0,008,236), 6 aspartic-type endopeptidases (GO:0,004,190), 6 aminopeptidases (GO:0,004,177), 4 carboxypeptidases (GO:0,004,180), and 3 metalloaminopeptidases (GO:0,070,006) (Fig. 3b). These results showed that in A. niger, in addition to regulating the main acid proteases (Punt et al. 2008), PrtT also controlled a broad range of other types of proteases.

The pepD gene detection and functional classification of the 32 proteolytic enzymes. a PCR amplification of pepD gene using cDNA and gDNA from 5 A. niger as the templates. b FungiFun enrichment analysis of the 32 differentially expressed protease genes in the delprtT, WT and OEprtT strains. The function corresponding to each GO term can refer to the Supplementary Table 5
The protease regulatory factor PrtT and its effector genes are inhibited by the amylolytic enzyme regulator AmyR
The significant conserved consensus sequence was screened in the 1000 bp upstream regions (relative to the start codon) of the 32 proteolytic genes by MEME (https://meme-suite.org/tools/meme). The putative PrtT binding consensus sequence 5′-CCGHCGG-3′ (H: A/C/T) was identified (Fig. 4a). Interestingly, the enrichment results for the regulatory motifs in the promoters of the 32 protease genes showed an AmyR binding motif (CGGN8CGG) in addition to the prtT core motif (Fig. 4a). We suspected that AmyR may participate in the regulation of proteases with PrtT. In support of this hypothesis, the EMSA results showed that AmyR binds to the promoters of the pepA and prtT genes (Fig. 4b). To further clarify the regulatory effect of AmyR on proteases, we cultured the WT strain in different carbon sources, including glucose, sucrose, starch, and maltose (Fig. 4c). The phenotypic analysis in Fig. 4c shows that the wild-type strain exhibited stronger proteolysis on plates with preferred carbon sources (glucose, sucrose) and that extracellular protease activity was significantly downregulated in secondary carbon sources (Fig. 4d). In addition, the transcription of prtT was significantly downregulated in secondary carbon sources (starch and maltose) compared to that on preferred carbon sources (glucose and sucrose) (Fig. 4e). These results suggested that the transcription factor PrtT was regulated by the carbon source metabolic regulator AmyR and competed with AmyR to regulate protease genes. Moreover, we knocked out amyR using homologous recombination. The qPCR results for the amyR knockout strain showed that the transcription level of the acid protease gene pepA increased by 228% (Fig. 4f). In addition, the prtT transcription level in the amyR knockout strain was 64.85% higher than that in the wild-type strain (Fig. 4f). These results suggested that knocking out amyR can abolish its inhibitory effect on protease activity and further confirmed that the carbon metabolism regulator AmyR is involved in the regulation of protease activity. However, protease activity analysis on delamyR and wild-type strain by phenotype, SDS-page, and protease activity assay showed that delamyR secreted fewer extracellular protein and had weaker protease activity in both glucose and starch media (Supplementary Fig. 1).

Analysis of the starch hydrolase regulator AmyR on protease activity and its regulatory factor PrtT. a The PrtT binding motif was identified for the 32 protease genes using MEME. b DNA binding of AmyR to the promoter regions of prtT and pepA. The "+" represents the addition of the indicated ingredient. The "−" represents no addition of the indicated ingredient. The “→” indicates the AmyR and DNA binding complex. c Colony morphology of the WT strain cultured in CD containing 1.5% SM and 0.05% Triton X-100 with different carbon sources. GLU glucose, SUC sucrose, STA starch, MAL maltose. d Determination of the extracellular protease activity of the wild-type strain cultured in different carbon sources. e qRT-PCR analysis of prtT expression in different carbon sources. f qRT-PCR analysis was performed on genes (amyR, prtT, pepA and glaA) in the amyR knockout strain. The glaA gene served as an effector gene for amyR deletion
DEGs involved in cell wall integrity in A. niger
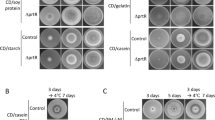
We identified 10 down-regulated DEGs linked to CWI in the group of delprtT-vs-WT (Supplementary Table 6). To further determine whether PrtT has a role in CWI, the stains were grown on the PDA medium added with 500 μg/mL CR and 100 μg/mL CFW (Fig. 5). Compared with the WT strain, PrtT did not have a significant effect on the growth of delprtT and OEprtT strains in PDA medium. However, CFW and CR had stronger inhibition effect on delprtT, while CR also inhibited WT and OEprtT. These results further verified PrtT plays an important role in CWI.

Cell wall integrity analysis of the delprtT, WT and OEprtT strains. a Colony morphology of the WT, delprtT and OEprtT strains grown on PDA medium, and PDA medium containing 500 ng/ml CR or 100 ug/ml CFW at 30 °C for 5 days, respectively. b Statistical analysis of the colony diameter of the WT, delprtT and OEprtT strains
Discussion
We analyzed the function of PrtT in A. niger by generating a homozygous prtT disruption mutant (delprtT) and an overexpression mutant (OEprtT). Phenotypic analysis and proteolytic activity assays showed that PrtT is a key protease regulator. Proteome sequencing of the A. fumigatus ∆prtT strain identified strongly reduced secretion of 22 proteases, including Lap1, Lap2, Cp1, Cp3 Alp1, Alp2, and Mep (Shemesh et al. 2017). Transcriptome sequencing of P. oxalicum showed that the expression of 17 proteases changed significantly between the parental strain 114–2 and the ΔprtT mutant (Chen et al. 2014). The differentially expressed protease profile obtained from comparative transcriptome analysis identified 32 differentially expressed protease genes, which suggested that A. niger contains more PrtT-regulated protease genes than other filamentous fungi. These differentially expressed protease genes included not only extracellular proteases but also intracellular proteases. This result was consistent with that in P. oxalicum (Chen et al. 2014). It is worth noting that even though studies by Punt et al. showed that pepD was significantly down-regulated in prtT-deficient strains, the gene did not appear in DEGs. This is because the pepD gene is scarcely transcribed in the A. niger strains (FPKM < 1 or RPKM < 1)(Pullan et al. 2014; van Munster et al. 2014; Borin et al. 2017; Gomaa et al. 2017; Wang et al. 2018), and this result was further verified by PCR in 5 different A. niger strains (Fig. 3a). Extracellular proteases hydrolyze proteins into smaller peptides and amino acids for subsequent absorption into cells, constituting a vital step in nitrogen metabolism. Intracellular proteases are important for various metabolic processes in cells, such as protein maturation and cell differentiation, and maintain the homeostasis of the cellular protein pool. Three upregulated protease genes were found in delprtT; this result corresponded with the transcriptome data for P. oxalicum (Chen et al. 2014), was responsible for the weak extracellular protease activity retained by the delprtT strain and provided a new perspective to engineer the A. niger host for heterologous protein expression.
MEME analysis showed that the binding motif for prtT in the region upstream of A. niger protease gene promoters was CCG(H)CGG (H: A/C/T). We speculate that the PrtT protein regulates the expression of protease genes through direct interaction with the core DNA motif. The results of EMSA for PrtT in P. oxalicum (Chen et al. 2014) support the hypothesis that PrtT directly regulates protease genes and that increased expression of PrtT is a good indicator of protease activity.
Protease activity was higher under the carbon-free conditions than on a preferred carbon source (Fig. 4c and d), an effect that is generally thought to be caused by CCR (Katz et al. 2000). Interestingly, protease activity under secondary carbon source culture conditions was lower than that in culture on preferred carbon sources (Fig. 4c and d). These results were also observed in P. oxalicum (Chen et al. 2014). Although CCR is thought to be an important factor regulating protease activity, under secondary carbon source culture conditions, CCR inhibition is relieved, and the major secondary carbon source regulator AmyR is activated (Gomi et al. 2000). AmyR is a major amylase regulator that recognizes CGGN8CGG motifs in the genome (Tani et al. 2001; Wang et al. 2012). In the results of MEME enrichment analysis of the 1000 bp upstream sequence in the 32 differentially expressed protease genes, a phenomenon was seen in which the AmyR and PrtT binding motifs overlap, suggesting that AmyR and PrtT have competitive binding effects on the regulation of downstream genes. The EMSA results verified that AmyR has a direct effect on the main acid protease pepA, and the relative quantification analysis of the amyR knockout showed that amyR deletion led to upregulated transcription of the acid protease pepA. These results demonstrate that AmyR represses transcription by directly binding to the promoter region of protease genes. In addition, the transcription of prtT is regulated by AmyR, and this result has not been found in previous studies. Although the delamyR showed up-regulation of pepA gene at the transcriptional level, the phenotype showed decreased extracellular protease activity. (Zhang et al. 2016) reported the same results, and they found that five putative protease genes were down-regulated in the ΔamyR strain by RNA-seq. We speculated that it was an indirect effect of AmyR by affecting growth because none of the 5 genes has an AmyR binding motif (Zhang et al. 2016).
PrtT also participated in a variety of biological regulatory functions, including secondary metabolic process, oxidoreductase activity, transporters, hydrolase activity and so on (Chen et al. 2014; Hagag et al. 2012; Shemesh et al. 2017). Our GO analysis further confirmed the previous results (Fig. 2). Interestingly, delprtT strain showed a deficiency in CWI. Among the 10 down-regulated CWI-related genes, wsc1, agsA, and acw-6 were related to MAPK pathway, glucan synthase, and chitinase, respectively. The main factors regulating the CWI of fungi include glucan synthase, chitinase, and MAPK signaling pathways (Pel et al. 2007), which might be responsible for the sensitivity of delprtT to CFW and CR.
In summary, we provided a genome-wide protease profile of the pathway-specific protease regulator PrtT, in which a delprtT-induced increase in protease expression provides a new target for engineering A. niger as a host. In addition, we showed for the first time that PrtT and the starch hydrolase regulator AmyR have a competitive regulatory effect on protease genes, which exposes new connections between carbon source and nitrogen source regulation in A. niger.
Abbreviations
- CCR:
-
Carbon catabolite repression
- CD:
-
Czapek–Dox medium
- DPY:
-
Dextrose-peptone-yeast extract medium
- EMSA:
-
Electrophoretic gel mobility shift assay
- qRT-PCR:
-
Quantitative real-time PCR
- DEGs:
-
Differential expression genes
- CWI:
-
Cell wall integrity
References
Ballester AR, Lopez-Perez M, de la Fuente B, Gonzalez-Candelas L (2019) Functional and pharmacological analyses of the role of Penicillium digitatum proteases on virulence. Microorganisms 7(7):E198
Bergmann A, Hartmann T, Cairns T, Bignell EM, Krappmann S (2009) A regulator of Aspergillus fumigatus extracellular proteolytic activity is dispensable for virulence. Infect Immun 77:4041–4050
Borin GP, Sanchez CC, de Santana ES, Zanini GK, Dos Santos RAC, de Oliveira PA, de Souza AT, Dal'Mas R, Riano-Pachon DM, Goldman GH, Oliveira JVC (2017) Comparative transcriptome analysis reveals different strategies for degradation of steam-exploded sugarcane bagasse by Aspergillus niger and Trichoderma reesei. BMC Genomics 18:501
Budak SO, Zhou M, Brouwer C, Wiebenga A, Benoit I, Di Falco M, Tsang A, de Vries RP (2014) A genomic survey of proteases in Aspergilli. BMC Genomics 15:523
Chen L, Zou G, Zhang L, de Vries RP, Yan X, Zhang J, Liu R, Wang C, Qu Y, Zhou Z (2014) The distinctive regulatory roles of PrtT in the cell metabolism of Penicillium oxalicum. Fungal Genet Biol 63:42–54
Gomaa OM, Selim NS, Wee J, Linz JE (2017) RNA Seq analysis of the role of calcium chloride stress and electron transport in mitochondria for malachite green decolorization by Aspergillus niger. Fungal Genet Biol 105:1–7
Gomi K, Akeno T, Minetoki T, Ozeki K, Kumagai C, Okazaki N, Iimura Y (2000) Molecular cloning and characterization of a transcriptional activator gene, amyR, involved in the amylolytic gene expression in Aspergillus oryzae. Biosci Biotechnol Biochem 64:816–827
Hagag S, Kubitschek-Barreira P, Neves GW, Amar D, Nierman W, Shalit I, Shamir R, Lopes-Bezerra L, Osherov N (2012) Transcriptional and proteomic analysis of the Aspergillus fumigatus DeltaprtT protease-deficient mutant. PLoS ONE 7:e33604
Jarai G, Buxton F (1994) Nitrogen, carbon, and pH regulation of extracellular acidic proteases of Aspergillus niger. Curr Genet 26:238–244
Katz ME, Masoumi A, Burrows SR, Shirtliff CG, Cheetham BF (2000) The Aspergillus nidulans xprF gene encodes a hexokinase-like protein involved in the regulation of extracellular proteases. Genetics 156:1559–1571
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform 12(1):323
Pel HJ, de Winde JH, Archer DB, Dyer PS, Hofmann G, Schaap PJ, Turner G, de Vries RP, Albang R, Albermann K, Andersen MR, Bendtsen JD, Benen JA, van den Berg M, Breestraat S, Caddick MX, Contreras R, Cornell M, Coutinho PM, Danchin EG, Debets AJ, Dekker P, van Dijck PW, van Dijk A, Dijkhuizen L, Driessen AJ, d'Enfert C, Geysens S, Goosen C, Groot GS, de Groot PW, Guillemette T, Henrissat B, Herweijer M, van den Hombergh JP, van den Hondel CA, van der Heijden RT, van der Kaaij RM, Klis FM, Kools HJ, Kubicek CP, van Kuyk PA, Lauber J, Lu X, van der Maarel MJ, Meulenberg R, Menke H, Mortimer MA, Nielsen J, Oliver SG, Olsthoorn M, Pal K, van Peij NN, Ram AF, Rinas U, Roubos JA, Sagt CM, Schmoll M, Sun J, Ussery D, Varga J, Vervecken W, van de Vondervoort PJ, Wedler H, Wosten HA, Zeng AP, van Ooyen AJ, Visser J, Stam H (2007) Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nat Biotechnol 25:221–231
Pullan ST, Daly P, Delmas S, Ibbett R, Kokolski M, Neiteler A, van Munster JM, Wilson R, Blythe MJ, Gaddipati S, Tucker GA, Archer DB (2014) RNA-sequencing reveals the complexities of the transcriptional response to lignocellulosic biofuel substrates in Aspergillus niger. Fungal Genet Biol 1:1–14
Punt PJ, Schuren FH, Lehmbeck J, Christensen T, Hjort C, van den Hondel CA (2008) Characterization of the Aspergillus niger prtT, a unique regulator of extracellular protease encoding genes. Fungal Genet Biol 45:1591–1599
Ruijter GJ, Visser J (1997) Carbon repression in Aspergilli. FEMS Microbiol Lett 151:103–114
Sharon H, Hagag S, Osherov N (2009) Transcription factor PrtT controls expression of multiple secreted proteases in the human pathogenic mold Aspergillus fumigatus. Infect Immun 77:4051–4060
Shemesh E, Hanf B, Hagag S, Attias S, Shadkchan Y, Fichtman B, Harel A, Kruger T, Brakhage AA, Kniemeyer O, Osherov N (2017) Phenotypic and proteomic analysis of the Aspergillus fumigatus DeltaPrtT, DeltaXprG and DeltaXprG/DeltaPrtT protease-deficient mutants. Front Microbiol 8:2490
Tani S, Itoh T, Kato M, Kobayashi T, Tsukagoshi N (2001) In vivo and in vitro analyses of the AmyR binding site of the Aspergillus nidulans agdA promoter; requirement of the CGG direct repeat for induction and high affinity binding of AmyR. Biosci Biotechnol Biochem 65:1568–1574
Tarazona S, Furio-Tari P, Turra D, Pietro AD, Nueda MJ, Ferrer A, Conesa A (2015) Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res 43:e140
van Munster JM, Daly P, Delmas S, Pullan ST, Blythe MJ, Malla S, Kokolski M, Noltorp ECM, Wennberg K, Fetherston R, Beniston R, Yu X, Dupree P, Archer DB (2014) The role of carbon starvation in the induction of enzymes that degrade plant-derived carbohydrates in Aspergillus niger. Fungal Genet Biol 72:34–47
Wang P, Kojima T, Kobayashi T, Nakano H (2012) Comprehensive analysis of the DNA-binding specificity of an Aspergillus nidulans transcription factor, AmyR, using a bead display system. Biosci Biotechnol Biochem 76:1128–1134
Wang B, Lv Y, Li X, Lin Y, Deng H, Pan L (2018) Profiling of secondary metabolite gene clusters regulated by LaeA in Aspergillus niger FGSC A1279 based on genome sequencing and transcriptome analysis. Res Microbiol 169:67–77
Zhang H, Wang S, Zhang XX, Ji W, Song F, Zhao Y, Li J (2016) The amyR-deletion strain of Aspergillus niger CICC2462 is a suitable host strain to express secreted protein with a low background. Microb Cell Fact 15:68
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Numbers 31871736 and 31870024) and the Guangdong Provincial Key Laboratory of Advanced Biofermentation Technology Enterprise in Flavoring & Food (Grant Number 2017B030302002).
Supplementary Figure 1
Extracellular protease activity analysis of delamyR strain. a Protease secretion phenotype of wild-type strains (1,2,3) and delamyR (a, b, c) under 10% glucose culture condition. b Protease secretion phenotype of wild-type strains (1,2,3) and delamyR (a, b, c) under 10% starch culture condition. c SDS-PAGE of extracellular protein from wild-type strain under glucose and starch fermentation medium. d SDS-PAGE of extracellular protein from delamyR strain under glucose and starch fermentation medium. e Determination of protease activity in wild-type strain and delamyR.
Supplementary Table 1. Primers used in this study.
Supplementary Table 2. Summary and QC of all RNA-seq libraries.
Supplementary Table 3. Complete list of all differentially expressed genes in the WT-vs-delprtT, WT-vs-OEprtT, delprtT-vs-OEprtT strains.
Supplementary Table 4. Enriched GO terms from all differentially up-regulated or down-regulated genes in the delprtT/WT and OEprtT/WT datasets.
Supplementary Table 5. Functional classification of the 32 proteolytic enzymes by FungiFun.
Supplementary Table 6. DEGs related to CWI in the WT-vs-delprtT group.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Huang, L., Dong, L., Wang, B. et al. The transcription factor PrtT and its target protease profiles in Aspergillus niger are negatively regulated by carbon sources. Biotechnol Lett 42, 613–624 (2020). https://doi.org/10.1007/s10529-020-02806-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-020-02806-3