
Abstract
Codon usage bias of coding sequences has been usually used for exploring the evolutionary factors that affect the variation of genes. We took 20 chloroplast genomes of Malus species into account to explore the codon usage patterns, including the composition, relationship between GC3s and effective number of codons, the parity rule two analyses, the relative synonymous codon usage, the codon adaptation index, the frequency of optimal codons, the codon bias index, etc., of their coding genes. The relationship between GC3 and the ENC values showed that when the separate genes are concerned, the distribution of their GC3 contents is relatively concentrated and the distribution of the ENC values are from 35 to 61 or so. The neutrality plot showed that the correlation coefficient between GC12 and GC3 is 0.095, revealing the mutation factor played a weak role in codon pattern formation. Correspondence analysis results revealed that the codon usage patterns in the chloroplast genomes of Malus species are similar. All these results showed that all Malus chloroplast genomes are AT rich ones, the third bases of the codons are affected by the natural selection pressure, the first two nucleotide base of the codon are affected by mutation pressure. Some genes, such as the rsp7, psbA and ycf2 are of lower codon usage divergences, while the rps12, rps16 and ndhD are of higher codon usage divergences. Codon usage bias exists in the Malus genomes could be used for exploring the evolutionary characteristics in chloroplast genomes and for further study on evolutionary phenomenon in other species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Plants of Malus species are of important economical genus, mainly distributed in the North Temperate Zone (Amandine et al. 2014). Among them, about twenty sub-species are endemic to China (Yan et al. 2019; Naizaier et al. 2019; Li et al. 2020). Malus species are well known for their edible value (Zhang et al. 2018), and medicinal functions of compounds in them. Furthermore, Malus species are of great significance to scientists for their excellent horticultural trait and ornamental value (Bao, et al. 2016; Xun, et al. 2021). Wildly cultivated Malus species appeared very often recently because they could be easily cultivated (Svetlana et al. 2013). Therefore, these traits make the Malus species as a series of model plants for study. As an important organelle, chloroplast genome has a quadripartite organization, which has been widely used in studying evolutionary traits. In addition, chloroplasts are maternally inherited organelles in all green plants (Liang et al. 2019). Studies have shown that most families of photosynthetic eukaryotes emerged due to the takeover of a free-living photosynthetic eukaryote by the host (Supriyo et al. 2020). Further, many factors have been verified to be the factors that affect the codon usage pattern, such as mutation pressure (Sharp et al. 1988), gene length (Tao et al. 2009), natural selection pressure (Shackelton et al. 2006), tRNA abundance, etc. (Pandey et al. 2020). Codon usage pattern, including the effective number of codons (ENC), the parity rule 2 (PR2) of G3 vs. A3, the relative synonymous codon usage (RSCU), the frequency of optimal codons (FoP), etc., of the chloroplast genomes may be useful for exploring their molecular evolution characteristics and predicting the expression level of a certain chloroplast gene (Mazumder et al. 2020; Yan et al. 2022).
The importance of codon usage pattern in chloroplast genomes had been emphasized by many studies (He et al. 2016; Challabathula et al. 2018; Gichira et al. 2019). Extensive studies on codon usage pattern of chloroplast genomes in the past have revealed that the variation in chloroplast genomes due to varying degrees of mutation pressure, selection pressure (Yang, et al. 2018; Haruo et al. 2016; Kong et al. 2017; Xu et al. 2011) or cultured pressure from humans. Comparative analyses have been used to examine the codon usage pattern of genomes within or between the two groups of plants (Liu et al. 2020b). Genetic diversity of Malus species had been studied by considering the microsatellite markers to assess their evolutionary range. However, sample quantities of most previous studies were about ten, meanwhile, evolution features of specific genes in chloroplast genomes had not been considered (Li et al. 2016). There are about 80 coding genes in a Malus chloroplast genome. It was believed that all genes in chloroplast genome may use universal genetic code (Nakamura et al. 2007), showing that a wide diversity may exist in Malus species (Mazumdar et al. 2017). Knowledge of the codon usage patterns of chloroplast genomes in Malus species would be very useful for exploring mechanism of environmental adaptation and molecular variation under human cultivation pressure. And now, there are still some issues, such as the codon usage diversity of each typical gene in chloroplast genomes of Malus species, the evolutionary pressure that affect the components in chloroplast genomes of Malus species, etc., need to be studied. In the present study, based on the previous studies, all 55 chloroplast genomes in the NCBI database, covering a total of 20 subgenus species of Malus, were considered and analyzed. We performed a comparative analysis on the codon usage pattern and their evolution of 20 Malus species. We calculated the ENC, Fop, CBI values, as well as the ENC-GC3s values, the G3%–A3% values, the RSCU values, etc. The correlation values among them were also explored in the present study. Furthermore, the genetic relationships among 20 Malus chloroplast genomes were conducted via correspondence analysis, and their characteristics were analyzed.
Materials & Methods
All searching results via indexing the keywords ‘Malus Chloroplast complete genome’ in the NCBI database were considered, getting a total of 55 chloroplast complete genomes (Supplementary Tab.S1), including 20 kinds of Malus sub-species. Among of them, all 20 kinds of Malus sub-species were selected, as their names and accession numbers were Malus angustifolia (MN061984.1), Malus baccata (KX499859.1), Malus coronaria (MN068247.1), Malus domestica (MK434916.1), Malus doumeri (KX499861.1), Malus florentina (KX499862.1), Malus halliana (MT246302.1), Malus hupehensis (NC_040170.1), Malus ioensis (MN062004.1), Malus micromalus (MF062434.1), Malus prattii (NC_043902.1), Malus prunifolia (NC_031163.1), Malus sieboldii (MT593044.1), Malus sylvestris (MK434921.1), Malus toringoides (MT483999.1), Malus transitoria (MK098838.1), Malus trilobata (NC_035671.1), Malus tschonoskii (KX499864.1), Malus x atrosanguinea (MN061983.1), Malus yunnanensis (MH394387.1). The genes of ycf1, ycf2, ycf3, psaA, psaB, psbA, psbB, psbC, psbD, rpoC1, rpoC2, rps3, rps8, rps14, rps18, cemA, and ccsA in each strain were studied and compared specifically. The criteria for choosing the sequences were: (1) longer than three hundred bases, (2) starting with ATG, (3) the count of the bases is divisible by three, and the sequences which contain ambiguous bases were excluded.
The parameters of componential content T, G, A and C of chloroplasts were counted. Furthermore, the second parity rule (PR2) of the third position of each separate genes, with AT-bias [A/(A + T)] as the y-axis and GC bias [G/(G + C)] as the x-axis in a graphical presentation (McLean et al. 1998; Sueoka, 1999), was used to evaluated the codon bias in all concerned coding sequences. Neutrality plot analysis was used to compare the role of mutation pressure and natural selection pressure (Sueoka, 1988). The effective number of codons, denoting the absolute codon usage pattern in coding sequences, was used to quantify the codon usage bias in the 20 Malus chloroplast complete genomes. The ENC of each gene was actually calculated via using the following formula in the present study:
where \(\overline{f}_{k}\)(k = 2, 3, 4, 6) denotes the average homozygosity for the amino acid class whose degree of codon degeneracy is k, it is the mean value of fk for the k-fold degenerate amino acids, and the fk is calculated by the following equation.
where ni is the total number of occurrences of the i-th codon for that amino acid. To elucidate the relationship between GC3s and ENC values, the expected ENC values for different GC3s are calculated as ENCexpected = 2 + s + {29/[s2 + (1−s)2]}, Where s represents the given GC3s. The RSCU values of all Malus coding sequences in chloroplast genomes were calculated by following the equation (Xu et al. 2017):
where gij is the observed number of the i-th codon for the j-th amino acid, which has ni kinds of synonymous codons. Further, the correspondence analysis (COA) was used to explore the principal component coefficients among 20 chloroplast complete genomes of Malus species based on the RSCU values (three stop codons and AUG, UGG were excluded). Evolutionary distance among 20 chloroplast complete genomes of Malus species was further studied via considering Euclidean distance among them. All the previous parameters in the present study were calculated on the Matlab 2010b software (Li et al. 2021a).
Results
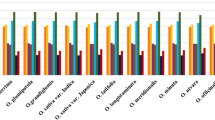
Many factors affect plant genomes in the evolutionary process (Saurabh et al. 2019). In the present study, the basic components of typical genes in all chloroplast genomes are counted (Supplementary Tab.S2). The ENC values of all genes in 20 chloroplast genomes of Malus species were calculated and plotted against GC3s (Fig. 1A), the PR2-bias (Fig. 1B) and the overall ENC values for each genome were plotted (Fig. 1C) to examine the evolutionary forces. The results showed that the ENC values of most genes are generally greater than 35 in chloroplast genomes of Malus species, the ENC values of the genes for ribosomal protein large subunit 16 (rpl16) in Malus prattii (NC_043902.1), Malus prunifolia (NC_031163.1), Malus micromalus (MF062434.1), Malus halliana (MT246302.1) and Malus hupehensis (NC_040170.1) are all equal to 34.931, and the ENC values of rpl16 codon genes in Malus trilobata (NC_035671.1), Malus yunnanensis (MH394387.1), Malus transitoria (MK098838.1), and Malus ioensis (MN062004.1) are equal to 34.581.

Evolutionary forces in 20 chloroplast genomes of Malus species. A The effective number of codons (ENC) vs. GC3s dot values of all genes in 20 chloroplast complete genomes of Malus species. The curve in the figure denotes the expected ENC values for genes without any evolutionary constraint. B The PR2-bias plot of all genes in 20 chloroplast complete genomes of Malus species. There are 1154 selected genes in total in 20 chloroplast complete genomes of Malus species. C Overall ENC values of chloroplast for each genome of Malus specie
In order to explore the biased codon choices, the relation between G and C content, and between A and T content at the third position of genes in the 20 chloroplast genomes of Malus species were shown by the PR2 bias plot (Fig. 1B). The results show that A and C are preferred bases, but it’s not obvious, showing that the bases at the third position are affected by mutations in general, and the pressure effect of natural selection is not dominant. From Fig. 1C, the overall ENC values of the 20 chloroplast genomes of Malus species, more codon usage bias exists in M. yunnanensis, M. ioensis, M.doumeri and M. florentina chloroplast genomes.
In order to evaluate the relationships among the codon usage pattern (such as the content of G, C, A and T, the ENC values, the codon adaption index, and the Fop values, etc.) of the genes in all 20 chloroplast genomes of Malus species, correlation analysis was performed as shown in Fig. 2. The content of GC3 exhibits strong positive correlation with gene length and the ENC values, suggesting that the sequence lengths may be an important factor contributes to codon usage bias in the chloroplast genomes, and that the GC3 may be a result of the codon selection in the evolutionary process of chloroplast genomes. The GC content in 20 chloroplast genomes of Malus species is mainly contributed by the GC12, the correlation value between them is 0.953, while the correlation value between GC and GC3 is 0.363.

Correlation analysis among the codon usage parameters
The CBI value of a gene was usually regarded as an effective measurement of codon bias (Deb et al. 2018). It could measure the extent of the usage pattern of a subset of optimal codons in a gene. In an extreme bias gene, the CBI value will be equal to 1 (Maldonado et al. 2018). Uncommonly, from Fig. 3A, in the chloroplast complete genomes of Malus species, it shows weak correlation between the CBI and the ENC values. This is mainly because the variability range of the ENC is much broader than that of the CBI. Most of the CBI values are distributed in the range of 0.25 to 0.4. In fact, the ENC values should be normalized to the scope of the range within CBI to reveal the explicit relationship between them as a similar method was used in our previous studies (Li et al. 2018). Here, if the scope of the ENC values were equalized to that of the CBI values, their correlation was ENC (Equalization) = − 0.296 × CBI, showing that there is a strong negative relationship between them (Supplementary Fig.S1). Further, there is also a strong positive correlation between codon bias index and the frequency of optimal codons, denoting that the codon usage pattern in the chloroplast complete genomes of Malus species may be shaped by the frequency of optimal codons in the process of evolution (Wei et al. 2014). The GC content of a gene plays an important role in determining the effects of base composition bias (Supriyo et al. 2017). The neutrality plot was also performed to explore the directional mutation pressure versus natural selection in chloroplast complete genomes of Malus species. The relationship between GC12 and GC3 for sequences is shown in Fig. 3B; each point shown in the figure represents a separate gene. It shows that the GC12 of genes were distributed to the range of 30 to 55%, and the GC3 of genes were distributed to the range of 20 to 40%. Rates of GC3 are less than GC12 in all genes. In Fig. 3C, content of GC, GC12, and GC3 in all chloroplast genomes of Malus species were displayed. The third bases of all codons are AT-rich. The overall GC rates for whole length of all genes are all less than 50%. The number of genes with GC12 content larger than 50% is 99. In Fig. 3D, relationships between protein length and GC content (both GC3 and GC12) were described. The GC3 contents are higher than the GC12 contents in all genes. Meanwhile, from the figure, it could be seen that the length of the sequences have no obvious relationships to their GC contents. From this perspective, the natural selection is a greater impact factor on the rates of GC contents in chloroplast genomes of Malus species.

CBI vs. ENC, neutrality of GC content, gene number vs. GC content, and the GC content vs. protein length analysis of the 20 chloroplast genomes of Malus species. A The effect of the ENC on the CBI of all genes in the 20 chloroplast genomes of Malus species. B The neutrality analysis. C Statistical analysis of the overall GC, GC12 and GC3. D Relationship between GC12 / GC3 ratio and protein lengths. The area graphs in the sub-figures denote number density of corresponding genes
o reveal the codon usage pattern of RSCU values in 20 Malus chloroplast genomes, we performed the RSCU analysis and counted the codon quantity of all genomes including the stop codons (UAA, UAG and UGA) and the one-dimensional degenerate codons (AUG and UGG) as shown in Fig. 4. The RSCU results show that the abundant codons with RSCU values more than 1.5 covering UUA, GUA, UCU, CCU, ACU, GCU, UAU, CAU, CAA, AAU, GAU, AGA, and GGA, and that the less-abundant codons with RSCU values less than 0.5 including CUG, CUC, GAC, AGC, UAC, GCG, GGC, CGC, GUC, ACG, CAC, CGG, CAG, AAC, and UGA. Among the stop codons, more than half of them select the UAA as their terminal codons. Meanwhile, among all the RSCU values, the RSCU values of UUA, GCU and AGA are the most preferred ones. Each codon quantities of concerned genes in 20 Malus chloroplast genomes were also counted and shown in Fig. 4, and the results showed that the most used codons in the chloroplast genomes are AAU (15,824), GAU (16,968), UUA (16,690), and UAU (14,943), etc. While the least used codons are UGC (1445), CGC (2104), CGG (2278), and AGC (2392), etc., except for stop codons UGA (192), UAG (283), and UAA (679) with no corresponding amino acid.

RSCU values of whole genomes of 20 chloroplast complete genomes of Malus species
Genetic differences among 20 chloroplast genomes of Malus species are shown in Fig. 5 via calculating the correspondence based on the RSCU distances of 20 chloroplast complete genomes of Malus species. The relative and cumulative inertia of correspondence analysis factors are also shown in the inner graph of Fig. 5. The previous four axis factors are 36.01%, 31.57%, 15.21% and 5.86%, it could be seen that the previous two axes would explain the evolutionary distances among the Malus species well. In the correspondence analysis, the terminal codons and the codons for Met and Trp were excluded.

Correspondence analysis for 20 chloroplast genomes of Malus species (Axis 1 and Axis 2). The inner graph shows the relative and cumulative inertia versus factors
The Euclidean distances of RSCU values among 20 chloroplast genomes of Malus species were used to explore their clustering characteristics (Fig. 6). There are four clusters when the Euclidean distances equals to 0.06 while all 20 Malus chloroplast genomes were considered. Here, for conformity and consistency of the data, the RSCU value of every genome contains the same gene sequences.

Clustering analysis for 20 Malus chloroplast genomes
All 20 coding genes in chloroplast genomes of Malus species were calculated, and the results were shown in the Tab.1. From the results, the genes of yeast cadmium factor 1 (ycf1) are of the largest divergences. The standard deviation of its codon usage parameters are all the largest ones. Among all the genes concerned, they differ greatly in codon usage preference, for instance, GC12 of psbB and rpl16 are greater than 50%, while other GC content, whether the GC12 content, overall GC or GC3 are less than 50%. Therefore, the genes of yeast cadmium factors, including ycf1, ycf2 and ycf3, are affected by mutations more obviously than other factors.
Summation of standard deviation values of all parameters, including A, G, C, T, G3, C3, A3, T3, GC12, GC3, CBI, ENC and Fop of each gene, as shown in Fig. 7, were used to explore their whole codon usage divergence properties. From the graph, codon usage pattern and basic components in rps7 in all genes are same to each other. Some other genes, such as the psbA, psbD, and ycf2 are relatively lower than other genes. The codon usage divergence of a gene is an indicator for estimating its diversity property. The codon usage divergence of rps12 is the largest one according to Fig. 7, however, from the Supplementary Tab.S1; the encoding sequences are of two different lengths, resulted the larger deviation value of the ENC. If the two kinds of sequences were considered separately, the codon usage divergence would be lower than it is shown in the present study.

Codon usage divergence of the specific genes in Mulas chloroplast genomes. Values of the bars in the graph represent the divergence degree of the genes, which equal to the summation of standard deviations for both the codon usage patterns and sequence compositions in the particular genes
Discussion
It has been proposed that codon usage pattern in coding sequences is very useful for the identification of plants (Hosokawa et al. 2004). DNA sequence data from rpl16 in chloroplasts were used to address phylogenetic relationships among the major lineages of the grass family for their non-conservative evolution (Zhang et al. 2000). In present study, all ENC values of ycf3 codon genes are equal to 61.122 except Malus coronaria (MN068247.1), which ENC value is equal to 60.206. According to the previous perspective, the rpl16 gene in chloroplast genomes of Malus species is greatly influenced by the selection pressure. On the contrary, the codon usage pattern in ycf3 genes is mainly affected by mutation pressure. The ycf3 protein is very important for accumulation of the photosystem I complex. Previous studies have shown that the sequence of ycf3 was conservative in its evolutionary process (Naver et al. 2001).
Four canonical bases-A, C, G and T should be used proportionally if mutation is the absolute cause of codon bias in a gene (Li et al. 2014). On the contrary, natural selection for codon choice in a gene would cause uneven use of G, C, A and T. In the present study, the chloroplast genomes of Malus prunifolia and M. micromalus show relative light codon usage bias in their genomes. The present study shows the component biases for the third bases between G3 and C3, and between A3 and T3 in 20 Malus chloroplast genomes are similar, all of them are affected slightly by both mutation pressure and natural selection. Actually, the mutational pressure was considered as a major factor in shaping the codon usage pattern compared with natural selection (Jenkins et al. 2003).
When the genetic diversity is concerned, it is more important to study the codon usage pattern of specific genes in the chloroplast genomes of Malus species, especially the differences of proteins that derived from the variation of nucleosides sequences (Li et al. 2022). The codon usage pattern of certain genes in chloroplast can reveal their conservative characteristics (Tan et al. 2020). According to the previous findings, the ycf1 could be used as a fragment to identify species of land plant, for some regions in the ycf1 gene were the most variable loci (Dong et al. 2015). Another previous research also revealed that the ycf1 gene was a vulnerable gene (Koh et al. 2006). Chloroplast genome has an independent, highly conserved genetic system (Xiong et al. 2009). According to the CBI values listed in Table.1, the subsequence of the ycf (except ycf1) and rps genes all show high adaption in the evolution process. In fact, the ycf1 in the samples are of two types with their lengths are 5640 bp and 1083 bp. On the contrary, the gens of psbB and psbC show lower codon adaption. In the present study, genes of rps8, rps18 and rpl16 are affected by natural selection during their evolutionary process; this is consistent with the results of FoP values listed in the table showing the corresponding genes have larger FoP values (Supplementary Tab.S2) (Debadin et al. 2019). Related studies also found that the Fop value would strong correlate with gene expression in P. glauca (Torre et al. 2015). The present study also shows that the standard deviations of the codon usage parameters are important for analyzing the phenotypic divergence and codon adaption.
Codons link nucleic acids to proteins even the function of the genes (Bastolla et al. 2017). Therefore, the codon usage patterns have been used for characterizing the evolutionary distance among the genomes (Liu et al. 2020a). The similar codon preference denotes the closer genetic relationship (Sophiarani et al. 2019). The chloroplast genomes of Malus species concerned in the present study would be divided into three groups. Among all the chloroplast genomes, the Malus angustifolia (MN061984.1), Malus coronaria (MN068247.1), Malus ioensis (MN062004.1) and Malus x atrosanguinea (MN061983.1) are all from the USA and three of them gathered together with some other strains, others are all from Chinese (Fig. 6). From this perspective, the geographic location dose not affects the evolutionary relationships obviously among those chloroplast genomes.
Phylogenetics depends greatly on the sequence alignment of genomes (Li et al. 2021b). The reliable sequences and effective analytical methods are very helpful for improving the reliability of the results (Zhang et al. 2017). Our present analyses produced largely identical deep relationships among the 20 chloroplast genomes of Malus species. The results highlight the importance of codon usage pattern in studying the diversity and the evolutionary distance among the plants via using their chloroplast genomes. All the diversity degree were calculated based on the ratio of the base pairs and the ratio of codons, therefore, the results shown in Fig. 7 depend strictly normalized data: on the nature of the genetic dispersion among the sequences, not the lengths of the gene sequences.
All data of chloroplast genomes of Malus species derived from the NCBI database were took into account, and 20 strains of them covering all kinds of the species were selected. Codon usage patterns both in overall genomes and in several certain typical genes, as well as evolutionary relationships among them, were calculated and analyzed in the present study. From the results, we found that, (1) all chloroplast genomes of Malus species are AT rich, bases of the sequences are affected by both the mutation and the natural selection pressure. The natural selection played a major role, especially on their third bases. (2) codon usage preferences in different genes are of significant differences although their overall codon usages are similar, and (3) the evolutionary characteristics of all genomes have not show obvious regional characteristics from the correspondence analysis. Overall, the codon usage pattern of the chloroplast genomes of Malus species will facilitate phylogenic and genetic research of plant species.
References
Amandine C, Tatiana G, Marinus JMS, Isabel R, Pierre G (2014) The domestication and evolutionary ecology of apples. Trends Genet 30(2):57–65. https://doi.org/10.1016/j.tig.2013.10.002
Bao L, Li K, Liu Z, Han M, Zhang D (2016) Characterization of the complete chloroplast genome of the Chinese crabapple Malus prunifolia (Rosales: Rosaceae: Maloideae). Conserv Genet Resour 8(3):227–229. https://doi.org/10.1007/s12686-016-0540-0
Bastolla U, Dehouck Y, Echave J (2017) What evolution tells us about protein physics, and protein physics tells us about evolution. Curr Opin Struct Biol 42:59–66. https://doi.org/10.1016/j.sbi.2016.10.020
Challabathula D, Zhang Q, Bartels D (2018) Protection of photosynthesis in desiccation-tolerant resurrection plants. J Plant Physiol 227:84–92. https://doi.org/10.1016/j.jplph.2018.05.002
Deb B, Uddin A, Mazumder GA, Chakraborty S (2018) Analysis of codon usage pattern of mitochondrial protein-coding genes in different hookworms. Mol Biochem Parasitol 219:24–32. https://doi.org/10.1016/j.molbiopara.2017.11.005
Debadin B, Subhasis M (2019) Comparative genomics of a few members of the family Aquificaceae on the basis of their codon usage profile. Gene Reports 14(10):54–64. https://doi.org/10.1016/j.genrep.2018.11.003
Dong W, Xu C, Li C, Sun J, Zuo Y, Shi S, Cheng T, Guo J, Zhou S (2015) ycf1, the most promising plastid DNA barcode of land plants. Sci Rep 5:8348. https://doi.org/10.1038/srep08348
Gichira AW, Avoga S, Li Z, Hu G, Wang Q, Chen J (2019) Comparative genomics of 11 complete chloroplast genomes of Senecioneae (Asteraceae) species: DNA barcodes and phylogenetics. Bot Stud 60(1):17. https://doi.org/10.1186/s40529-019-0265-y
Haruo S, Brian RM (2016) Codon adaptation of plastid genes. PLoS One 11(5):e0154306. https://doi.org/10.1371/journaLpone.0154306
He Y, Xiao H, Deng C, Xiong L, Yang J, Peng C (2016) The complete chloroplast genome sequences of the medicinal plant Pogostemon cablin. Int J Mol Sci 17:820. https://doi.org/10.3390/ijms17060820
Hosokawa K, Shibata T, Nakamura I, Hishida A (2004) Discrimination among species of papaver based on the plastid rpl16 gene and the rpl16–rpl14 spacer sequence. Forensic Sci Int 139(2–3):195–199. https://doi.org/10.1016/j.forsciint.2003.11.001
Jenkins GM, Holmes EC (2003) The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res 92(1):1–7. https://doi.org/10.1016/s0168-1702(02)00309-x
Koh EJ, Song WY, Lee Y, Kim HK, Kim K, Chung N, Lee KW, Hong SW, Lee H (2006) Expression of yeast cadmium factor 1 (YCF1) confers salt tolerance to Arabidopsis thaliana. Plant Sci 170(3):534–541. https://doi.org/10.1016/j.plantsci.2005.10.007
Kong WQ, Yang JH (2017) The complete chloroplast genome sequence of Morus cathayana and Morus multicaulis, and comparative analysis within genus Morus L. PeerJ 8(5):e3037. https://doi.org/10.7717/peerj.3037
Li S, Yang J (2014) System analysis of synonymous codon usage biases in archaeal virus genomes. J Theor Biol 355:128–139. https://doi.org/10.1016/j.jtbi.2014.03.022
Li N, Sun M, Jiang Z, Shu H, Zhang S (2016) Genome-wide analysis of the synonymous codon usage patterns in apple. J Integr Agr 15(5):983–991. https://doi.org/10.1016/S2095-3119(16)61333-3
Li G, Ren Y, Pan H, Zhang L (2018) Comprehensive analysis and comparison on the codon usage pattern of whole Mycobacterium Tuberculosis genome from different area. Biomed Res Int 2018:3574976. https://doi.org/10.1155/2018/3574976
Li Y, Liu Y, Wu P, Zhou S, Wang L, Zhou S (2020) The complete chloroplast genome sequence of Malus toringoides (Rosaceae). Mitochondrial DNA B 5(3):2790–2792. https://doi.org/10.1080/23802359.2020.1780977
Li C, Cai C, Tao Y, Sun Z, Jiang M, Chen L, Li J (2021a) Variation and evolution of the whole chloroplast genomes of Fragaria spp (Rosaceae). Front Plant Sci 12:754209
Li G, Zhang L, Xue P (2021b) Codon usage pattern and genetic diversity in chloroplast genomes of Panicum species. Gene 802:145866. https://doi.org/10.1016/j.gene.2021.145866
Li G, Zhang L, Xue P (2022) Codon usage divergence of important functional genes in Mycobacterium tuberculosis. Int J Biol Macromol 209:1197–1204. https://doi.org/10.1016/j.ijbiomac.2022.04.112
Liang C, Wang L, Lei J, Duan B, Ma W, Xiao S, Qi H, Wang Z, Liu Y, Shen X, Guo S, Hu H, Xu J, Chen S (2019) A comparative analysis of the chloroplast genomes of four salvia medicinal plants. Engineering 5(5):907–915. https://doi.org/10.1016/j.eng.2019.01.017
Liu M, Dong H, Wang M, Liu Q (2020a) Evolutionary divergence of function and expression of laccase genes in plants. J Genet 99:23. https://doi.org/10.1007/s12041-020-1184-0
Liu XY, Li Y, Ji KK, Zhu J, Ling P, Zhou T, Fan LY, Xie SQ (2020b) Genome-wide codon usage pattern analysis reveals the correlation between codon usage bias and gene expression in Cuscuta australis. Genomics 112(4):2695–2702. https://doi.org/10.1016/j.ygeno.2020.03.002
Maldonado LL, Stegmayer G, Milone DH, Oliveira G, Rosenzvit M, Kamenetzky L (2018) Whole genome analysis of codon usage in Echinococcus. Mol Biochem Parasitol 225:54–66. https://doi.org/10.1016/j.molbiopara.2018.08.001
Mazumdar P, Binti Othman R, Mebus K, Ramakrishnan N, Ann Harikrishna J (2017) Codon usage and codon pair patterns in non-grass monocot genomes. Ann Bot 120(6):893–909. https://doi.org/10.1093/aob/mcx112
Mazumder GA, Uddin A, Chakraborty S (2020) Analysis of codon usage pattern of mitochondrial ND genes in Platyhelminthes. Mol Biochem Parasitol 238:111294. https://doi.org/10.1016/j.molbiopara.2020.111294
McLean MJ, Wolfe KH, Devine KM (1998) Base composition skews, replication orientation, and gene orientation in 12 prokaryote genomes. J Mol Evol 47(6):691–496. https://doi.org/10.1007/pl00006428
Naizaier R, Qu Z, Wu S, Tian X (2019) The complete chloroplast genome of Malus sieversii (Rosaceae), a wild apple tree in Xinjiang. China Mitochondrial DNA B 4(1):983–984. https://doi.org/10.1080/23802359.2019.1581108
Nakamura M, Sugiura M (2007) Translation efficiencies of synonymous codons are not always correlated with codon usage in tobacco chloroplasts. Plant J 49(1):128–134. https://doi.org/10.1111/j.1365-313X.2006.02945.x
Naver H, Boudreau E, Rochaix JD (2001) Functional studies of Ycf3: its role in assembly of photosystem I and interactions with some of its subunits. Plant Cell 13(12):2731–2745. https://doi.org/10.1105/tpc.010253
Pandey S, Prasad A, Sharma N, Prasad M (2020) Linking the plant stress responses with RNA helicases. Plant Sci 299:110607. https://doi.org/10.1016/j.plantsci.2020.110607
Saurabh P, Mehanathan M, Namisha S, Vaishali C, Priya D, Shweta S, Sarita J, Saloni M, Manoj P (2019) Characterization of DEAD-box family of RNA helicases in tomato provides insights into their roles in biotic and abiotic stresses. Environ Exp Bot 158:107–116. https://doi.org/10.1016/j.envexpbot.2018.11.018
Shackelton LA, Parrish CR, Holmes EC (2006) Evolutionary basis of codon usage and nucleotide composition bias in vertebrate DNA viruses. J Mol Evol 62(5):551–563. https://doi.org/10.1007/s00239-005-0221-1
Sharp PM, Cowe E, Higgins DG, Shields DC, Wolfe KH, Wright F (1988) Codon usage patterns in Escherichia coli, Bacillus subtilis, Saccharomyces cerevisiae, Schizosaccharomyces pombe, Drosophila melanogaster and Homo sapiens; a review of the considerable within-species diversity. Nucleic Acids Res 16(17):8207–8211. https://doi.org/10.1093/nar/16.17.8207
Sophiarani Y, Arif U, Supriyo C (2019) Deciphering codon usage patterns and evolutionary forces in chloroplast genes of Camellia sinensis var. assamica and Camellia sinensis var. sinensis in comparison to Camellia pubicosta. J Integr Agr 18(12):2771–2785. https://doi.org/10.1016/S2095-3119(19)62716-4
Sueoka N (1988) Directional mutation pressure and neutral molecular evolution. P Natl Acad Sci Usa 85(8):2653–2657. https://doi.org/10.1073/pnas.85.8.2653
Sueoka N (1999) Translation-coupled violation of parity rule 2 in human genes is not the cause of heterogeneity of the DNA G+C content of third codon position. Gene 238(1):53–58. https://doi.org/10.1016/s0378-1119(99)00320-0
Supriyo C, Debojyoti N, Tarikul HM, Arif U (2017) Codon usage pattern and prediction of gene expression level in Bungarus species. Gene 604:48–60. https://doi.org/10.1016/j.gene.2016.11.023
Supriyo C, Sophiarani Y, Arif U (2020) Analysis of codon usage bias of chloroplast genes in Oryza species. Planta 252(4):67. https://doi.org/10.1007/s00425-020-03470-7
Svetlana VN, Duccio C, Vadim RVG (2013) Phylogenetic analysis of 47 chloroplast genomes clarifies the contribution of wild species to the domesticated apple maternal line. Mol Biol Evol 30(8):1751–1760. https://doi.org/10.1093/molbev/mst092
Tan W, Gao H, Zhang H, Yu X, Tian X, Jiang W, Zhou K (2020) The complete chloroplast genome of Chinese medicine (Psoralea corylifolia): molecular structures, barcoding and phylogenetic analysis. Plant Gene 21:100216. https://doi.org/10.1016/j.plgene.2019.100216
Tao P, Dai L, Luo M, Tang F, Tien P, Pan Z (2009) Analysis of synonymous codon usage in classical swine fever virus. Virus Genes 38(1):104–112. https://doi.org/10.1007/s11262-008-0296-z
Torre AR, Lin YC, Van de Peer Y, Ingvarsson PK (2015) Genome-wide analysis reveals diverged patterns of codon bias, gene expression, and rates of sequence evolution in picea gene families. Genome Biol Evol 7(4):1002–1015. https://doi.org/10.1093/gbe/evv044
Wei L, He J, Jia X, Qi Q, Liang Z, Zheng H, Ping Y, Liu S, Sun J (2014) Analysis of codon usage bias of mitochondrial genome in Bombyx mori and its relation to evolution. BMC Evol Biol 14:262. https://doi.org/10.1186/s12862-014-0262-4
Xiong AS, Peng RH, Zhuang J, Gao F, Zhu B, Fu XY, Xue Y, Jin XF, Tian YS, Zhao W, Yao QH (2009) Gene duplication, transfer, and evolution in the chloroplast genome. Biotechnol Adv 27(4):340–347. https://doi.org/10.1016/j.biotechadv.2009.01.012
Xu C, Cai X, Chen Q, Zhou H, Cai Y, Ben A (2011) Factors affecting synonymous codon usage bias in chloroplast genome of oncidium gower ramsey. Evol Bioinform 7:271–278. https://doi.org/10.4137/EBO.S8092
Xu X, Fei D, Han H, Liu H, Zhang J, Zhou Y, Xu C, Wang H, Cao H, Zhang H (2017) Comparative characterization analysis of synonymous codon usage bias in classical swine fever virus. Microb Pathog 107:368–371. https://doi.org/10.1016/j.micpath.2017.04.019
Xun M, Song J, Shi J, Li J, Shi Y, Yan J, Zhang W, Yang H (2021) Genome-wide identification of sultr genes in Malus domestica and low sulfur-induced MhSultr3;1a to increase cysteine-improving growth. Front Plant Sci 12:748242. https://doi.org/10.3389/fpls.2021.748242
Yan M, Zhao X, Zhou J, Huo Y, Ding Y, Yuan Z (2019) The complete chloroplast genome of cultivated apple (Malus domestica Cv. ‘Yantai Fuji 8’). Mitochondrial DNA B 4(1):1213–1216. https://doi.org/10.1080/23802359.2019.1591182
Yan L, Wang H, Huang X, Li Y, Yue Y, Wang Z, Tang S (2022) Chloroplast genomes of genus Tilia: comparative genomics and molecular evolution. Front Genet 13:925726. https://doi.org/10.3389/fgene.2022.925726
Yang Y, Zhu J, Feng L, Zhou T, Bai G, Yang J, Zhao G (2018) Plastid genome comparative and phylogenetic analyses of the key genera in Fagaceae: highlighting the effect of codon composition bias in phylogenetic inference. Front Plant Sci 9:82. https://doi.org/10.3389/fpls.2018.00082
Zhang W (2000) Phylogeny of the grass family (Poaceae) from rpl16 intron sequence data. Mol Phylogenet Evol 15(1):135–146. https://doi.org/10.1006/mpev.1999.0729
Zhang SD, Jin JJ, Chen SY, Chase MW, Soltis DE, Li HT, Yang JB, Li DZ, Yi TS (2017) Diversification of rosaceae since the late cretaceous based on plastid phylogenomics. New Phytol 214(3):1355–1367. https://doi.org/10.1111/nph.14461
Zhang X, Rong C, Qin L, Mo C, Fan L, Yan J, Zhang M (2018) Complete chloroplast genome sequence of Malus hupehensis: genome structure, comparative analysis, and phylogenetic relationships. Molecules 23:2917. https://doi.org/10.3390/molecules23112917
Acknowledgements
The authors of this manuscript sincerely appreciate the great efforts of all researchers who have contributed the data to the public databases of GenBank.
Funding
The authors have not any funding support.
Author information
Authors and Affiliations
Contributions
GL conceived, designed, and supervised the overall study. LZ, PX, and MZ conducted data processing and computational analyses. GL plotted the figures and drafted the manuscript, MZ revised the manuscript. All authors contributed to the article and agreed to the submitted version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, G., Zhang, L., Xue, P. et al. Comparative Analysis on the Codon Usage Pattern of the Chloroplast Genomes in Malus Species. Biochem Genet 61, 1050–1064 (2023). https://doi.org/10.1007/s10528-022-10302-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10528-022-10302-8