
Abstract
The phytochemical epigallocatechin gallate (EGCG) has been reported to alleviate age-associated immune disorders and organ dysfunction. However, information regarding the mechanistic role of EGCG in the suppression of cellular senescence is limited. The present study thus assessed the effects and underlying mechanisms of EGCG in the inhibition of senescence as well as its potential to selectively eliminate senescent cells (senolytics) using 3T3-L1 preadipocytes. Premature senescence was established in cells by repeated exposure of H2O2 at a sub-lethal concentration (150 μM). H2O2 treated cells showed characteristic senescence-associated features including increased cell size, senescence-associated β-galactosidase activity (SA-β-gal), development of senescence-associated secretory phenotype (SASP), activation of reactive oxygen species (ROS) and pathways, DNA damage as well as induction of cell cycle inhibitors (p53/p21WAF1/p16INK4a). In addition, a robust activation of PI3K/Akt/mTOR and AMPK pathways was also observed in H2O2 treated cells. Presence of EGCG (50 and 100 μM) showed significant downregulation of PI3K/Akt/mTOR and AMPK signaling along with the suppression of ROS, iNOS, Cox-2, NF-κB, SASP and p53 mediated cell cycle inhibition in preadipocytes. In addition, EGCG treatment also suppressed the accumulation of anti-apoptotic protein Bcl-2 in senescent cells thereby promoting apoptosis mediated cell death. Our results collectively show that EGCG acts as an mTOR inhibitor, SASP modulator as well as a potential senolytic agent thereby indicating its multi-faceted attributes that could be useful for developing anti-aging or age-delaying therapies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cellular senescence is a state of irreversible growth arrest characterized by a specific set of physio-chemical changes and cellular functions. A gradual accumulation of senescent cells and SASP is considered as a causative agent leading to familiar macroscopic consequences of tissue dysfunction and aged phenotype (Bhatia-Dey et al. 2016). Cellular senescence can be induced by a variety of factors, but the age-associated accumulation of ROS is a potent modulator of inflammatory and nutrient sensing pathways that ultimately culminate in the acceleration of cellular senescence program (Davalli et al. 2016). Hydrogen peroxide (H2O2) is a major ROS within the cells and a key source of mitochondrial damage which is closely associated with premature aging. Experimental evidence has shown that sub-cytotoxic concentration of H2O2 can induce senescence in a variety of cells which display several characteristic features associated with replicative senescence (Chen et al. 2007). As such, in recent times, the cellular model of H2O2-induced premature senescence has been extensively used to understand the mechanisms of senescence as well as in identification of potential modulators of senescence.
EGCG is a major component of green tea with several purported health beneficial effects. We and others have previously shown that EGCG is also effective in improving age-associated disorders such as immunosenescence (Sharma et al. 2017), inflammation and organ functions (Niu et al. 2013), as well as extending lifespan in experimental animals (Brown et al. 2006; Zhang et al. 2009). The mechanisms governing these effects of EGCG are multi-faceted and largely unclear. It has been shown that EGCG can suppress replicative senescence in human dermal fibroblasts by inhibiting p53 acetylation (Han et al. 2012). However, there appears to be a distinct dearth of studies pertaining to understanding the anti-senescence mechanisms of EGCG and, in particular, its influence in modulating nutrient sensing pathways vis-à-vis senescence and SASP. In this regard, PI3K/Akt/mTOR nutrient signaling pathway is considered central to aging and with mechanisms still not fully understood, mTOR activation in various tissues during aging is considered a major driving force of senescence (Blagosklonny 2008; Kennedy and Lamming 2016). Apart from inhibiting the onset of senescence, another novel approach is to identify compounds that can selectively induce apoptosis in senescent cells thereby preventing their accumulation. Such compounds are called ‘senolytics’ which appear to target multiple anti-apoptotic pathways related to Bcl-2/Bcl-XL, PI3K/Akt, p53/p21/serpines, dependence receptors/tyrosine kinases, and HIF-1α (Kirkland and Tchkonia 2017). However, very few compounds have been recognized with senolytic activity thus far and there is no information whether EGCG can also influence senescent cells by modulating apoptotic pathways. Therefore, keeping the foregoing discussion in view, in the present study we hypothesized that EGCG treatment could inhibit the progression of H2O2-induced premature senescence in murine preadipocytes by modulating mTOR, ROS and SASP pathways. Further, we theorized that senescent preadipocytes would be sensitive to EGCG-induced cell death by modulation of Bax/Bcl-2 signaling. To test our hypothesis, an H2O2-induced model of premature cell senescence was established in 3T3-L1 cell line and anti-senescence as well as senolytic attributes of EGCG were tested using a range of phenotypic, biochemical and molecular approaches. Our findings indicate that EGCG can inhibit the development of stress-induced senescence by countering DNA damage, cell cycle arrest and SASP production aided by the suppression of mTOR pathway. Further, EGCG appeared to act as a mild senolytic as it suppressed the accumulation of anti-apoptotic protein Bcl-2 in senescent cells resulting in augmentation of apoptosis and senescent cell death. Overall, the present work indicates multi-faceted anti-senescence potential of EGCG.
Materials and methods
Cell culture and conditions
3T3-L1 preadipocytes (ATCC, Manassas, VA, USA) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, 11965092; Thermo, USA) supplemented with 10% fetal bovine serum and 100 µg/ml of penicillin–streptomycin (15140122; Invitrogen, USA) at 37 °C in a 5% CO2 incubator. Studies were performed with cells in initial passage number (~ 3–5). This particular cell line was chosen since it has been shown that senescence in preadipocytes not only impairs its functional attributes, but also hampers neighboring non-senescent preadipocytes (Zoico et al. 2010; Trabucco and Zhang 2016).
Senescence induction and EGCG treatment
Cells were seeded into 6-well culture plates at density of 3000 cells/cm2 and cultured till ~ 80% confluency. The concentration of H2O2 was determined spectrophotometrically using molar extinction coefficient of 43.6 M−1cm−1. The experimental approach for inducing senescence and EGCG treatment is outlined in Fig. 1. Briefly, after each exposure to H2O2, cells were washed with PBS and resuspended in complete DMEM. After final H2O2 exposure, cells were washed and kept for three additional days in complete media for the development of senescent features and subsequent cellular analyses. EGCG was isolated, identified and characterized from fresh Kangra green tea shoots as described previously (Sharma et al. 2017). On the first day of H2O2 treatment, EGCG was dissolved in PBS and co-exposed with H2O2 to cells at a final concentration of 50 or 100 μM. After washing for removal of H2O2, cells were kept in EGCG at respective concentrations overnight and next day, H2O2 was directly applied to these cells. After 3 h of H2O2 treatment and subsequent washing, fresh EGCG was again added and the same treatment was continued for the third day. Post-third day, cells were kept in complete DMEM media only.

Schematic study design for investigating the effect of EGCG on H2O2-induced premature senescence in preadipocytes
Morphology and cell size
The changes in cell morphology against various treatments was observed using EVOS FL Auto 2 Imaging system (Thermo Scientific, USA). Senescence-induced changes in cell size were measured using the ‘area’ feature in brightfield channel of AMNIS ImageStream®X Mark II Imaging Flow Cytometer (Merck Millipore, Germany) as previously described (Biran et al. 2017).
Senescence-associated β-galactosidase assay
Cellular senescence was identified by measuring activity of β-galactosidase using β-galactosidase assay kit (K802; Biovision, USA) as per manufacturer’s protocol. In brief, at the end of experiment, cells were washed with PBS to remove culture media and then fixed with fixative solution. Cells were again washed and treated with staining solution followed by overnight incubation in dark at 37 °C in absence of CO2. Next day, cells were observed under EVOS FL Auto 2 Imaging System (Thermo Scientific, USA) for the appearance of blue color as a marker of senescence-associated β-galactosidase activity. Several images were taken and cells were manually counted for enumeration of percentage senescent cells.
Intracellular ROS assay
After respective experimental treatments, cells were stained with 10 μM H2DCFDA (D399; Molecular Probes, USA) for 30 min as per manufacturer’s protocol. Oxidation of the probe to 2′,7′-diichlorofluorescein (DCF) was measured by AMNIS ImageStream®X Mark II Imaging Flow Cytometer (Merck Millipore, Germany). Median fluorescence intensity was calculated and analyzed by INSPIRE ImageStream system software.
Cell proliferation assay
The proliferative potential of EGCG in H2O2 induced senescent cells was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; M2128, Sigma Aldrich, USA) based assay as previously described (Sharma et al. 2014). Briefly, after treatment regimen as per Fig. 1, cells were trypsinized, re-seeded in complete DMEM and cultured for 72 h followed by the addition of MTT (5 mg/ml) and further incubation for 4 h at 37 °C in a humidified CO2 incubator. Spent media was pipetted out, and 100 μL of acid isopropanol (0.1 N HCl in anhydrous isopropanol) was mixed to dissolve formazan crystals and absorbance was read at 540 nm. Proliferation rate (%) was calculated relative to control (proliferative) cells.
Senolytic assay and EGCG treatment
For analysis of senolytic attributes of EGCG, preadipocyte cells were seeded at a density of 3000 cells/cm2 followed by induction of H2O2 mediated senescence. Subsequently, senescent cells were treated with 50 or 100 μM of EGCG for 96 h and cellular morphology was assessed using the EVOS FL Auto 2 Imaging System (Thermo Scientific, USA). Cells were then trypsinized and numbers of live and dead cells were enumerated by trypan blue exclusion method using a hemocytometer. Further, total cellular protein was isolated and stored at − 80 °C till use. Appropriate proliferative cell controls as well as treatment controls were utilized for accurate inferences.
Annexin V assay
Senolytic activity of EGCG was further validated by analyzing the surface expression of phosphatidylserine in apoptotic cells using FITC-conjugated annexin V (556547; BD Biosciences, USA) as per manufacturer’s instructions. Briefly, cells were trypsinized and incubated with annexin V for 15 min at 37 °C in a CO2 incubator following which the fluorescent cells were captured using the AMNIS ImageStream®X Mark II Imaging Flow Cytometer (Merck Millipore, Germany) and data were analyzed by INSPIRE ImageStream system software. Results are expressed as percent annexin V positive cells.
RNA isolation, reverse transcription, and real-time PCR
Total cellular RNA was isolated was isolated by Qiagen RNeasy mini kit (74104, Qiagen, Germany) in accordance with manufacturer’s protocol. RNA was quantified and the integrity of RNA was assessed by identifying 18s and 23s RNA bands on agarose gel. One step qRT-PCR was performed using QuantiFast® SYBR® Green PCR kit (204054; Qiagen, Germany) as per manufacturer’s instruction. To normalize mRNA expression GAPDH (glyceraldehyde-3-phosphate dehydrogenase), expression was used as a house-keeping control. The PCR reactions were performed in 96-well plates (Applied Biosystems, USA) using the Step one Plus™ Real-Time PCR system (Applied Biosystems, USA). The ΔΔCt method was used to calculate the mRNA relative quantitation (Sharma et al. 2017). The primer sequences used for mRNA examination are provided in Supplementary Table 1.
Protein isolation and Western blotting
At the end of experiment, cells were washed twice with ice cold PBS (10 mM) and lysed with RIPA buffer (R0278; Sigma Aldrich, USA) containing protease inhibitor cocktail. Lysates were collected and centrifuged at 12,000×g for 10 min at 4 °C. Protein quantity was determined by Bradford assay (Bradford 1976). Furthermore, 50 µg of total protein from each sample was subjected to SDS-PAGE and transferred onto polyvinylidene difluoride membrane (GE Healthcare Life Sciences, Europe,) membrane with semidry trans-blotter (BioRad, USA) at 15 V for 90 min. The membranes were blocked with 5% non-fat dried milk for 1 h at room temperature. Washed thrice in PBST and were probed with following primary antibodies overnight at 4 °C with gentle shaking: anti-p53 (13-4100; 1:500) and anti-p21WAF1 (AHZ0422; 1:250) were procured from Thermo Fisher Scientific, USA while anti-PI3K (4257; 1:500), anti-Akt (9272; 1:500), anti-p-Akt (9271S; 1:250); anti-mTOR (2983; 1:500), anti-p-mTOR (5536; 1:500), anti-ATM/ATR substrate (2851; 1:500), anti-NF-κB (8242; 1:500), anti-Cox-2 (12282; 1:500), anti-Nrf-2 (12721; 1:500), anti-iNOS (13120; 1:500), anti-AMPKα (2603; 1:500) and anti-p-AMPKα (2535; 1:1000) were obtained from Cell signaling technologies, USA. Anti-Bcl-2 (sc-7382; 1:250), anti-Bax (sc-7480; 1:250) and anti-β-tubulin (sc-58882; 1:500) antibodies were purchased from Santa Cruz Biotechnology, USA. After incubation, the blots were again washed thrice (15 min/wash) in PBST and further incubated with a 1:5000 dilution of the HRP-conjugated anti-rabbit secondary antibody (7074S; Cell Signaling Technology, USA) with gentle shaking for 1 h. Subsequently, membranes were washed and protein bands were detected using ECL substrate on X-ray films. β-Tubulin (sc-58882; Santa-Cruz, USA) expression was analyzed as a loading control. The relative protein expression was quantified using the ImageJ software.
Statistical analyses
Data were analyzed using GraphPad Prism (Version 7) software. Experimental results are presented as mean ± standard error of the mean (SEM). Each value is the mean of the data from an assay performed in triplicate. Data were subjected to analysis of variance (ANOVA) and the Tukey test was used to separate the means (p < 0.05), which were considered statistically significant.
Results
EGCG inhibits the development of H2O2-induced senescent cells and senescence-associated secretory phenotype
The optimal concentration of H2O2 required for inducing senescence without affecting the cell viability was initially assessed by trypan blue exclusion method. It was observed that application of H2O2 at a final concentration of 400 μM and above caused significant (p < 0.05) cell death (Supplementary Fig. 1A). Based on this and subsequent standardization for senescence, exposure of H2O2 at a concentration of 150 μM for 3 h and for three consecutive days was selected as it induced irreversible cell senescence without significant cytotoxic effects. Further, cytotoxicity analysis of EGCG did not reveal any significant adverse effects at the concentrations of EGCG used in present study (50 and 100 μM) (Supplementary Fig. 1B). Hydrogen peroxide treatment to cells resulted in a heterogeneous morphology, with irregularly enlarged and flattened cells being observed within 3 days of the stress (Fig. 2a–d). The median cell size showed a significant (p < 0.05) and stark increase in H2O2 treated cells as compared to normal cells while EGCG treated cells appeared to counter this effect, albeit in a statistically non-significant manner (Fig. 2e). SA-β-gal activity significantly (p < 0.05) enhanced in H2O2 treated cells with over 60% cells appearing senescent as compared to control group, which was significantly (p < 0.05) abrogated on treatment with EGCG (Fig. 3a–e). Development of SASP in cells was assessed by qRT-PCR of inflammatory cytokines and protein expression of NF-κB. It was observed that H2O2 treatment immensely (p < 0.05) enhanced the gene expression of TNF-α, IL-6 and iNOS as compared to control (Fig. 4a–c). Similarly, NF-κB protein expression also significantly (p < 0.05) enhanced in H2O2 treated cells (Fig. 4d–e). Conversely, EGCG treatment significantly (p < 0.05) downregulated transcription of SASP factors as well as NF-κB protein expression, suggesting robust suppression of SASP components (Fig. 4a–e). However, EGCG application did not result in any significant changes in gene expression of TNF-α, IL-6 and iNOS in the proliferative controls (Fig. 4a–c).

EGCG alleviates H2O2-induced morphological changes in preadipocytes. Premature senescence was induced in cells after treatment with H2O2 for 3 h (150 μM) and for three consecutive days in presence or absence of EGCG. a Control cells, b cells treated with H2O2 treated alone, c, d cells supplemented with 50 and 100 μM EGCG respectively in addition to H2O2. Scale bar: 500 μm. e Median cell size as determined by flow cytometry. Values are mean ± SEM of three independent experiments. *Represents significant difference at p < 0.05 with respect to control cells

EGCG prevents activation of senescence-associated β-galactosidase. Premature senescence was induced in cells after treatment with H2O2 for 3 h (150 μM) and for three consecutive days in presence or absence of EGCG. a Control group, b H2O2 treated alone, c, d cells supplemented with 50 and 100 μM EGCG respectively in addition to H2O2. Scale bar: 500 μm. e Percentage of cells positive for SA-β-gal activity. Values are mean ± SEM of three independent experiments. **Represents significant difference at p < 0.01 with respect to H2O2 treated cells

EGCG treatment inhibits activation of SASP components. Premature senescence was induced in cells after treatment with H2O2 for 3 h (150 μM) and for three consecutive days in presence or absence of EGCG. Relative gene expression of selected SASP markers a TNF-α, b IL-6 and c iNOS. Relative protein expression of (d) NF-κB. e Representative western blot images. Values are mean ± SEM of three independent experiments. **Represents significant difference at p < 0.01 with respect to proliferative untreated control. ##Represents significant difference at p < 0.01 with respect to H2O2 treated cells
EGCG downregulates the expression of H2O2-induced cell cycle inhibitors
Activation of cell cycle inhibitory pathways including p53/p21WAF1 and p16Ink4a is considered as hallmark of senescence. It was observed that H2O2-induced senescence caused a significant (p < 0.05) and striking increase in p53 mRNA (Fig. 5a) and protein expression (Fig. 5d, f) as compared to control while EGCG treatment significantly (p < 0.05) attenuated both mRNA (Fig. 5a) and protein expression of p53 (Fig. 5d, f). Expression of p21WAF1 showed a non-significant increase in mRNA expression (Fig. 5b) while a significant (p < 0.05) increase in protein expression (Fig. 5e, f) was observed in H2O2-induced senescent cells. This upregulation was also significantly (p < 0.05) inhibited by EGCG treatment (Fig. 5b, e, f). In addition, p16INK4a mRNA expression also showed a significant (p < 0.05) increase which was again significantly (p < 0.05) attenuated by EGCG treatment (Fig. 5c). However, transcription analysis of these cell cycle inhibitors in proliferative controls revealed no discernible trend on account of EGCG application (Fig. 5a–c). Further analysis of cellular proliferative capacity validated the apparent EGCG mediated suppression of cell cycle inhibitors in senescent cells as significantly (p < 0.01) enhanced proliferation rate was observed in EGCG treated cells as compared to H2O2 treated cells (Table 1).

EGCG treatment inhibits the accumulation of cell cycle inhibitors associated with H2O2-induced premature senescence. Cells were treated with H2O2 for 3 h (150 μM) and for three consecutive days in presence or absence of EGCG. Relative gene expression of a p53, b p21, and c p16. Relative protein expression of d p53 and e p21. f Representative western blot images. Values are mean ± SEM of three independent experiments. ** and ***Represents significant difference at p < 0.01 and p < 0.001 respectively with respect to proliferative untreated control. # and ##Represents significant difference at p < 0.05 and p < 0.01 respectively with respect to H2O2 treated cells
EGCG protects against H2O2-induced ROS production and DNA damage
It was observed that H2O2 treated senescent cells suffered severe and significant (p < 0.05) oxidative damage as indicated by strong accumulation of intracellular ROS, which appeared to be slightly alleviated in presence of EGCG (Fig. 6a). Concomitantly, H2O2 treated cells showed a strong and significant (p < 0.05) increase in protein expression of iNOS, Cox2 and Nrf-2 as compared to control which further signifies presence of acute oxidative stress (Fig. 6b–e). In contrast, EGCG treated cells showed a robust and significant (p < 0.05) inhibition of ROS, iNOS, Cox and Nrf-2 suggesting minimal presence of ROS and subsequent stimulatory pathways in H2O2 treated cells (Fig. 6a–e). DNA damage in H2O2 treated cells was confirmed by the presence of phosphorylated ATM/ATR substrate which was also dose-dependently attenuated by EGCG (Fig. 6e).

EGCG treatment prevents oxidative damage associated with H2O2-induced premature senescence. Cells were treated with H2O2 for 3 h (150 μM) and for three consecutive days in presence or absence of EGCG. a Intracellular ROS production, b relative protein expression of iNOS, c relative protein expression of Cox2, d relative protein expression of Nrf-2, e Representative western blot images. Values are mean ± SEM of three independent experiments. * and **Represents significant difference at p < 0.05 and p < 0.01 respectively with respect to H2O2 treated cells
EGCG downregulates H2O2-induced activation of PI3K/Akt/mTOR pathway
Analysis of mTOR pathway revealed that H2O2-induced senescence significantly (p < 0.05) enhanced PI3K and Akt protein expression as compared to control (Fig. 7a, b, d). Similarly, ratio of phosphorylated mTOR to its native form showed a significant (p < 0.05) increase in H2O2-induced senescent cells (Fig. 7c, d). Conversely, EGCG treatment significantly (p < 0.05) downregulated expression of both PI3K and Akt (at 50 and 100 μM) as well as p-mTOR (albeit at 100 μM only) suggesting its role in modulation of stress-induced mTOR signaling pathway (Fig. 7a–d). A suppression in the gene expression of translation initiation factors (4E-BP1 and eIF4E) downstream of mTOR pathway was also observed that further indicated the inactivation of mTOR pathway in the presence of EGCG (Fig. 7e).

EGCG treatment inhibits H2O2-induced activation of mTOR pathway. Cells were treated with H2O2 for 3 h (150 μM) and for three consecutive days in presence or absence of EGCG. a Relative protein expression of PI3K, b relative protein expression of Akt, c p-mTOR/mTOR ratio, d Representative western blot images and e Relative gene expression of downstream targets of mTOR pathway. Values are mean ± SEM of three independent experiments. * and **Represents significant difference at p < 0.05 and p < 0.01 respectively with respect to H2O2 treated cells
EGCG inhibits senescence-induced AMPK activation
Analysis of protein levels of AMPK and p-AMPK revealed a strong and significant (p < 0.05) increase in AMPK and p-AMPK expression in H2O2-induced senescent cells while no significant change in p-AMPK/AMPK ratio could be observed (Fig. 8a–d). On the other hand, EGCG treatment significantly (p < 0.05) downregulated the expression of both AMPK, p-AMPK and their relative ratios suggesting an energetically dynamic state in EGCG treated cells (Fig. 8a–d).

EGCG treatment inhibits AMPK expression and activation in senescent preadipocytes. Premature senescence was induced in cells after treatment with H2O2 for 3 h (150 μM) and for three consecutive days in presence or absence of EGCG. a Relative protein expression of AMPK, b relative protein expression of p-AMPK, c p-AMPK/AMPK ratio, d representative western blot images. Values are mean ± SEM of three independent experiments. **Represents significant difference at p < 0.01 with respect to H2O2 treated cells
EGCG promotes apoptosis and induces senescent cell death
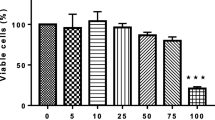
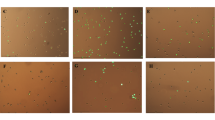
EGCG treatment to senescent cells showed dose-dependent prevalent signs of blebbing and the presence of apoptotic bodies (Fig. 9a–c). No such morphological changes were observed in young (proliferating) cells treated with EGCG (data not shown). Further, application of EGCG caused over threefolds significant increase (p < 0.05) in annexin V positive cells as compared to senescent control cells thereby suggesting the induction of apoptosis program in EGCG treated cells (Fig. 9d). EGCG treatment also caused a moderate dose-dependent decrease in survival of senescent cells as compared to proliferating cells thereby suggesting its senolytic attributes (Fig. 9e).

EGCG acts as a senolytic in senescent preadipocytes. Premature senescence was induced in cells after treatment with H2O2 for 3 h (150 μM) and for three consecutive days followed by EGCG treatment for 96 h. a–c Representative morphological images showing presence of apoptotic bodies and blebbing. d % Annexin V positive cells, e % Viable cells. Values are mean ± SEM of three independent experiments. *, ** and ***Represents significant difference at p < 0.05, p < 0.01 and p < 0.001 respectively with respect to control
EGCG acts as a senolytic by inhibiting Bcl-2 expression in senescent cells
Assessment of underlying molecular mechanism for apparent senolytic effects of EGCG revealed a strong upregulation of Bcl-2 protein expression in senescent cells which was dose dependently and significantly (p < 0.05) abrogated by EGCG treatment (Fig. 10b–d). The expression of pro-apoptotic protein Bax also appeared to increase in all treated cells with a highest expression in EGCG (50 μM) treated cells (Fig. 10a, d) indicating that EGCG could induce pro-apoptotic pathways in senescent cells. However, analysis of Bax/Bcl-2 ratio clearly revealed a significant (p < 0.05) decrease in senescent cells while a corresponding increase was observed in EGCG treated cells (both at 50 and 100 μM) signifying that application of EGCG augmented apoptosis in senescent cells by modulation of pro-and anti-apoptotic pathways (Fig. 10c).

The senolytic activity of EGCG is mediated by the inhibition of Bcl-2 pathway. Relative protein expression of a Bax and b Bcl-2. c Ratio of Bax to Bcl-2, d representative western blot images. Values are mean ± SEM of three independent experiments. **Represents significant difference at p < 0.01 with respect to H2O2 treated cells
Discussion
Gradual loss of cell proliferative capacity is an important barrier to tumorigenesis. However, in the context of aging, this loss of proliferation is recognized as a pivotal contributor to the development of cell senescence (Bhatia-Dey et al. 2016). In pursuit of strategies aimed at lifespan extension, nutraceuticals have been recognized as key modifiable factors that can influence aging (Verburgh 2015). In present study, we reveal that the bioactive tea component EGCG has multiple effects on amelioration of H2O2-induced cell senescence which is mediated by suppression of mTOR pathway along with selective elimination of senescent cells by modulating apoptotic regulators Bax/Bcl-2.
The oxi-inflamm-aging theory proposes that age-associated oxidative stress is a major driving factor of senescence (De la Fuente and Miquel 2009). Consistent with previous studies, we observed that application of sub-lethal concentration of H2O2 can activate senescence program in cells and exhibit characteristic senescence-associated features such as increase in cell size, expression of SA-β-gal, and NF-κB mediated development of SASP (Zhou et al. 2015; Zhu et al. 2017). In particular, the SASP, which includes plethora of cytokines such as TNF-α, IL-6 and IL-1β is detrimental for surrounding cells and tissue functions by causing inflammatory aggravation and immune cell activation. Further, it was observed that H2O2 exposure activated cell cycle inhibitor pathways p53/p21WAF1/p16INK4a and strongly suppressed cell proliferative capacity as also reported previously (Choo et al. 2014). On the other hand, EGCG supplemented cells were characterized by attenuated SA-β-gal activity as well as striking down-regulation of genes associated with SASP which could be attributed to EGCG mediated NF-κB inhibition. In addition, EGCG treatment appeared to counter senescence-associated cell cycle arrest by inhibiting the accumulation of cell cycle suppressors p53 and p21WAF1 which was further validated as EGCG treated cells showed improved proliferative capacity in comparison to H2O2 treated cells. Interestingly, in the absence of H2O2 treatment, application of EGCG to proliferative cells did not result in any appreciable changes in the expression of cell cycle inhibitory genes or measured SASP factors thereby indicating selectivity in cellular effects of EGCG. Together, these preliminary investigations indicated that EGCG treatment inhibited the development of oxidative stress induced senescence-associated features as well as cell cycle arrest. Previously, several polyphenols such as curcumin, genistein, apigenin and quercetin have shown anti-senescence attributes in vitro including the attenuation of SA-β-gal activity, morphological changes, suppression of some components of SASP and inhibition of p53/p21WAF1 (Mária and Ingrid 2017). However, in case of EGCG, a preliminary study has indicated that treatment with EGCG (at 100 μM) can inhibit replicative senescence by inhibition of p53/p21WAF1 and SA-β-gal activity (Han et al. 2012).
To further assess the mechanism(s) of senescence induction and ascertain the influence of EGCG, we analyzed stress-associated oxidative pathways and DNA damage response. It was observed that 3 days-post H2O2 treatment, the senescent cells were characterized by a strong elevation in the levels of intracellular ROS as well as protein levels of iNOS, Cox-2, NF-κB and Nrf-2. Acute oxidative stress can enhance the expression of pro-inflammatory genes such as Cox-2 and iNOS by altering redox signaling pathways such as NF-κB, which not only contributes to the development of SASP but also further enhances ROS production thereby aiding in the development and maintenance of the senescent phenotype (Chung et al. 2011). Together, it is reasonable to conclude that the transient H2O2 treatment instigated permanent damage to redox balance of the cells resulting in pro-oxidant and pro-inflammatory behavior. As a result of this sustained intracellular oxi-inflammatory stress, DNA damage would be imminent which was indeed confirmed by the activation of ATM via its phosphorylated substrates. The presence of DNA damage could have directly contributed to activation of cell cycle inhibitor pathways. On the other hand, EGCG treated cells categorically suppressed ROS, iNOS, Cox-2, NF-κB, as well as phosphorylated ATM substrates in a dose-dependent manner thereby indicating a milieu of redox and inflammatory homeostasis in EGCG treated cells. Previously, it has been shown that pre-treatment of EGCG for 6 h, and before application of H2O2, confers anti-senescence effects in mesenchymal stem cells mediated by the activation of Nrf-2 (Sadowska-Bartosz and Bartosz 2014). However, in present study, Nrf-2 over-activation was observed in H2O2 treated senescent cells only (thereby suggesting prevalent robust oxidative stress) while co-treatment of EGCG actually caused attenuation of Nrf-2 expression which was at par with the control. Given the fact that EGCG treatment concomitantly attenuated ROS, iNOS, Cox-2 and NF-κB, it appears that apparent anti-senescence/anti-oxidative effects of EGCG may not be exclusively dependent on the direct up-regulation of antioxidant defensive pathways (mediated by Nrf-2), but could involve alternate signaling mechanisms depending upon the type and duration of exposure in experimental designs.
We next hypothesized that PI3K/Akt/mTOR pathway could be involved in governing the observed effects of EGCG. Emerging evidence has shown that cell growth as well as cell senescence both are related to mTOR signaling. In fact, it has been theorized that senescence and aging are a consequence of overactivation (due to elements such as ROS) of growth promoting signaling pathways such as mTOR, which represents an evolutionary perspective of aging (Blagosklonny 2008; Laplante and Sabatini 2012; Campisi 2013). Although in vivo experiments have shown that inhibition of Akt/mTOR signaling prolongs lifespan (Johnson et al. 2013), however, causes and consequences of persistent mTOR signaling during senescence are still not clear (Carroll and Korolchuk 2017). Nonetheless, it is suggested that dysregulated ROS production during senescence is responsible for observed growth factors-independent activation of mTOR pathway (Nacarelli et al. 2015). Corroborating this, in present study, we observed that H2O2 mediated premature senescence is accompanied by a strong activation of PI3K, Akt and mTOR signaling. Persistent ROS in cells can directly stimulate PI3K which leads to downstream activation of Akt and mTOR (Zhang et al. 2016). In turn, it has been shown that Akt activation can enhance ROS production by increasing oxygen consumption thereby sensitizing Akt-expressing cells to ROS-dependent premature senescence (Nogueira et al. 2008). Further, there is evidence that Akt stimulation directly induces senescence by mTORC1 dependent up-regulation of p53 translation and stabilization as well as SA-β-gal activity (Swami 2008). In addition, activated mTOR has been shown to maintain SASP phenotype via enhanced NF-κB transcription thereby maintaining oxidative and inflammatory stress (Bent et al. 2016). Thus, it appears that after initial H2O2-induced ROS aggravation, activation of mTOR signaling could have contributed to sustained SASP and ROS generation resulting in activation of DNA damage response that ultimately culminated in the induction of cellular senescence mediated by p53/p21 pathway (Fig. 11). On the other hand, a dose-dependent inhibition of both PI3K and Akt was observed in EGCG treated groups while a strong inhibition of p-mTOR was also observed in 100 μM EGCG supplemented group. Together, it is reasonable to conclude that EGCG mediated suppression of PI3K/Akt/mTOR may have contributed to the apparent attenuation of SASP and ROS production in cells which could have contributed to the observed suppression of senescence (Fig. 11). Previously, EGCG has been shown to inhibit mTOR pathway and induce apoptosis albeit in cancerous cell lines only. This, however, is the first report implicating that EGCG mediated anti-senescence attributes under oxidative stress could be linked to its inhibitory effects on PI3K/Akt/mTOR signaling. Senescent cells display altered metabolic features characterized by a shift towards pro-glycolytic but less energetic state which is associated with increases in adenosine diphosphate (ADP) and adenosine monophosphate (AMP) relative to adenosine triphosphate (ATP) (Zwerschke et al. 2003). The increased ADP:ATP and AMP:ATP ratios activate AMPK which is a master regulator of cellular responses to energy stress. Sensing energetic stress, AMP kinase can activate p53/p21 cell cycle inhibitors by at least two known mechanisms thereby inducing cell senescence (Wiley and Campisi 2016). Considering this, we sought to analyze how EGCG treatment influences metabolic and energy status of senescent cells. It was observed that expression of AMPK and p-AMPK were upregulated in senescent cells but were inhibited by EGCG treatment which could have directly contributed in suppression of p53 accumulation and subsequent cellular senescence. Thus, this study provides evidence of role of EGCG in maintaining energy homeostasis in senescent cells mediated by AMPK suppression.

Schematic diagram of the observed effects and proposed anti-senescence mechanism of EGCG in present study. H2O2 treatment resulted in oxidative damage that stimulated DNA damage response and activated PI3K/Akt/mTOR pathway. The stimulated mTOR resulted in multiple effects including ROS generation, SASP development and accumulation of cell cycle inhibitors thereby developing senescent phenotype. EGCG treatment attenuated PI3K/Akt/mTOR pathway resulting in suppression of ROS, SASP and cell cycle inhibitors. EGCG acted as senolytic by suppressing the accumulation of Bcl-2 resulting in inhibition of pro-survival pathways in senescent cells and thereby causing selective cell death
Encouraged by the apparent robust anti-senescence attributes of EGCG, we next assessed whether EGCG could also act as a senolytic. Several proteins control the process of apoptosis, including pro-apoptotic regulator Bax and anti-apoptotic regulator Bcl-2. Bax regulates the activities of caspases and endonucleases by extricating cytochrome c from mitochondria, while Bcl-2 prevents the release of cytochrome c and encourages cell survival (Green and Reed 1998). It has been shown that senescent cells are resistant to apoptosis due to overexpression of Bcl-2 family proteins and inhibition of these proteins by senolytics can induce selective death (Kirkland and Tchkonia 2017). In present study, we observed that EGCG treatment to senescent cells dose-dependently induced apoptosis that caused a moderate decrease in cell viability. Further analysis revealed that Bcl-2 expression was highly upregulated in senescent cells which could have induced apoptotic resistance. On the other hand, EGCG treatment dose-dependently repressed Bcl-2 expression and enhanced Bax/Bcl-2 ratio suggesting the inhibition of pro-survival pathways and induction of apoptosis program in senescent cells. The apparent senolytic property of EGCG could have also contributed to its anti-senescence attributes, since it is plausible that deeply developing senescent cells may have been eliminated by EGCG treatment resulting in the abolition of SA-β-gal activity and SASP production. Thus, this interplay could have eventually aided in suppressing the escalation of senescence to neighboring cells resulting in the observed senescence abrogating attributes of EGCG.
In conclusion, the present work justifies our hypothesis and demonstrates that EGCG has multi-faceted inhibitory influence on the development and severity of cellular senescence. Recent research identifies that inhibition of development of senescent cells or attenuation of SASP or selective elimination of senescent cells (senolytics) are potential targets of anti-senescence therapies. Our results provide compelling evidence that EGCG exhibits all these attributes thereby indicating that consumption of EGCG could be useful in attenuating age-associated disorders by mitigating stress-associated functional damage in various cell types including adipocytes. The senolytic attributes of EGCG could further eliminate senescent cell burden in tissues rendering robust functional capacity. However, from a therapeutic perspective, it would be interesting to assess whether application of EGCG could also reverse or attenuate the senescent phenotype (replicative as well as stress-induced) using post-treatment experimental system(s), especially in human cell lines. It also needs to be considered that concentrations of EGCG used in present study are relatively higher than those generally attained after oral or intraperitoneal treatments due to known low bioavailability of catechins (Lambert et al. 2003). Thus, directions for future research may also include investigations of the physiological relevance of present findings as well as strategies for improving the bioavailability of EGCG so as to fully exploit its anti-aging attributes.
Abbreviations
- EGCG:
-
Epigallocatechin gallate
- mTOR:
-
Mechanistic target of rapamycin
- H2O2 :
-
Hydrogen peroxide
- SASP:
-
Senescence-associated secretory phenotype
- ROS:
-
Reactive oxygen species
- iNOS:
-
Inducible nitric oxide synthase
- Cox-2:
-
Cyclooxygenase-2
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- AMPK:
-
Adenosine monophosphate-activated protein kinase
References
Bent EH, Gilbert LA, Hemann MT (2016) A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses. Genes Dev 30(16):1811–1821
Bhatia-Dey N, Kanherkar RR, Stair SE, Makarev EO, Csoka AB (2016) Cellular senescence as the causal nexus of aging. Front Genet 7:13
Biran A, Zada L, Abou Karam P, Vadai E, Roitman L, Ovadya Y, Porat Z, Krizhanovsky V (2017) Quantitative identification of senescent cells in aging and disease. Aging Cell 16(4):661–671
Blagosklonny MV (2008) Aging: ROS or TOR. Cell Cycle 7(21):3344–3354
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Brown MK, Evans JL, Luo Y (2006) Beneficial effects of natural antioxidants EGCG and alpha-lipoic acid on life span and age-dependent behavioral declines in Caenorhabditis elegans. Pharmacol Biochem Behav 85(3):620–628
Campisi J (2013) Aging, cellular senescence, and cancer. Annu Rev Physiol 75:685–705
Carroll B, Korolchuk VI (2017) Dysregulation of mTORC1/autophagy axis in senescence. Aging 9(8):1851–1852
Chen JH, Ozanne SE, Hales CN (2007) Methods of cellular senescence induction using oxidative stress. Methods Mol Biol 371:179–189
Choo KB, Tai L, Hymavathee KS, Wong CY, Nguyen PN, Huang CJ, Cheong SK, Kamarul T (2014) Oxidative stress-induced premature senescence in Wharton’s jelly-derived mesenchymal stem cells. Int J Med Sci 11(11):1201–1207
Chung HY, Lee EK, Choi YJ, Kim JM, Kim DH, Zou Y, Kim CH, Lee J, Kim HS, Kim ND, Jung JH, Yu BP (2011) Molecular inflammation as an underlying mechanism of the aging process and age-related diseases. J Dental Res 90:830–840
Davalli P, Mitic T, Caporali A, Lauriola A, D’Arca D (2016) ROS, cell senescence, and novel molecular mechanisms in aging and age-related diseases. Oxid Med Cell Longev 2016:3565127
De la Fuente M, Miquel J (2009) An update of the oxidation-inflammation theory of aging: the involvement of the immune system in oxi-inflamm-aging. Curr Pharm Des 15(26):3003–3026
Green DR, Reed JC (1998) Mitochondria and apoptosis. Science 281:1309–1312
Han DW, Lee MH, Kim B, Lee JJ, Hyon SH, Park JC (2012) Preventive effects of epigallocatechin-3-O-gallate against replicative senescence associated with p53 acetylation in human dermal fibroblasts. Oxid Med Cell Longev 2012:850684
Johnson SC, Rabinovitch PS, Kaeberlein M (2013) mTOR is a key modulator of ageing and age-related disease. Nature 493(7432):338–345
Kennedy BK, Lamming DW (2016) The mechanistic target of rapamycin: the grand conductor of metabolism and aging. Cell Metab 23(6):990–1003
Kirkland JL, Tchkonia T (2017) Cellular senescence: a translational perspective. EBioMedicine 21:21–28
Lambert JD, Lee MJ, Lu H, Meng X, Hong JJ, Seril DN, Sturgill MG, Yang CS (2003) Epigallocatechin-3-gallate is absorbed but extensively glucuronidated following oral administration to mice. J Nutr 133(12):4172–4177
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149:274–293
Mária J, Ingrid Ž (2017) Effects of bioactive compounds on senescence and components of senescence associated secretory phenotypes in vitro. Food Funct 8(7):2394–2418
Nacarelli T, Azar A, Sell C (2015) Aberrant mTOR activation in senescence and aging: a mitochondrial stress response? Exp Gerontol 68:66–70
Niu Y, Na L, Feng R, Gong L, Zhao Y, Li Q, Li Y, Sun C (2013) The phytochemical, EGCG, extends lifespan by reducing liver and kidney function damage and improving age-associated inflammation and oxidative stress in healthy rats. Aging Cell 12(6):1041–1049
Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, Unterman T, Hay N (2008) Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell 14(6):458–470
Sadowska-Bartosz I, Bartosz G (2014) Effect of antioxidants supplementation on aging and longevity. Biomed Res Int 2014:404680
Sharma R, Kapila R, Haq MR, Salingati V, Kapasiya M, Kapila S (2014) Age-associated aberrations in mouse cellular and humoral immune responses. Aging Clin Exp Res 26(4):353–362
Sharma R, Sharma A, Kumari A, Kulurkar PM, Raj R, Gulati A, Padwad YS (2017) Consumption of green tea epigallocatechin-3-gallate enhances systemic immune response, antioxidative capacity and HPA axis functions in aged male swiss albino mice. Biogerontology 18(3):367–382
Swami M (2008) Akt: a double-edged sword. Nat Rev Cancer 2008(9):76. https://doi.org/10.1038/nrc2586
Trabucco SE, Zhang H (2016) Finding Shangri-La: limiting the impact of senescence on aging. Cell Stem Cell 18(3):305–306
Verburgh K (2015) Nutrigerontology: why we need a new scientific discipline to develop diets and guidelines to reduce the risk of aging-related diseases. Aging Cell 14(1):17–24
Wiley CD, Campisi J (2016) From ancient pathways to aging cells-connecting metabolism and cellular senescence. Cell Metab 23(6):1013–1021
Zhang L, Jie G, Zhang J, Zhao B (2009) Significant longevity-extending effects of EGCG on Caenorhabditis elegans under stress. Free Radic Biol Med 46(3):414–421
Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y, Dong W (2016) ROS and ROS-mediated cellular signaling. Oxid Med Cell Longev 2016:4350965
Zhou L, Chen X, Liu T, Gong Y, Chen S, Pan G, Cui W, Luo ZP, Pei M, Yang H, He F (2015) Melatonin reverses H2O2-induced premature senescence in mesenchymal stem cells via the SIRT1-dependent pathway. J Pineal Res 59(2):190–205
Zhu X, Yue H, Guo X, Yang J, Liu J, Liu J, Wang R, Zhu W (2017) The preconditioning of berberine suppresses hydrogen peroxide-induced premature senescence via regulation of sirtuin 1. Oxid Med Cell Longev 2017:2391820
Zoico E, Di Francesco V, Olioso D, Fratta Pasini AM, Sepe A, Bosello O, Cinti S, Cominacini L, Zamboni M (2010) In vitro aging of 3T3-L1 mouse adipocytes leads to altered metabolism and response to inflammation. Biogerontology 1:111–122
Zwerschke W, Mazurek S, Stöckl P, Hütter E, Eigenbrodt E, Jansen-Dürr P (2003) Metabolic analysis of senescent human fibroblasts reveals a role for AMP in cellular senescence. Biochem J 376(Pt 2):403–411
Acknowledgements
Authors are grateful to the Director, CSIR-IHBT for constant encouragement and support. This work was supported by grants from Department of Science and Technology, Government of India under the INSPIRE Faculty scheme (IFA17-LSPA79) and CSIR in-house project MLP0204.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kumar, R., Sharma, A., Kumari, A. et al. Epigallocatechin gallate suppresses premature senescence of preadipocytes by inhibition of PI3K/Akt/mTOR pathway and induces senescent cell death by regulation of Bax/Bcl-2 pathway. Biogerontology 20, 171–189 (2019). https://doi.org/10.1007/s10522-018-9785-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10522-018-9785-1