
Abstract
Antibody drug conjugates (ADCs) have emerged as a viable option in targeted delivery of highly potent cytotoxic drugs in treatment of solid tumors. At the time of writing, only two ADCs have received regulatory approval with >40 others in clinical development. The first generation ADCs suffered from a lack of specificity in amino acid site-conjugations, yielding statistically heterogeneous stoichiometric ratios of drug molecules per antibody molecule. For the second generation ADCs, however, site-specific amino acid conjugation using enzymatic ligation, introduction of unnatural amino acids, and site-specific protein engineering hold promise to alleviate some of the current technical limitations. The rapid progress in technology platforms and antibody engineering has introduced novel linkers, site-specific conjugation chemistry, and new payload candidates that could possibly be exploited in the context of ADCs. A search using the Clinical Trial Database registry (www.clinicaltrials.gov), using the keyword ‘antibody drug conjugate’, yielded ~270 hits. The main focus of this article is to present a brief overview of the recent developments and current challenges related to ADC development.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
With the introduction of engineered humanized and fully human monoclonal antibodies (mAbs), targeted cancer therapy has reached a new level of sophistication (Sliwkowski and Mellman 2013; Gharwan and Groninger 2016). However, while therapeutic mAbs have had a high success rate in treatment of hematologic tumors, targeting solid tumors has been relatively difficult because of their lack of sufficient permeability (Choi et al. 2013). Most solid tumors are derived from epithelial cells and their tissues pose significant barriers to drug penetration due to their high interstitial fluid pressure (IFP), high cell density, excessive deposition of extracellular matrix (ECM), and physical barriers composed of stroma proteins. In order to overcome the above challenges, passive and active drug targeting models have successfully been introduced (Lammers et al. 2012).
In passively targeted drug delivery, the objective is to reach drug accumulation in the vicinity of the tumor, often by increasing its half-life, and eventually to achieve an “enhanced permeation and retention (EPR)” effect (Bae and Park 2011; Khawar et al. 2015). The leaky nature of the tumor’s vascular system coupled with its poor lymphatic drainage, enableS drug accumulation within the tumor mass, contributing to the EPR effect. Examples of passive drug targeting include the use of pegylation, artificial phospholipid vesicles such as liposomes, polymeric micelles, and other nanoparticles for drug delivery. Some of the barriers to passive spontaneous drug delivery include the heterogeneous vascularization of the tumor tissues, higher IFP, and possibly areas of necrosis (Torchilin 2014).
In contrast, actively targeted drug delivery involves the interaction between a tumor cell specific antigen (e.g., a surface receptor) and the drug or the drug carrier. Since certain tumor-associated antigens are over-expressed (e.g., a high copy number is considered >105 surface antigens per cell) on the surface of the cancer cells, one can harness the specificity, high affinity, and relatively longer half-life of a mAb to selectively bind to high density surface receptors; however, binding alone may not often lead to cytotoxicity. Fortunately, combining the potency and cytotoxicity of a chemotherapeutic agent with tumor-specificity of a mAb has the potential to exploit an effective approach in intracellular drug delivery. Clinical applications of antibody drug conjugates (ADC) represent an option in the treatment of solid tumors using actively targeted drug delivery. An ADC is designed to take advantage of the potency of a cytotoxic agent and specificity of a mAb, in order to induce anti-tumor activity, while minimizing systemic toxicity of the free drug. The first ADC product was gemtuzumab ozogamicin (Mylotarg), an anti-CD33 humanized IgG4 antibody calicheamicin conjugate, that was approved by the United States Food and Drug Administration (US-FDA) in 2000 (Sievers and Senter 2013) for patients suffering from acute myeloid leukemia, but was withdrawn in 2010. Although earlier granted an ‘orphan’ status, the European Medicines Agency rejected Mylotarg in 2008, on the basis of criticism of its supporting clinical study designs (Makuch and Shi 2014). Brentuximab vedotin (Adcetris), an anti-CD30 antibody linked to a monomethyl auristatin E, and ado-trastuzumab-emtansine conjugate (Kadcyla) were approved by the FDA, respectively, in 2011 and 2013. The re-emergence of ADCs as a novel class of drugs in oncology has resulted in more than 40 candidates in clinical development (Mack et al. 2014; Perez et al. 2014; Kim and Kim 2015; Polakis 2015). In this article, key attributes of an effective ADC, mode-of-action, product characterization, and recent progress will be briefly discussed.
Anatomy of an ADC
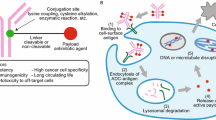
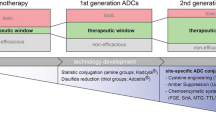
An ADC consists of three key components: a drug or payload, a linker, and an antibody. The so-called first-generation ADC platforms experienced several drawbacks including inadequate payload potency, linker chemistry, tumor biology, insufficient tumor internalization, lower than expected tumor accumulation rates in human subjects, heterogeneous, and immunogenicity (Chari 2008). In contrast, introduction of second-generation ADCs has alleviated some of the above hurdles, leading to the regulatory approval of ado-trastuzumab-emtansine and brentuximab vedotin ADCs (Table 1). The design and development attributes of ADCs (Fig. 1) present a relatively more complex biochemical and analytical characterizations as well as formulation challenges. Currently, the ADC research and development is experiencing a rapid growth in understanding and optimization of the mAb, the linker, conjugation chemistry, and the payload design. A brief description of each of the ADC components is presented in the following sections.

A simplified representation of an ADC architecture with some of the desired attributes
Monoclonal antibody selection
Monoclonal antibodies can be engineered to be extremely specific in antigen binding for a wide array of oncology and immunology related indications (Bakhtiar 2012; Liu 2015). Majority of approved therapeutic mAbs are based on the IgG1 isotype with a few engineered using IgG2 and IgG4 scaffolds. IgG3 has not yet attracted much attention due to its relatively short half-life and allotypic polymorphism, obtained from serological studies, that could affect CH3–CH3 inter-domain interactions leading to possible variations in C1q binding (e.g., complement activation) (Vidarsson et al. 2014).
In treating solid tumors, initial studies showed a heterogeneous distribution of mAbs within the tumor (Juweid et al. 1992). The non-uniform distribution of high affinity mAbs within tumor tissues has been partly due to the so-called “binding site barrier”, which is the result of high non-specific protein binding within tumors. Therefore, the mAb alone will have limited therapeutic efficacy in treating solid tumors unless titrated to high doses, in order to achieve saturation of target binding, which could present safety and tolerability concerns. An alternative would be to either use a therapeutic mAb in combination with a small cytotoxic molecule such as in combination therapies, or use the former as a delivery vehicle for the latter.
The ideal mAb in an ADC would have the following features: (1) it utilizes receptor-mediated endocytosis or other appropriate intracellular trafficking via the endosome and lysosomal systems (Fig. 2); (2) it is engineered against a surface antigen that is highly expressed on the target cell (Fig. 2); (3) the cell-surface receptor shedding is minimal to reduce circulatory binding; (4) it is fine-tuned with respect to its binding affinity, specificity, and internalization kinetics; (5) it produces a low immune response in humans; (6) the payload conjugation does not affect the mAb’s stability, internalization, binding, and overall pharmacokinetics; (7) if the therapeutic indication requires it, the mAb triggers Fc effector functions such as complement-dependent cytotoxicity (CDC), antibody-dependent cell-mediated cytotoxicity (ADCC), and/or antibody-dependent cell-mediated phagocytosis (ADCP); and (8) it has a long half-life to allow significant accumulation in a tumor cell. Therefore, the specificity and affinity of a mAb to the surface target antigen are amongst key determinants in efficient receptor-mediated endocytosis (e.g., micropinocytosis, clathrin-mediated endocytosis, or caveolin-mediated endocytosis) (Correia 2010; Hogarth and Pietersz 2012; Jackson and Stover 2015).

A simplified general scheme for the mechanism of ADC activity
The mAb in gemtuzumab ozogamicin was a recombinant humanized IgG4 kappa antibody which was generated in mammalian cell suspension culture using a myeloma NS0 cell line. Gemtuzumab ozogamicin targeted the CD33 antigen that was a sialic acid-dependent adhesion transmembrane receptor expressed on cells of myeloid lineage. IgG4 and IgG2 subclasses have a reduced affinity for a number of Fcγ receptors (RI, RII, and RIII) and C1q activation which translates to lower ADCC and CDC activities. Therefore, IgG4 subclass is preferred for immunotherapy where activation of the host effector function is not beneficial. The challenge in using a wild-type IgG4 scaffold is dynamic Fab-arm exchange with endogenous IgG4 molecules yielding bispecific Abs, unable to cross-link cognate antigen, and hence, reduced efficacy (van der Neut et al. 2007). In order to minimize IgG4s’ Fab-arm exchanges, manufacturers often stabilize the core-hinge region by site-directed mutagenesis (as in the case of gemtuzumab ozogamicin), where the serine amino acid at position 228 is replaced by proline, known as the S228P mutation (Cys-Pro-Ser-Cys-Pro to Cys-Pro-Pro-Cys-Pro).
The mAb in brentuximab vedotin (Adcetris) is a heterotetrameric chimeric IgG1 with two kappa light chains and two gamma one heavy chains against CD30. CD30 is a member of the tumor-necrosis factor (TNF) receptor superfamily. Activated immune cells such as T and B cells show expression of CD30 as do systemic anaplastic large cell lymphoma (sALCL), Hodgkin’s lymphoma (HL), mature T cell lymphomas, and B cells formed from non-Hodgkin’s lymphoma (NHL) (Scott et al. 2012). The mAb in ado-trastuzumab-emtansine conjugate (Kadcyla) is a humanized anti-HER2 IgG1, which has been well-characterized (Van den Mooter et al. 2015). Similar to most other therapeutic mAbs and ADCs, the pharmacokinetics of ado-trastuzumab-emtansine is non-linear, characterized by a two-compartment model with first-order elimination from the central compartment (Dhillon 2014). Unlike small molecule drugs, therapeutic mAbs and ADCs often exhibit linear and non-linear pharmacokinetics at high- and low-doses, respectively (Han and Zhao 2014). Target-mediated clearance is one of the reasons for non-linear elimination of mAbs (Vugmeyster et al. 2012). Trastuzumab undergoes facile internalization subsequent to target binding on the cell surface; and hence, the non-linearity of its pharmacokinetics is due to receptor-mediated drug disposition. Therefore, trastuzumab’s mean half-life increases and its clearance decreases with increases in dose, presumably due to saturation of the above elimination route (Tang et al. 2004).
Payload selection
Payload selection is another critical factor that defines the success of an ADC. An ideal ADC payload is a highly potent small molecule with lack of specificity. Currently, there are two broad categories of payloads for conjugation to a mAb. The first category is referred to as radionuclide antibody conjugates (RACs), where a radionuclide emitting radiation penetrates into the targeted cells of the solid tumor to induce a sufficient lethal response with no or minimal damage to the surrounding healthy cells. Initially, there were two approved RACs, namely 131I-tositumomab (Bexxar) and 90Y-ibritumomab (Zevalin) used in treatment of B cell lymphoma, HL, NHL, or multiple myeloma (Kitson et al. 2013). Bexxar was a mouse IgG2a anti-CD20 mAb labelled with I-131 which emitted both beta and gamma radiations with a half-life of approximately eight days. Zevalin was a mouse IgG1 anti-CD20 mAb labelled with a beta emitter, Y-90, with a 64 h half-life. However, Bexxar was voluntarily withdrawn due to its decline in sales, partly attributed to its complex dosing preparation (Prasad 2014; AlDeghaither et al. 2015). Further discussion on RACs is beyond the scope of this manuscript but details can be found elsewhere (Navarro-Teulon et al. 2013).
The second and a major category for antibody payloads include high potency synthetic or natural product small molecules. ADC payloads approved or under development are for the most part cytotoxic agents with picomolar or sub-picomolar potencies (e.g., 100–2000 fold more potent than doxorubicin, vinca alkaloids, or taxanes). The mechanisms-of-action of these payloads are often either interference with the tumor cell mitotic cycle by inhibition of tubulin polymerization, yielding G2/M phase cell cycle arrest or disruption of DNA by alkylation, cleavage (Lambert 2012; Singh et al. 2015). Some of these payloads, mainly natural products, such as monomethyl auristatin E are extremely toxic (potency ~10−11–10−9 M) to healthy cells and cannot be administered as mono-therapy to cancer patients. For instance, maytansine, which inhibits microtubule polymerization when administered alone, can lead to dose-limiting neuropathy, fatigue, and diarrhea (Wong and Hurvitz 2014; Ho and Chien 2014). Other examples of ADC payloads include calicheamicins, duocarmycins, tomaymycin, maytansinoids, pyrrolobenzodiazepine dimers, dolastatin 10, and tubulysins (Dosio et al. 2014; Maderna and Leverett 2015; Kamath and Iyer 2016).
A higher drug-antibody ratio or an average drug-to-antibody molar ratio of highly potent cytotoxins used as ADC payloads yields greater potency. However, a higher drug-antibody ratio is also associated with deterioration of certain ADC attributes such as increased systemic clearance, reduction in therapeutic efficacy, lower stability under stressed conditions (Adem et al. 2014), heterogeneity, and higher propensity to aggregation, presumably due to the hydrophobic nature of the payloads (Beckley et al. 2013). Some adverse clinical effects including immunogenicity have been attributed to protein aggregation (Moussa et al. 2016). Based on extensive research, a drug-antibody ratio of about 4 is an optimal threshold for anti-tumor activity (Hamblett et al. 2004; McDonagh et al. 2006). Promising preliminary data on the use of site-specific conjugation to yield drug-antibody ratio values of 6 and 8, for tackling low-expression tumor antigens and slower tumor cell internalization kinetics, while achieving a high therapeutic index, has been reported (Strop et al. 2015).
Linker design
There are two broad categories of ADC linkers, namely cleavable and non-cleavable linkers (Blencowe et al. 2011; Nolting 2013; Jain et al. 2015). The former can be divided into several sub-types including:
-
Acid-labile linkers such as a hydrazine linker which undergoes hydrolysis in endosomes (pH 5–6.5) and lysosomes (pH 4.5–5) environments (e.g., gemtuzumab ozogamicin). For example, hydrazones have half-life values of 183 and 4.4 h at pH 7 and 4.4, respectively (McCombs and Owen 2015). Also, lower cellular pH conditions can be due to tumor hypoxia resulting from an imbalance of oxygen delivery and rise in lactic acid production (the Warburg effect) (Chiche et al. 2010).
-
Protease cleavable linkers like a valine-citrulline (Val-Cir) dipeptide that can be cleaved by cathepsin B, a cysteine protease, under lysosomal acidic environment (e.g., brentuximab vedotin).
-
Disulfide linkers that rely on the high level of cellular reduced glutathione to release their payload.
In contrast, the non-cleavable linkers have higher blood stability and often rely on internalization kinetics, facile lysosomal delivery, and ensuing ADC degradation to yield cancer cell apoptosis (e.g., ado-trastuzumab-emtansine). Upon internalization, the free payload is released with the linker attached to an amino acid from the mAb. To date, most non-cleavable linkers used have been thioether-based bonds (Hamilton 2015). Non-cleavable linkers account for about 20 % of the ADCs in clinical testing. Clearly, there is inter-dependency between the linker and conjugation chemistry. The choice of linker-conjugation chemistry affects ADC’s stability, efficacy, pharmacokinetics, homogeneity, and biophysical integrity (Chari et al. 2014). There are several conjugation strategies:
-
Conjugation through lysine amino acids. An IgG scaffold has 80–90 lysines, with about 20 being solvent accessible and hence easily amenable to conjugation. This approach could lead to a wide spectrum of drug-antibody ratios and requires batch-to-batch consistency. Ado-trastuzumab-emtansine uses Ab-lysine modification.
-
Conjugation via cysteine residues by reducing ‘inter-chain’ native disulfide bonds. An IgG has 12 intra-chain and 4 inter-chain disulfide bonds. Reduction of the latter yields 8 cysteine residues with partial reduction leading to about 4. Brentuximab vedotin uses partial cysteine reduction of native inter-chains.
-
Increasing the homogeneity of the ADC by site-specific conjugation via genetically engineered amino acid alteration. One example of such an approach is using the THIOMAB platform, which results in a more uniform drug-antibody ratio and contains thiol-maleimide linkages. However, cysteine-engineered mAbs require additional down-stream steps such as partial reduction and re-oxidation (Chari et al. 2014).
-
Enzyme-mediated conjugation is another approach using ligating enzymes with high specificity for a given substrate. One route is to incorporate glutamine residues and coupling of an acyl acceptor payload using microbial transglutaminase (Kline et al. 2015).
-
The incorporation of an unnatural amino acid such as para-acetylphenylalanine instead of alanine using an orthogonal amber suppressor tRNA/aminoacyl-tRNA synthetase (aaRS) pair, followed by subsequent payload coupling (Kline et al. 2015).
Generally, payload coupling approaches that allow specificity, narrower drug-antibody ratio distribution, and higher ADC homogeneity hold promise to optimize the product target profile. Furthermore, site-specific coupling will facilitate production of linking other novel payloads (Behrens and Liu 2014; Deonarain et al. 2015).
Production and characterization
The production of the mAb component of an ADC is similar to that used to produce the traditional therapeutic mAbs. However, there are exceptions when dealing with site-specific amino acid engineering, partial reduction of cysteine amino acids, or enzymatic coupling of the payload. One of the common challenges in manufacturing ADCs is handling of highly cytotoxic payloads or high-potency active pharmaceutical ingredients (HPAPIs: defined as biological activity at about 150 μg/kg body weight or below in humans). The manufacturing facility should be able to accommodate steam-in-place and clean-in-place capabilities. Some key requirements to ensure staff safety could include occupational exposure limits set at or below a specified limit (at or less than 30 ng/m3) in air for Category 4 compounds (SafeBridge criteria) as an 8 h time-weighted average, airlocks, appropriate personal protective equipment, adequate ventilation, long-term storage, transport, filtration, negative pressure rooms, de-contamination procedures, compliance to current good manufacturing practice, etc.
In addition to the standard handling techniques for HPAPIs and antibody components, the large scale conjugation chemistry step requires careful process control. Depending on the sponsor’s bioprocess, cysteine residues would need to undergo partial reduction, reacted with a compatible functional moiety on a linker (e.g., maleimide-activated peptide), and followed by HPAPI coupling. Alternatively, the primary amine on lysine residues could be coupled to a bi-functional amide linker (e.g., using a N-hydroxysuccinimide-activated ester), with subsequent reaction of the second linker’s reactive site with HPAPI. Regardless of the route of conjugation, free HPAPI and organic solvents must not be in the final drug substance. Thus, multiple chromatographic steps, ultrafiltration, and diafiltration are used to remove any unconjugated cytotoxin by >99.5 %. A recent excellent review on ADCs formulation, physicochemical stability, and characterization discusses some of the above challenges in more detail (Singh et al. 2015).
As expected, due to the complex nature of ADCs, there are a series of tests to ensure conformance to specifications, characterize physicochemical properties, potency, impurities (product- and process-related), comparability, and stability for the payload, mAb, and ADC itself. The product specifications for the mAb portion of the ADC are the same as the traditional therapeutic mAbs, including antigen binding, glycosylation, charge variants, higher-order structure, effector function, aggregation, host-cell proteins, viral clearance, endotoxin presence, bioburden, and others as appropriate. Intermediates such as the cytotoxic drug, the linker, and the drug-linker combination are tested for purity where structural determination could be warranted for impurities higher than 0.1 %. In addition to drug-antibody ratio, drug load, free drug, linker, and residual solvent levels are determined. Moreover, container closure, intravenous infusion bag (if applicable), transport, and storage stabilities are rigorously tested (Wakankar et al. 2011; Luo et al. 2016; Ross and Wolfe 2016).
Generally, the pharmacokinetics of ADCs are similar to therapeutic mAbs, which means long half-lives and low clearance values due to the human neonatal Fc receptor recycling. Conversely, the pharmacokinetics-pharmacodynamics properties of ADCs vary significantly from those of small molecule entities. Depending on the linker chemistry, in vivo de-conjugation of ADC can take place in the systemic circulation yielding lower drug-antibody ratios. In addition, the production of the ADC can also lead to certain degree of heterogeneity resulting from different drug-antibody ratios which can affect its pharmacokinetics profile (Perez et al. 2014). Therefore, often a combination of ELISA and LC–MS/MS are used to quantify the payload, mAb, possibly the linker (e.g., if novel or first-in-class), linker-drug combination, and ADC levels in the systemic circulation during pre-clinical and clinical development (Liu et al. 2015). Other drug metabolism-related experiments, such as reaction phenotyping, passive/active transport, cytochrome p450 inhibition/induction, plasma protein binding, and in vitro plasma or serum stability, should be considered on a case-by-case basis (Kraynov et al. 2016). In addition to safety assessment studies (Donaghy 2016), an immunogenicity screening assay to detect anti-drug antibodies (ADAs) using an appropriate assay cut-point, domain specificity characterization, and a neutralizing Ab assay need to be designed and validated (Hock et al. 2015).
Adverse events and resistance mechanisms
An adverse event is any unfavorable experience related to the use of a medical product. The Common Terminology Criteria for Adverse Events (CTCAE) contains a grading scale for severity of an adverse event. The CTCAE recommends Grades 1 through 5 with specific clinical description where 1 is considered mild and 5 is death related to an adverse event. Grades 2, 3, and 4 are considered moderate, severe, and life-threatening, respectively. In general, the determinants of ADC-related adverse events could originate from four distinct entities, payload (e.g., off-target), mAb (e.g., non-antigen mediated uptake, cross-reactivity, target-induced), normal cell or the so-called “bystander toxicity”, linker (e.g., stability issues), and/or product attributes (e.g., drug-antibody ratio, formulation) (de Goeij and Lambert 2016). Currently, the most common ADC-related adverse events are also observed with some of the standard Ab monotherapies (Hansel et al. 2010), such as thrombocytopenia (platelet count of lower than 150,000 per microliter of blood), neutropenia (neutrophil count of less than 1500 per microliter of blood), fatigue, liver toxicity, and nausea, with increase in severity at the maximum tolerated dose.
Similar to small molecule therapeutic agents and mAbs, ADCs also have to face innate or acquired resistance, the mechanisms of which are not fully understood (Diamantis and Banerji 2016). Contributing factors to ADC resistance include target antigen down-regulation, inefficient internalization of the complex, drug efflux proteins (e.g., matansinoids are substrates of MDR1 p-glycoproteins), defective intra-cellular trafficking of the ADC, tumor heterogeneity, and upregulation of ADC recycling to the cell surface (Barok et al. 2014). Several possible strategies are under evaluation to improve efficacy and ameliorate ADC resistance. These include combination therapy, linker modification to minimize MDR1-mediated resistance, combination of multiple types of payload per and mAb (Shefet-Carasso and Benhar 2015).
Conclusions
There is renewed interest in the development of ADCs, with > 40 currently in clinical development (Donaghy 2016; Schumacher et al. 2016). Up to now, approved ADCs have been confined to the oncology therapeutic area, but this therapeutic type may also show promise in inflammatory and infectious diseases in the future. Currently, there are two established ADC technologies, as practiced by Seattle Genetics and Immunogen, but more controlled approaches to couple new payload molecules, new linker technologies, and site-specific attachment will lead to narrower drug-antibody ratio distribution, superior efficacy, and lower toxicity. In some instances, lack of efficacy due to low receptor occupancy, poor tumor penetration, low target antigen expression, T-cell response induction, and/or antigen mutation(s) remain to be fully addressed (Scott et al. 2012). Use of in-house manufacturing facilities or partnerships with qualified contract manufacturing organizations will demand closer scrutiny due to the handling of high-potency active pharmaceutical ingredients and specific conjugation chemistry.
References
Adem YT, Schwarz KA, Duenas E, Patapoff TW, Galush WJ, Esue O (2014) Auristatin antibody drug conjugate physical instability and the role of drug payload. Bioconj Chem 25:656–664
AlDeghaither D, Smaglo BG, Weiner LM (2015) Beyond peptides and mAbs—current status and future perspectives for biotherapeutics with novel constructs. J Clin Pharmacol 55:S4–S20
Bae YH, Park K (2011) Targeted drug delivery to tumors: myths, reality and possibility. J Control Release 153:198–205
Bakhtiar R (2012) Therapeutic recombinant monoclonal antibodies. J Chem Educ 89:1537–1542
Barok M, Joensuu H, Isola J (2014) Trastuzumab amtansine: mechanisms of action and drug resistance. Breast Cancer Res 16:209
Beckley NS, Lassareschi KP, Chih H-W, Sharma VK, Flores HL (2013) Investigation into temperature-induced aggregation of an antibody drug conjugate. Bioconj Chem 24:1674–1683
Behrens CR, Liu B (2014) Methods for site-specific drug conjugation to antibodies. MAbs 6:1–8
Blencowe CA, Russell AT, Greco F, Hayes W, Thornthwaite DW (2011) Self-immolative linkers in polymeric delivery systems. Polym Chem 2:773–790
Chari RVJ (2008) Targeted cancer therapy: conferring specificity to cytotoxic drugs. Acc Chem Res 41(1):98–107
Chari RVJ, Miller ML, Widdison WC (2014) Antibody-drug conjugates: an emerging concept in cancer therapy. Angew Chem Int Ed 53:3796–3827
Chiche J, Brahimi-Horn MC, Pouyssegur J (2010) Tumour hypoxia induces a metabolic shift causing acidosis: a common feature in cancer. J Cell Mol Med 14:771–794
Choi I-K, Strauss R, Richter M, Yun C-O, Lieber A (2013) Strategies to increase drug penetration in solid tumors. Front Oncol 3(Article 193):1–18
Correia IR (2010) Stability of IgG isotypes in serum. MAbs 2:221–231
de Goeij BECG, Lambert JM (2016) New develoipments for antibody-drug conjugate-based therapeutic approaches. Curr Opin Immunol 40:14–23
Deonarain MP, Yahioglu G, Stamati I, Marklew J (2015) Emerging formats for next-generation antibody drug conjugates. Expert Opin Drug Discov 10:463–481
Dhillon S (2014) Trastuzumab emtansine: a review of its use in patients with HER2-positive advanced breast cancer previously treated with trastuzumab-based therapy. Drugs 74:675–686
Diamantis N, Banerji U (2016) Antibody-drug conjugates-an emerging class of cancer treatment. Br J Cancer 114:362–367
Donaghy H (2016) Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs, In Press (DOI:10.1080/19420862.2016.1156829)
Dosio F, Stella B, Cerioni S, Gastaldi D, Arpicco S (2014) Advances in anticancer antibody-drug conjugates and immunotoxins. Recent Patents Anti-Cancer Drug Discov 9:35–65
Gharwan H, Groninger H (2016) Kinase inhibitors and monoclonal antibodies in oncology: clinical implications. Nat Rev Clin Oncol 13:209–227
Hamblett KJ, Senter PD, Chace DF, Sun MMC, Lenox J, Cerveny CG, Kissler KM, Bernhardt SX, Kopcha AK, Zabinski RF, Meyer DL, Francisco JA (2004) Effects of drug loading on the antitumor activity of a monoclonal antibody conjugate. Clin Cancer Res 10:7063–7070
Hamilton GS (2015) Antibody-drug conjugates for cancer therapy: the technological and regulatory challenges of developing drug-biologics hybrids. Biologicals 43:318–332
Han TH, Zhao B (2014) Absorption, distribution, metabolism, and excretion considerations for the development of antibody-drug conjugates. Drug Metab Dispos 42:1914–1920
Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJT (2010) The safety and side effects of monoclonal antibodies. Nat Rev Drug Discov 9:325–338
Ho RJY, Chien J (2014) Trends in translational medicine and drug targeting and delivery: new insights on an old concept—targeted drug delivery with antibody-drug conjugates for cancers. J Pharm Sci 103:71–77
Hock MB, Thudium KE, Carrasco-Triguero M, Schwabe NF (2015) Immunogenicity of antibody drug conjugates: bioanalytical methods and monitoring strategy for a novel therapeutic modality. AAPS J 17:35–43
Hogarth PM, Pietersz GA (2012) Fc receptor-targeted therapies for the treatment of inflammation, cancer and beyond. Nat Rev Drug Discov 11:311–331
Jackson D, Stover D (2015) Using the lessons learned from the clinic to improve the preclinical development of antibody drug conjugates. Pharm Res 32:3458–3469
Jain N, Smith SW, Ghone S, Tomczuk B (2015) Current ADC linker chemistry. Pharm Res 32:3526–3540
Juweid M, Neumann R, Paik C, Perez-Bacete MJ, Sato J, van Osdol W, Weinstein JN (1992) Micropharmacology of monoclonal antibodies in solid tumors: direct experimental evidence for a binding site barrier. Cancer Res 52:5144–5153
Kamath AV, Iyer S (2016) Challenges and advances in the assessment of the disposition of antibody-drug conjugates. Biopharm Drug Dispos 37:66–74
Khawar IA, Kim JH, Kuh H-J (2015) Improving drug delivery to solid tumors: priming the tumor microenvironment. J Control Release 201:78–89
Kim EG, Kim KM (2015) Strategies and advancement in antibody-drug conjugate optimization for targeted cancer therapeutics. Biomol Ther 23(6):493–509
Kitson SL, Cuccurullo V, Moody TS, Mansi L (2013) Radionuclide antibody-conjugates, a targeted therapy towards cancer. Curr Radiopharm 6:57–71
Kline T, Steiner AR, Penta K, Sato AK, Hallam TJ, Yin G (2015) Methods to make homogeneous antibody conjugates. Pharm Res 32:3480–3493
Kraynov E, Kamath AV, Walles M, Tarcsa E, Deslandes A, Iyer RA, Datta-Mannan A, Sriraman P, Bairlein M, Yang JJ, Barfield M, Xiao G, Escandon E, Wang W, Rock DA, Chemuturi NV, Moore DJ (2016) Current approaches for absorption, distribution, metabolism, and excretion characterization of antibody-drug conjugates: an industry white paper. Drug Metab Dispos 44:617–623
Lambert JM (2012) Durg-conjugated antibodies for the treatment of cancer. Br J Clin Pharmacol 76:248–262
Lammers T, Kiessling F, Hennink WE, Storm G (2012) Drug targeting to tumors: principles, pitfalls and (pre-) clinical progress. J Control Release 161:175–187
Liu L (2015) Antibody glycosylation and its impact on the pharmacokinetics and pharmacodynamics of monoclonal antibodies and Fc-fusion proteins. J Pharm Sci 104:1866–1884
Liu A, Kozhich A, Passmore D, Gu H, Wong R, Zambito F, Rangan VS, Myler H, Aubry A-F, Arnold ME, Wang J (2015) Quantitative bioanalysis of antibody-conjugated payload in monkey plasma using a hybrid immunocapture LC–MS/MS approach: assay development, validation, and a case study. J Chromatogr B 1002:54–62
Luo Q, Chung HH, Borths C, Janson M, Wen J, Joubert MK, Wypych J (2016) Structural characterization of a monoclonal antibody–maytansinoid immunoconjugate. Anal Chem 88:695–702
Mack F, Ritchie M, Sapra P (2014) The next generation of antibody drug conjugates. Semin Oncol 41(5):637–652
Maderna A, Leverett CA (2015) Recent advances in the development of new auristatins: structural modifications and application in antibody drug conjugates. Mol Pharm 12:1798–1812
Makuch RW, Shi R (2014) Comparison of drug approvals in Europe versus the United States: an analysis of discrepancies between drug products reviewed by EMA and FDA. Ther Innov Regul Sci 48:362–366
McCombs JR, Owen SC (2015) Antibody drug conjugates: design and selection of linker, payload and conjugation chemistry. AAPS J 17:339–351
McDonagh CF, Turcott E, Westendorf L, Webster JB, Alley SC, Kim K, Andreyka J, Stone I, Hamblett KJ, Francisco JA, Carter P (2006) Engineered antibody-drug conjugate with defined sites and stoichiometries of drug attachment. Prot Eng Des Sel 19:299–307
Moussa EM, Panchal JP, Moorthy BS, Blum JS, Joubert MK, Narhi LO, Topp EM (2016) Immunogenicity of therapeutic protein aggregates. J Pharm Sci 105:417–430
Navarro-Teulon I, Lozza C, Pelegrin A, Vives E, Pouget J-P (2013) General overview of radioimmunotherapy of solid tumors. Immunotherapy 5:467–487
Nolting B (2013) Linker technologies for antibody conjugates. Method Mol Biol 1045:71–100
Perez HL, Cardarelli PM, Deshpande S, Gangwar S, Schroeder GM, Vite GD, Borzilleri RM (2014) Antibody-drug conjugates: current status and future directions. Drug Discov Today 19:869–881
Polakis P (2015) Antibody drug conjugates for cancer therapy. Pharmacol Rev 68:3–19
Prasad V (2014) The withdrawal of drugs for commercial reasons: the incomplete story of tositumomab. J Am Med Assoc Intern Med 174:1887–1888
Ross PL, Wolfe JL (2016) Physical and chemical stability of antibody drug conjugates: current status. J Pharm Sci 105:391–397
Schumacher D, Hackenberger CPR, Leonhardt H, Helma J (2016) Current status: site-specific antibody drug conjugates. J Clin Immunol. doi:10.1007/s10875-016-0265-6
Scott AM, Wolchok JD, Old LJ (2012) Antibody therapy of cancer. Nat Rev Cancer 12:278–287
Shefet-Carasso LR, Benhar I (2015) Antibody-targeted drugs and drug resistance-challenges and solutions. Drug Resist Updates 18:36–46
Sievers EL, Senter PD (2013) Antibody-drug conjugates in cancer therapy. Annu Rev Med 64:15–29
Singh SK, Luisi DL, Pak RH (2015) Antibody-drug conjugates: design, formulation, and physicochemical stability. Pharm Res 32:3541–3571
Sliwkowski MX, Mellman I (2013) Antibody therapeutics in cancer. Science 341:1192–1198
Strop P, Delaria K, Foletti D, Witt JM, Hasa-Moreno A, Poulsen K, Casas MG, Dorywalska M, Farias S, Pios A, Lui V, Dushin R, Zhou D, Navaratnam T, Tran T-T, Sutton J, Lindquist KC, Han B, Liu S-H, Shelton DL, Pons J, Rajpal A (2015) Site-specific conjugation improves therapeutic index of antibody drug conjugates with high drug loading. Nat Biotechnol 33:694–696
Tang L, Persky AM, Hochhaus G, Meibohm B (2004) Pharmacokinetic aspects of biotechnology products. J Pharm Sci 93:2184–2204
Torchilin VP (2014) Multifunctional, stimuli-sensitive nanoparticulate systems for drug delivery. Nat Rev Drug Discov 13:813–827
Van den Mooter T, Teuwen L-A, Rutten A, Dirix L (2015) Trastuzumab emtansine in advanced human epidermal growth factor receptor 2-positive breast cancer. Expert Opin Biol Ther 15:749–760
van der Neut Kolfschoten M, Schuurman J, Losen M, Bleeker WK, Martinez-Martinez P, Vermeulen E, den Bleker TH, Wiegman L, Vink T, Aarden LA, De Baets MH, van de Winkel JGJ, Aalberse RC, Parren PWHI (2007) Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science 317:1554–1557
Vidarsson G, Dekkers G, Rispens T (2014) IgG subclasses and allotypes: from structure to effector functions. Front Immunol 5:1–17
Vugmeyster Y, Xu X, Theil F-P, Khawli LA, Leach MW (2012) Pharmacokinetics and toxicology of therapeutic proteins: advances and challenges. World J Biol Chem 3:73–92
Wakankar A, Chen Y, Gokarn Y, Jacobson FS (2011) Analytical methods for physicochemical characterization of antibody drug conjugates. MAbs 3:161–172
Wong DJL, Hurvitz SA (2014) Recent advances in the development of anti-HER2 antibodies and antibody-drug conjugates. Ann Transl Med 2:122
Acknowledgments
Helpful discussions with Dr. James Chovan are greatly appreciated.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Bakhtiar, R. Antibody drug conjugates. Biotechnol Lett 38, 1655–1664 (2016). https://doi.org/10.1007/s10529-016-2160-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-016-2160-x