
Abstract
Objectives
To establish a positive cloning system with a zero background for high-throughput DNA cloning purpose.
Results
The cloning vector, pRI857, and the genomic-library construction vector, pRI857-BAC, were constructed based on the mechanism of expression of the thermo-sensitive cI857 repressor gene that can stringently repress the PR promoter and kanamycin resistance gene (PR-kan R) at 30 °C, but have no effect on PR-kan R gene at 37 °C or at higher temperatures. When the pRI857 vectors were transformed into E. coli with or without a target foreign DNA fragment inserted at the BfrBI site of the cI857 gene, only colonies with the foreign DNA fragment survive. We extended this method to construct a pRI857-BAC vector for genomic library cloning which displays an efficiency of ~107 cfu per µg of genomic DNA, with no empty vectors detected.
Conclusions
Cloning by indirect activation of resistance marker gene represents a novel DNA-capturing system, which can be widely applied for high-throughput DNA cloning.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gene cloning is an essential bio-technique tool and it is desirable to generate recombinant clones with zero or low background of false-positive rate. The lacZ-based blue/white screening method is the most widely used approach that allows researchers to readily distinguish the recombinants from the empty vectors by inspecting colors of the colonies (Holton and Graham 1991). Unfortunately, the false-positive rate of this method is not negligible due to the inefficient T-tailing of vector DNA and defects of X-gal staining (Guo and Bi 2002). The alternative method using counter-selection markers such as ccdB and sacB became popular for DNA cloning due to easy handling and good transform efficiency. However, there are some limitations because a large number of mutations can lead to ccdB tolerance, and the sacB gene can cause the cell toxin levan to accumulate in the periplasm in addition to a high sucrose resistance (Gay et al. 1983; Lepesant et al. 1974; Mitchell and Michaelis 1998).
From a practical point of view, recombinants selection by activating an antibiotic resistance marker is more logical and reliable. Therefore, we have engineered the temperature-sensitive repressor promoter system cI857-PR for positive generation of recombinants. The expression of the positive selection marker kanamycin resistance gene (kan R) was stringently repressed by the cI857-PR at 30 °C or lower, but gradually recovered as the temperature was increased above 30 °C (Breitling et al. 1990; Villaverde et al. 1993). Here, we report a novel cloning system pRI857 containing the cI857-PR-kan R cassette in which the target DNA fragment was inserted at the BfrBI site of the cI857 gene upon DNA cloning, resulting in the disruption of the cI857 repressor. Consequently, only the recombinant colonies rather than the transformants carrying the empty vectors can survive in the presence of kanamycin at 30 °C due to the transcriptional activation of the kan R gene.
Materials and methods
Materials
The bacterial strains and plasmids used in this study are listed in Supplementary Table 1. The sequence of primers are listed in Supplementary Table 2 and DNA sequencing were carried out by Invitrogen (Shanghai, China) or Genewiz (Suzhou, China). Ampicillin (amp), bleomycin (ble), gel purification kits and plasmid mini-preparation kits were purchased from Invitrogen. Restriction enzymes, T4-ligases, CIAP (calf intestinal alkaline phosphatase) and Gibson assembly kits were purchased from New England Biolabs (NEB). DNA marker, 1-kb DNA ladder was purchased from Takara. DNA manipulation and E. coli transformations were performed using established methods. E. coli strains and Pseudomonas syringae HS191 (Hurlbert and Gross 1983) were respectively cultured at 37 °C and 30 °C in lysogeny broth with appropriate antibiotics (ampicillin at 100 μg ml−1, kanamycin at 50 μg ml−1 and bleomycin at 50 μg ml−1 unless otherwise noted).
Construction of pRI857-based vectors
The three essential genetic components of the pRI857 vector were PCR amplified separately from their original plasmids, and then assembled simultaneously using the Gibson assembly method. In brief, the PR promoter was PCR amplified from pCP20 (Datsenko and Wanner 2000) using the primers P1/P2 and then assembled into the upstream of kan R gene in the PCR amplified vector pBBR1MCS-2 (Kovach et al. 1995) with primers P3/P4, resulting in an intermediate plasmid named pBBR1MCS-PR. The cI857 gene was PCR amplified from pCP20 using the primers P5/P6 and then inserted into the downstream of the Plac promoter in pUC19 with primers P7/P8, resulting in an intermediate plasmid named pUC-cI857. Finally, the PR-kan R (amplified using pBBR1MCS-PR as the template vector with primers P9/P10), Plac-cI857 (amplified using pUC-cI857 with primers P11/P12), and ColE1-dbl terminator (amplified using pKV6 (Petersen et al. 2013) with primers P13/P14) were simultaneously assembled and transformed into E. coli DH5α. The resulting plasmid DNA was confirmed by sequencing and designated pRI857 (Fig. 1). The cI857-PR-kan R cassette derived from plasmid pRI857 using the primers P15/P16 was then ligated into the pCC1BAC vector treated with restriction enzymes PciI and ScaI, by means of Gibson assembly. The newly constructed plasmid DNA was confirmed by sequencing and designated pRI857-BAC (Supplementary Fig. 1).

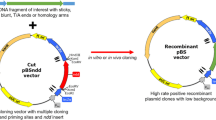
Flowchart of the pRI857 vector construction
Thermal sensitivity assay
Thermal sensitivity was tested by plating 200 µl E. coli DH5α/pRI857 on LB agar containing kanamycin in an overnight incubation at 30, 33, 37 and 39 °C. Cell survival rates were determined from the c.f.u. values. In addition, the growth of E. coli DH5α/pRI857 was measured from the OD600 values.
Results and discussion
Principles of the pRI857-based cloning system
The mechanism of recombiannt generation using pRI857 vector relies on the cI857-PR-kan R cassette, in which the kan R can be activated at 37 °C but is strongly inhibited by the thermo-sensitive repressor CI857 at 30 °C. Consequently, the empty vector transformants can proliferate in the presence of kanamycin at 37 °C but not at 30 °C. In the scenario of insertion of the foreign DNA of interest into the BfrBI site of the cI857 gene resulting in the disruption of the repressor gene, PR-kan R was activated. Therefore, only the recombinant plasmids can survive in the presence of kanamycin at 30 °C but not the empty vector, which gives a zero-background cloning effect. The flowchart of the stepwise protocol to construct the pRI857 vector is shown in Fig. 1.
The transformation experiment indicated that DH5α/pRI857 grew well at 37 °C, but not at 30 °C in the presence of kanamycin (Fig. 2a). Further, the growth curves showed that the DH5α/pRI857 cells incubated at higher temperatures gradually restored to normal growth. However, growth rate at 39 °C was slightly slower than that at 37 °C, probably due to the growth inhibition at high temperature (Fig. 2b). The stringency and feasibility of the cI857-PR-kan R system demonstrated the great potential of pRI857 vectors in zero-background cloning.

Thermo-sensitivity studies of cI857-PR system. a E. coli DH5α/pRI857 was plated on LB plate containing kanamycin and incubated at 30, 33, 37 and 39 °C, respectively. b The growth of E. coli DH5α/pRI857 was measured as the at OD600 value from 30 to 39 °C
In our earlier attempts, we directly integrated kan R gene to the downstream of promoter PR in plasmid pCP20 in order to repress kan R expression at 30 °C and re-activated it at higher temperatures via the interaction of the already existed CI857 with the promoter PR in pCP20 (Cherepanov and Wackernagel 1995). However, undifferentiated growth effects of E. coli DH5α/pCP-kan transformants were observed within the temperature range from 28 to 39 °C. Sequence analysis data later revealed that there was no promoter gene for cI857 in the parent pCP20 vector resulting in a constitutive expression of the PR-kan R gene. In order to control the expression of kan R, a Plac promoter together with a canonical ribosome binding sequence AGGA were used to drive the expression of cI857 as mentioned in the Materials and Methods section.
Applications of DNA cloning and genomic-library construction by using pRI857-based vectors
To test the gene-cloning efficiency of the pRI857-based vectors, the PCR fragment of AraC-Pbad-mCherry from the parent plasmid pCMrfp (Bi et al. 2013) with primers P17/18 was inserted into the BfrBI site of vector pRI857 using T4 ligase, and then the ligation mixture was transformed into E. coli DH5α, followed by plating on LB agar containing kanamycin and incubating at 30 °C. The colony PCR reactions were performed for the randomly picked colonies with primers P19/20 to verify that they contained the correct target DNA fragments (Fig. 3a). We also tested the robustness of the cI857-PR-kan R cassette by inserting ble R (Supplementary Fig. 2), PluxI-sfGFP (Supplementary Fig. 3) and 16S rRNA of P. syringae HS191 (Supplementary Fig. 4) fragments, respectively, into the pRI857 vector. The recombinant transformants were verified by colony PCR.

Application of pRI857 vectors in gene cloning and genomic library construction. a PCR analysis of 6 randomly selected transformants of the ligation mixture of pRI857 vector and the araC-Pbad-mCherry PCR product. The expected size of the PCR product was 2126 bp. b BamHI restriction analysis of 10 randomly selected BAC clones from the genomic library of strain P. syringae HS191 using the pRI857-BAC vector
The pRI857-BAC vector allows for positive selection of recombinants by activating resistance markers during the cloning processes. As an assessment of large DNA fragment cloning, the pRI857-BAC vector was used to clone the genomic fragments of strain P. syringae HS191.The resulting library yielded approximately 9000 CFU using 0.86 ng of genomic DNA, and 100 recombinants were end-sequenced with no empty vectors detected (Fig. 3b). Furthermore, the pRI857-BAC based genomic library construction efficiency achieved ~107 cfu per µg of genomic DNA.
Conclusion
Compared with traditional cloning vectors, pRI857 provides a simple and efficient approach for DNA cloning. The desired transformants can be directly selected with kanamycin at 30 °C, requiring no specific host strain or additional reagents like arabinose, IPTG or X-gal. Furthermore, the empty vector cannot survive on kanamycin plate, thus sparing the tedious and time-consuming screening procedures. The pRI857-based vectors are especially useful for the high-throughput DNA cloning in the post-genomic era, which holds a great potential in biotechnology, structural biology and other high-throughput demanding fields.
References
Bi CH, Su P, Müller J, Yeh YC, Chhabra SR, Beller HR, Singer SW, Hillson NJ (2013) Development of a broad-host synthetic biology toolbox for Ralstonia eutropha and its application to engineering hydrocarbon biofuel production. Microb Cell Factories 12:1–10
Breitling R, Sorokin AV, Behnke D (1990) Temperature-inducible gene expression in Bacillus subtilis mediated by the cI857-encoded repressor of bacteriophage lambda. Gene 93:35–40
Cherepanov PP, Wackernagel W (1995) Gene disruption in Escherichia coli: Tc R and Km R cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14
Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97:6640–6645
Gay P, Le Coq D, Steinmetz M, Ferrari E, Hoch JA (1983) Cloning structural gene sacB, which codes for exoenzyme levansucrase of Bacillus subtilis: expression of the gene in Escherichia coli. J Bacteriol 153:1424–1431
Guo B, Bi Y (2002) Cloning PCR products. An overview. Methods Mol Biol 192:111–119
Holton TA, Graham MW (1991) A simple and efficient method for direct cloning of PCR products using ddT-tailed vectors. Nucleic Acid Res 19:1156
Hurlbert RE, Gross DC (1983) Isolation and partial characterization of the cell wall of Pseudomonas syringae pv. syringae HS191: comparison of outer membrane proteins of HS191 with those of two plasmidless derivatives. Microbiology 129:2241–2250
Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM, Peterson KM (1995) Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176
Lepesant JA, Lepesant-Kejzlarova J, Pascal M, Kunst F, Billault A, Dedonder R (1974) Identification of the structural gene of levansucrase in Bacillus subtilis Marburg. Mol Gen Genet 128:213–221
Mitchell KM, Michaelis EK (1998) Multimembrane carbon fiber electrodes for physiological measurements of nitric oxide. Electroanalysis 10:81–88
Petersen KV, Martinussen J, Jensen PR, Solem C (2013) Repetitive, marker-free, site-specific integration as a novel tool for multiple chromosomal integration of DNA. Appl Environ Microbiol 79:3563–3569
Villaverde A, Benito A, Viaplana E, Cubarsi R (1993) Fine regulation of cI857-controlled gene expression in continuous culture of recombinant Escherichia coli by temperature. Appl Environ Microbiol 59:3485–3487
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No. 21271104 and 21671103 for Yi J, and 31300111 for Wang Y). The strain Pseudomonas syrigae HS191 was a gift from Dr. Dennis C Gross (Texas A&M University).
Supporting information
Supplementary Table 1—The bacterial strains and plasmids used.
Supplementary Table 2—Sequence of primers used.
Supplementary Fig. 1—Plasmid map of the pRI857-BAC vector.
Supplementary Fig. 2—Cloning test of the ble R gene using pRI857 vector.
Supplementary Fig. 3—Cloning test of the PluxI-sfGFP gene using pRI857 vector.
Supplementary Fig. 4—Cloning test of the 16S rRNA gene from strain P. syringae HS191 using pRI857 vector.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Research involving human and animal rights
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Accession Numbers
The sequences reported in this article were deposited in the GenBank database [accession numbers: KX774470 (pRI857), KX774471 (pRI857-BAC)].
An erratum to this article is available at http://dx.doi.org/10.1007/s10529-017-2316-3.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, K., Su, H., Yang, M. et al. Construction of a thermo-sensitive pRI857 vector for efficient DNA capturing in Escherichia coli . Biotechnol Lett 39, 905–909 (2017). https://doi.org/10.1007/s10529-017-2313-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-017-2313-6