
Abstract
We conducted whole-genome sequencing of four inbred mouse strains initially selected for high (H1, H2) or low (L1, L2) open-field activity (OFA), and then examined strain distribution patterns for all DNA variants that differed between their BALB/cJ and C57BL/6J parental strains. Next, we assessed genome-wide sharing (3,678,826 variants) both between and within the High and Low Activity strains. Results suggested that about 10% of these DNA variants may be associated with OFA, and clearly demonstrated its polygenic nature. Finally, we conducted bioinformatic analyses of functional genomics data from mouse, rat, and human to refine previously identified quantitative trait loci (QTL) for anxiety-related measures. This combination of sequence analysis and genomic-data integration facilitated refinement of previously intractable QTL findings, and identified possible genes for functional follow-up studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Anxiety disorders are among the most common mental disorders observed in the United States, affecting about 31.3% of all adults at least one time in their lifespan, with 19.1% of United States adults experiencing an anxiety disorder in the last year alone (National Institute of Mental Health 2017). Furthermore, those who are suffering from some type of anxiety disorder are 3–5 times more likely to visit a doctor than those who are not (Anxiety and Depression Association of America 2018), and these disorders are highly comorbid with other diseases (Meier and Deckert 2019). Anxiety disorders are estimated to cost ~ $45 billion annually, representing around 30% of total expenditures for all mental illnesses in the United States (DeVane et al. 2005).
There is clear evidence that risk for anxiety disorders is determined by a fine interplay between genetics and environment. Heritability estimates for anxiety disorders range between 30 and 50%, based on epidemiological, twin, and family studies (Shimada-Sugimoto et al. 2015) with small contributions from common familial environments in females and the same predisposing genes across sexes (Gottschalk and Domschke 2017).
Although treatment options such as psychotherapy, pharmacotherapy, and homeopathic interventions are currently available, many of these lead to unwanted side effects and may not have a substantial beneficial effect for patients [reviewed in (Bandelow et al. 2017)]. For example, quetiapine has high efficacy but is not licensed for use in most countries due to the substantial negative side effects (Bandelow et al. 2017). Given the lack of effective treatments, there is an urgent need to identify underlying biological mechanisms contributing to anxiety behaviors. In this paper, we advance this goal using a genetic mouse model combined with bioinformatic analyses of new and existing publicly available functional genomics data.
High and Low Activity selected strains as a model
Given the complex genetics of anxiety disorders in humans, mouse models provide a convenient system within which to clarify and identify genetic influences on anxiety. Previous research has demonstrated that neuronal circuitry involved in human anxiety disorders is also implicated in rodent models of anxiety-related behaviors, e.g. hyperactivation of the hippocampus, which is linked to anxiety behaviors in rodents and humans (Duval et al. 2015). Mouse models offer many opportunities for insight into the neurobiological mechanisms of anxiety including a well-characterized genome, controlled experiments to establish causality, and established measures of anxiety-like behavior. Open-field activity (OFA) is commonly used as a measure of anxiety in mice, where higher activity and increased exploration reflect lower anxiety and vice versa (Seibenhener and Wooten 2015). In a classic bidirectional selection experiment for open-field activity, DeFries et al. (1978) differentially selected two replicate lines of mice for 30 generations. The selection experiment began with an F3 generation derived from an initial cross between the inbred BALBc/J and C57BL/6J strains. From each of ten F3 generation litters that contained at least two males and two females, the most active male and most active female within each litter were selected. These ten selected males and ten selected females were then mated at random to become the progenitors of the H1 line. In a corresponding manner, the least active male and least active female from each of these same litters were selected and mated at random to become the progenitors of the L1 line. From a different sample of ten F3 litters, one male and one female within each litter were chosen at random to become the progenitors of the C1 line. From two additional sets of ten F3 litters, a similar procedure was used to establish the replicate H2, L2, and C2 lines (Turri, Henderson et al. 2001a, b). During 30 generations of subsequent within-litter selection, the two H lines gradually diverged from the two L lines, eventually manifesting a 30-fold difference in open-field activity scores (Fig. 1). These lines were subsequently maintained by random mating within line for 18 generations, and then inbred using brother-sister matings. As outlined below, F2 crosses of the H and L inbred strains have been utilized for genetic mapping experiments.

Response to 30 generations of bi-directional selection for high and low open-field activity reproduced, with permission, from (DeFries et al. 1978)
The unique H1 and L1 strains were studied in the early 1990s to perform the first quantitative trait loci (QTL) mapping study of a behavioral trait using an F2 intercross of the High and Low Activity strains (Flint et al. 1995). In addition to open-field activity, the investigators also confirmed differences between strains in the expected direction for other mouse measures of anxiety-like defensive behavioral responses, including Y-maze, elevated plus-maze (EPM), and defecation scores. QTL on chromosomes 1, 12, and 15 were identified to contain genes linked to “emotionality” across all four testing measures.
Both the behavioral and QTL results were subsequently replicated in a separately derived F2 population, and the light–dark box (LDB) measure of anxiety was added to the behavioral phenotypes. The same three loci (chromosomes 1, 12, and 15) as well as four more loci on chromosomes 4, 7, 18, and X were linked with anxiety phenotypes. However, based on the overlap among identified QTL for multiple phenotypes, it was unclear whether each QTL contributed equally to certain anxiety-related behaviors, or whether specific regions were unique to particular phenotypes (Turri et al. 2001a, b).
To address the question of whether the same set of genes impacts the behaviors observed in the different tests, data from mice tested in Turri et al. (2001a, b) were re-analyzed to evaluate an expanded set of anxiety-related behaviors in five tests: OFA, EPM, LDB, mirror chamber, and square maze (Turri et al. 2001a, b). Consistent with both of the previously described studies, loci on chromosomes 1, 4, and 15 had statistically significant LOD scores; however, they also reported QTL on chromosomes 7, 12, 14, 18, and X as having some influence on anxiety-related phenotypes, but to a lesser extent. Loci on chromosome 15 appeared to influence every phenotype while loci on chromosome 1, 4, 12, 18, and X influenced many but not all. They also reported high LOD scores for loci on chromosome 7. Finally, analyses revealed that avoidance behaviors were associated with loci on chromosome 15, generalized activity was associated with loci on chromosome 4, and exploratory behaviors were associated with loci on chromosome 1. Loci on chromosome 1 were estimated to account for 10% of phenotypic variance (Turri et al. 2001a, b).
Finally, a follow-up analysis by Henderson et al. (2004) combined data across the previous studies and used factor analysis to evaluate detailed phenotypic aspects to further characterize which QTL are associated with specific features of the anxiety-related behaviors. Their goal was to investigate potential consequences of certain choices for phenotypes in mapping studies. This comprehensive analysis provided a complete overview of genetic effects on over 100 endophenotypes related to anxiety-related behaviors. Using this approach, one additional chromosomal locus was linked to these phenotypes on chromosome 8, while replicating previously identified QTL (1, 4, 5, 7, 12, 15, 18, and X) consistent across the four mapping studies. Altogether these loci account for between 20 and 36% of the phenotypic variance for measures of anxiety-like defensive behavioral responses. Chromosomes 1 and 15 show the largest influence on these measures. A benefit of the diverse set of measures used is that it allows for more specific interpretation of the behavioral mechanisms involved. Upon further evaluation of the loci on chromosomes 1 and 15, it was discovered that their contributions to anxiety-related phenotypes are slightly different. Chromosome 1 appeared to play a role in fear and hiding. Chromosome 15 was associated with modulation of latency to move from one area of a testing apparatus to another, suppression of rearing behavior, and no contribution whatsoever to autonomic activity (Henderson et al. 2004). In summary, the experiments described above with the High and Low Activity mice have shown there is a strong influence of loci on chromosomes 1 and 15 on anxiety-like behavior, with smaller contributions coming from chromosomes 4, 5, 7, 8, 12, 14, 15, 18, and X. These mice have not been studied since these early QTL studies, but have been continuously inbred at the Institute for Behavioral Genetics, and were recently retested to confirm the dramatic differences in anxiety-related behaviors, including sub-measures of OFA (e.g., center activity in the open-field, and time spent in the center of the open-field), as well as other well-established tests for anxiety-related behaviors such as light–dark box, elevated plus-maze, and novel object recognition (Booher et al. submitted).
Other genetic models of anxiety
In addition to the High and Low Activity strains, other genetic populations have been used to study anxiety-like phenotypes. In BXD (derived from C57BL/6J and DBA/2J mice) recombinant inbred (RI) mice, (Philip et al. 2010) identified significant QTL on chromosomes 9, 10, and 13 and suggestive QTL on chromosomes 1, 4, 5, 6, 8, 9, 10, 11, and 13. In a separate study of habituation, characterized by a reduction in activity in a novel environment, chromosome 15 was identified using BXD RI lines, but suggest it may be more involved in anxiety-related behaviors than activity based on similar findings in other studies (Bolivar and Flaherty 2003). Another cross-species analysis using BXD recombinant inbred mice was carried out to assess candidate genes for anxiety-related behaviors (Ashbrook et al. 2015). Using BXD recombinant inbred mice, QTL on chromosomes 1 and 13 were linked with elevated zero-maze measures and open-field activity, respectively (Ashbrook et al. 2015). Chromosome 1 is consistent with results from the High and Low Activity mice results, and the two QTL overlap slightly. Another experiment involving 1063 advanced intercross line (AIL) mice (derived from LG/J and SM/J mice), a multigenerational outbred population, identified novel associations between QTL, expression QTL (eQTL) and locomotor activity (Gonzales et al. 2018). Integrating Genome Wide Association Study (GWAS) results with QTL data, four chromosomes were identified as being important for locomotor activity in mice: 4, 6, 8, and 17 (Gonzales et al. 2018). Chromosomes 4 and 8 are consistent with previous studies discussed for having an influence in anxiety-like phenotypes. Finally, an older review summarized findings from studies of four different crosses (C57BL/6J (B6) X DBA/2J (D2), B6 X C3H/HeJ (C3H), ILS/Ibg X ISS/Ibg (ISS) and C57BL/6By (B6y) X BALB/cBy (BALB)) and identified a QTL-rich region on chromosome 1 (QRR1) implicated in emotionality and other neurobehavioral phenotypes (Mozhui et al. 2008). This QRR1 hotspot is between 172.5 and 177.5 Mb in mice, which overlaps with the chromosome 1 QTL region in the High and Low Activity mice between 172.5 and 177.3 Mb (Mozhui et al. 2008).
Whole genome sequencing to assess effects of selection, coupled with bioinformatics to identify gene candidates
Mapping in selected mouse lines allows identification of QTL that account for a greater percentage of the genetic variance contributing to the phenotype (Parker et al. 2014; Zhou et al. 2019), compared to natural populations, such as humans, where high polygenicity and small effect sizes reduce statistical power to identify associated loci. However, these studies, particularly early QTL mapping studies, require substantial follow up and refinement to identify causal variants and the gene products they influence. Whole genome sequence (WGS) data from selected mouse strains can be coupled with expanding informatics databases and tools to create a system for prioritizing genes through the aggregation and integration of data across species. This bioinformatics approach represents a powerful tool for examining legacy data from historic gene mapping experiments where QTL intervals could not be narrowed to specific genes. In the current study, there were two main objectives directly related to the High and Low Activity strains. First, we assessed the effects of selection and subsequent inbreeding on genomic architecture in the High and Low Activity strains through whole genome sequencing, by identifying regions that were selected repeatedly in the two High or two Low Activity lines. Second, genes within these regions specific to the High and Low Activity strains were prioritized using current genomics databases and tools for cross-species analyses. As a broader third goal, we applied this cross-species analytical approach across ALL genetic studies related to anxiety, thereby allowing identification of genes most represented in the field as a whole.
Methods
Animals
Male and female High (H1 and H2) and Low (L1 and L2) Activity mice were bred and housed in the specific pathogen-free Jennie Smoly Caruthers Biotechnology Building at the University of Colorado Boulder on a 12-h light:dark cycle (lights on at 7:00 AM). The mice were housed in standard 30 cm × 13 cm × 17 cm polycarbonate cages with ad libitum access to food (Envigo Teklad 2914 irradiated rodent diet, Harlan, Madison, WI, USA) and water. Room temperature was maintained between 23 and 24.5 °C.
DNA sequencing, assembly, and variant calling, and high/low strain comparison
For each of the four strains, DNA was isolated from one male and one female using the QIAmp Fast DNA Tissue Kit (Qiagen, Inc, Hilden, Germany) per the manufacturer’s instructions for tail tissue. Isolated DNA samples were sent to Novogene, Inc for library construction using the NEB Next Ultrall DNA Library Prep Kit, 350 bp insert library. Sequencing was performed in China on an Illumina HiSeqX Ten instrument using a paired-end 150 bp sequencing strategy.
We aligned the short reads to the mouse GRCm38 reference genome using BWA-MEM 0.7.17 (Li and Durbin 2009; Li 2013) with default parameters, and sorted the resulting Sequence Alignment/Map (SAM) file and converted it to BAM format with samtools 1.9 (Li et al. 2009). We then jointly called variants across all eight individuals using bcftools (Li 2011; Danecek et al. 2014) mpileup (-e 20 -F 0.002 -h 100 -L 250 -m 1 -o 40), call (-m –keep-alts), and filter (-e ‘DP < 2’ -Oz). We restricted the called variants using the -targets-file call command to sites polymorphic between the C57BL/6J and BALB/cJ progenitor strains, identified through the Wellcome Sanger Mouse Genomes Project (ftp://ftp-mouse.sanger.ac.uk/current_snps/strain_specific_vcfs/BALB_cJ.mgp.v5.snps.dbSNP142.vcf.gz).
We removed indels and multi-allelic sites, and those in which read depth was below 20 or above 120, mapping quality was below 35, and/or quality score was below 20. Because all four lines (H1, H2, L1, and L2) had been inbred over a number of generations, we expected little to no autosomal variation still segregating within strains. Therefore, we further restricted our analysis to sites in which no individuals were heterozygous and where males and females of the same strains had exactly matching genotypes, retaining a total of 3,678,826 biallelic positions across the 19 autosomes. We note that this likely excluded some small number of real heterozygous positions that have arisen within strains through de novo mutation. However, we were interested primarily in the variants segregating among progenitor strains that influence anxiety-like behaviors, and there were unlikely to be many true new mutations compared to sequencing and variant calling errors. Future work may address the influence of de novo mutations on selected strains. We used the Mouse Map Converter from The Jackson Laboratory (Mouse Map Converter (n.d.)) to assign genetic map position based on physical positions, and assigned alleles as either of BALB/cJ (B) or C57BL/6J (C) origin (the two progenitor strains) by comparison to the sequenced genomes available through the Sanger Institute (Mouse Genomes Project-Sanger Institute (n.d.)).
High/low strain genome comparison
We performed two primary analyses to compare the strain distribution patterns of the selected strains. First, we estimated the proportion of variants possibly associated with OFA. Let V equal the number of variants that are associated with OFA, U equal the number that are not associated with OFA, P equal the proportion of variants associated with OFA, i.e., P = V/(V + U), and w, x, y, and z equal the observed number of variants in the four combinations of possible parental variants in the High and Low selected strains: BB, BC, CB, CC. The expected (w + z) = 0.5U, while the expected (x + y) = V + 0.5U. Therefore, the expected [x + y − (w + z)] = V and the expected total number of variants is (w + z + y + z) = V + U. P can then be calculated for each pair of H1 and L1 (P1), and H2 and L2 (P2). We estimated P1 and P2 across the whole genome and for each autosome separately.
Second, we estimated ϕ, the coefficient of association (Sokal and Rohlf 1995; Rieseberg et al. 2003) which varies from − 1 to 1, and compares the genomic distribution of progenitor strain alleles in each of the pairs of descendant lines with the expected distribution of alleles under selection. Namely, we expected there to be a strong positive association in H1-H2 and L1-L2 comparisons, but 0-to-negative association of alleles when comparing high to low strains. We performed this analysis in 1 cM windows to localize signals of association, and used a block jackknife to estimate the genome-wide mean and standard error of ϕ, which accounts for linkage disequilibrium across the genome (Reich et al. 2009). We also assessed the impact of window size by applying 5 cM and 10 cM windows.
Our initial analyses revealed remarkably consistent estimates for the L1 and L2 strains and for the H1 and H2 strains (see below). Although strong selection for a highly polygenic character may have yielded these results, it is possible that some inadvertent cross-contamination may have occurred between the two L strains and/or between the two H strains. Following a mouse hepatitis virus epidemic in our main colony during 2009–2010, newborn pups from these lines were transferred to a nucleus colony room and cross-fostered to clean mothers. Because it is possible that an inadvertent cross of the replicate strains could have occurred during that transfer, we regard our between-replicate analyses (i.e., H1 vs H2 and L1 vs L2) as being only exploratory.
Identification of genes within narrowed QTL
Using the whole genome sequence data from the H1, H2, L1, and L2 mice, we identified regions, which will be referred to as “regions of difference” in which High Activity strains and Low Activity strains had opposite alleles, i.e., H1 & H2 shared the same allele, and L1 & L2 shared the opposite allele. We overlaid the QTL found by Henderson et al. (2004), using these High and Low Activity strains with the regions of difference from our genotyping, resulting in narrowed regions on chromosomes 1, 4, 7, 8, 14, 15, and 18 based on their genetic map positions. We used the Mouse Genome Database (MGI-Mouse Genome Informatics 2019) to identify genes within the overlapping intervals, and considered these to be candidate genes for anxiety phenotypes in subsequent analyses.
Integrative network analysis
We performed a bioinformatics analysis to further prioritize the positional candidates using aggregate data from functional genomics experiments in the GeneWeaver 2.0 system. GeneWeaver is a suite of microservices that can be accessed via a web browser to perform customized analysis of gene sets selected from its data repository of hundreds of thousands of gene sets derived from genomic databases, publications, and user submissions on 10 different species (Baker et al. 2011). Genes are mapped onto one another using public sources of orthology information, obtained from Alliance of Genome Resources, homologene, and other identifier mapping information. Selections from the data repository are integrated by gene orthology and analyzed with customizable workflows based on its suite of analytic tools that support various set comparison and statistical operations (Baker et al. 2011).
In GeneWeaver, gene lists derived from studies of anxiety-related behaviors were identified using the search terms “anxiety”, “open-field behavior”, “open-field”, “elevated plus-maze”, “elevated zero-maze”, “light–dark box”, light/dark box”, and “social interaction test”. This revealed 55 viable publication sources across mouse, rat, and human studies on anxiety phenotypes. Gene sets involving the usage of drugs of any kind were excluded to reduce possible confounding variables. For human genetic studies, we only included appropriately powered genome-wide associations (no less than 1700 participants) to reduce possible bias from candidate gene studies with small sample sizes. Genome-wide association studies, rather than targeted candidate gene studies allow an agnostic survey of the genome allowing genes that are supported across species and data types to be identified. Several of the gene sets that arose were from the same publication but focused on different brain regions, etc., and these gene sets were combined using the Boolean Algebra Union tool within GeneWeaver to create one combined gene set consisting of the union of all list members from the same publication to eliminate over counting duplicate genes. To reduce the possible false negatives, we manually evaluated each paper and gene list to ensure there was no duplication by brain region from the same study or dataset.
The Boolean Algebra Intersection tool allows for genes appearing in a user defined number of gene sets to be highlighted for reference in subsequent analyses using the “Emphasis Genes” feature. These Emphasis Genes were determined for mice and rats individually using the Boolean Intersection tool. Emphasis genes were defined as those that appeared in our gene set from the High and Low Activity mice and at least three other gene sets. Finally, the collection of rat, mouse, and the High and Low Activity mouse QTL was compared against the Human GWAS data to identify candidate genes and visualized using the GeneSet Graph tool. This tool was set to a minimum of six connections meaning that only those genes that appeared in at least six independent studies across the species listed would appear. We further examined a larger list of genes, present in five independent studies, to determine whether they appeared to cluster in specific functional pathways, using PANTHER (Mi et al. 2009).
Results
Genomic comparison of High and Low strains
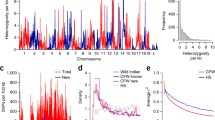
Across the four lines sequenced, we identified 3,678,826 high-quality biallelic positions across the 19 autosomes. For each position, we identified the strain of origin for each allele as either BALB/cJ or C57BL/6J (Table 1, Fig. 2, Supplementary Fig. 1).

ϕ coefficient of association, estimated in 1 cM windows, across the genome. Mean and 95% confidence interval using a block jackknife shown at left
We estimated, P, the genome-wide estimated proportion of variants associated with OFA, to be 0.103 and 0.073 for the two replicates (Table 1). These estimates varied substantially among chromosomes, with the among-chromosome standard deviation of the estimate to be 0.24 and 0.31 for the two replicates (Supplementary Table I). Because of the possible inadvertent crossing of the replicates, their corresponding estimates of P may not be independent.
The association of alleles among strains, ϕ, was strong and positive for comparisons of H1-H2 and L1-L2 (ϕ > 0.8), but weak for high activity vs. low activity strain comparisons (ϕ < 0.11; Fig. 2, Table 2). These estimates were highly and significantly different from each other, as shown by non-overlapping 95% confidence intervals (Table 2). Thus, lines under similar directional selection were more similar to one another than to strains under opposite directional selection. Estimates of ϕ were largely unaffected by the size of the window (1, 5, or 10 cM; Supplementary Fig. 2, Supplementary Table II). These analyses identified numerous genomic windows in which High Activity strains were nearly perfectly concordant, Low Activity strains were nearly perfectly concordant, and in which High and Low strains were highly dissimilar. These regions provided the basis for subsequent gene set analysis.
Identification of genes within narrowed QTL
Regions of interest were located on every chromosome with previously-identified QTL with the exception of chromosomes 12 and X. 247 genes were found on chromosome 1 within the narrowed QTL (71.0–87.0 cM). Chromosome 4 had 379 genes in the narrowed region of 26.0–52.0 cM (Supplementary Fig. 1). Chromosome 7 produced 661 genes between the region of 33.0 and 66.0 cM (Fig. 3). Chromosome 8 had 543 genes in the 22.0–65.0 cM region. Chromosome 14 had 517 genes between the 11.0 and 29.0 cM positions. Interestingly, chromosome 15 only had four coding genes within the region from 18.0–20.0 cM. Chromosome 18 had 77 genes in the region between 16.0 and 35.0 cM (Supplemental Table III. Genes found in QTL regions of High_Low mice). This list of 2428 genes was condensed into a single list and entered into GeneWeaver for comparison against other publications.

Assigned strain-of-origin for each allele carried by the high and low activity line (H1, H2, & L1, L2, respectively) on chromosome 7. The dashed line represents the coat color locus TYR, and we note that low activity mice are albino while high activity mice are brown
Prioritization of positional candidates with functional evidence in GeneWeaver
After collecting publications (Supplemental Table IV. GeneWeaver Publications) across the three species (mouse, rat, human) we identified eight mouse QTL studies containing a total of 17,554 positional candidate genes and seven mouse differential expression studies, which contained 1456 genes. For rats we found 11 QTL studies containing 11,979 positional candidate genes and six differential expression studies containing 3437 genes. Thirteen human GWAS studies were represented with only 280 genes based on genome-wide statistically significant GWAS hits. Upon completion of running the Boolean Intersection tool to find genes that were represented on at least 4 gene sets, 128 genes from the mouse data and 31 genes from the rat data were labeled as Emphasis Genes. "GeneSet Graph" tool was then run in order to enumerate the overlap among multiple publications, species, and genes. From this graph tool, five genes (Acyd8, Cstc, Vmn1r1, Vcam1, Slc6a15) appeared as most highly connected to all publications being compared (Fig. 4). These five genes each appeared in seven publications (Table 3, bold gene names). From those five genes, only two (Cstc and Vmn1r1) had come from our High and Low Activity sequencing study. 59 genes were discovered to be the second most highly connected genes from the publications, present in at least six publications. Thirteen of these were located with High/Low QTL regions as emphasis genes (see column 2 in Table 3).

Most highly connected genes amongst gene sets obtained using GeneWeaver. Furthest right are genes with higher connections amongst gene sets while genes on the left-hand side are less connected but still prevalent among the gene sets obtained
The GeneSet graph (Fig. 4, Supplementary Figs. 3 and 4) presents the bipartite graph of genes and gene sets with the most highly connected genes on the far right of the figure; Acyd8, Cstc, Vmn1r1, Vcam1, Slc6a15. The left side of the figure shows the 59 genes (of which seventeen are in High/Low QTL regions) that appear in the minimum number of publications as set by the user, in this case, six publications. These genes are shown in “Table 3. Highly Connected Genes”; with the most highly connected genes listed first and in bold.
Gene ontology and pathways implicated using PANTHER
There were 344 genes present in at least five studies, which was large enough to conduct PANTHER ontology and pathway analyses. The 2428 genes present in the High and Low QTL regions were used as the reference genes list to evaluate over/under representation and fold enrichment in the 344 genes present in five studies. Results from this analysis are presented in Supplementary Table V (those in red show fold-enrichment less than 3). In particular, the pathway analysis revealed over-representation of genes involved in glutamate signalling.
Discussion
The replicate strains of the High and Low Activity mice provide a unique opportunity to study the genetic architecture of response to behavioral selection, given the current availability of high-throughput, cost-effective sequencing technology.
The first major goal of the project was to assess how strongly selection affects allele distribution at a genome-wide level. From whole-genome sequence data, we estimated the ϕ coefficients within two High and Low strains to be very high, at 0.828 and 0.857, respectively. Conversely, ϕ was drastically lower (< 0.13) when comparing High to Low strains. These extreme differences between strains compared to high similarity within strains illustrate the strong power of selection on complex behavioral traits to affect the distribution of variants derived from two single progenitor strains of mice. Importantly, the proportion of variants associated with OFA was estimated at 0.103 within H1 and L1 strains, and 0.073 in the H2 and L2 strains, confirming the high polygenicity of the trait. Additionally, these similarities and differences were spread across every single chromosome, and replicated twice in each selected direction, indicating the genome-wide architecture of these selected behaviors. These results parallel selection experiments in other rodent models for behavioral traits, such as wheel running in mice (Waters et al. 2013) and alcohol consumption in rats (Lo et al. 2016), demonstrating the utility of bidirectional selection experiments in elucidating the genomic and mechanistic underpinnings of complex behavioral phenotypes.
Despite this large number of loci influencing these behaviors, and complete genome sequence data for the four selected strains, the specific genes underlying OFA cannot be narrowed down through sequence data alone due to the relatively few recombinations within the inbred strains. Individual regions consistent in their strain distribution pattern are relatively large, containing many known genes within them. However, by combining these exhaustive sequence data with results from previous mapping studies in these and other strains, specific, prioritized genes may be identified.
Historically, large QTLs were resolved through the breeding of congenic mice, backcrossing, and selecting for the QTL region until recombinations of the QTL against a pure background could be tested to identify more precisely the location of the causal variant or variants. Even with the advances in genotyping and marker assisted matings, this is an arduous process. Alternatively, aggregating new and existing functional genomic evidence has the potential to more rapidly identify plausible candidates for follow-up study, as was recently done for alcohol-related genes, which were confirmed in a mouse knock-out model (Bubier et al. 2016). We applied this same strategy to identify genes of interest, putatively influencing anxiety-like behaviors, by aggregating the narrowed genomic regions from whole genome sequence data with High and Low Activity QTL regions using cross-species ‘omics’ data available in GeneWeaver.
These analyses narrowed the large number of possible regions and genes of interest to two high priority genes with a connection to seven previous studies on anxiety phenotypes: Ctsc and Vmn1r1. Ctsc codes for a lysosomal protease, cathepsin C, involved in the activation of other proteases within bone marrow-derived cells, which perform functions such as cell-mediated phagocytosis, cytotoxicity, and activation of inflammatory mediators (Bloch-Zupan et al. 2012). Mutations in this gene have been associated with severe forms of periodontitis (Genetics Home Reference 2020), which may be a risk factor for neuropsychiatric disease (Hashioka et al. 2019; Wadhawan et al. 2020). Its role in coordinating activation of serine proteases in the immune system is interesting considering the connection between the immune system and anxiety and stress (Miller and Raison 2016); indeed, bone marrow-derived cells are thought to be an important determinant of stress-induced anxiety-like states and affective disorders. In the brain, cathepsin C is upregulated in microglia in association with neuroinflammation (Fan et al. 2012). Vmn1r1, which is found on chromosome 1, is a vomeronasal receptor and member of the V1R gene family. Little information is available about this particular receptor subtype, and a reported human homolog has not been identified. The VIR gene family is involved in pheromone and semiochemical detection, which contributes to communication between animals of the same species, including social and reproductive behaviors (Jiao et al. 2019). Given the lack of a human homolog, Vmn1r1 is a lower priority for immediate future study, but the fact that it emerged from the GeneWeaver analysis emphasizes the importance of smell or pheromone detection for anxiety-related behaviors in rodents, which has been well documented (Chen et al. 2019).
Several other genes, also highly connected amongst gene sets, were identified as interesting candidates based on previous biological and genetic evidence: Ndn, Cckbr, Kcnj10, and Slc12a2. Ndn (necdin) is located in the Prader-Willi syndrome deletion region on chromosome 15. It has been implicated in suppressing cell growth in postmitotic neurons, and it facilitates the entrance to cell cycle arrest (Chapman and Knowles 2009). Cckbr (cholecystokinin B receptor) encodes a G protein-coupled receptor for gastrin and cholecystokinin, which are regulatory peptides in the brain and gastrointestinal tract. It was previously identified as a top candidate for anxiety disorders with the use of comparative functional genomics (Le-Niculescu et al. 2011), which is supported by our results. In humans, a pharmacological agent targeting CCKBR, cholecystokinin-tetrapeptide (CCK-4), is a commonly used approach to induce panic attacks in otherwise healthy participants for study of neuronal functions in various brain imaging studies (Eser et al. 2009). Kcnj10 (potassium inwardly rectifying channel subfamily J member 10) is located on chromosome 1 and highly expressed in the brain. Loss of function of this gene has connections to epilepsy, ataxia, and sensorineural deafness (Bockenhauer et al. 2009).
Likewise, Slc12a2 encodes a sodium-potassium-chloride cotransporter and is necessary for inner ear function that leads to hyperactivity in a mouse model (Antoine 2017). Gene ontology and pathway analyses comparing genes present in at least five studies to all genes present in the High and Low Activity QTL regions revealed over-representation of genes involved in glutamate signaling. This is consistent with previous research demonstrating an important role of glutamatergic signaling in anxiety and stress-related disorders (Peterlik et al. 2016).
Recently, four well-powered human GWAS studies of anxiety-related measures have been reported, which each identified several loci reaching genome-wide significance thresholds (Meier et al. 2019; Levey et al. 2020; Purves et al. 2019; Ward 2019). Genes from these studies were included in our GeneWeaver analysis, but none emerged as any of the top genes, mentioned above. Human GWAS studies are limited by the fact that many of the top SNPs associated with the trait are located in intergenic/non-coding regions, so it is difficult to assign the variants to a specific “gene”. As more data become available through ongoing gene expression and epigenetic mapping approaches, the resulting data can be further integrated into GeneWeaver for improved integration of functional genomics analyses.
There were several limitations to this study. First, the gene sets defined for the High and Low Activity QTL were defined based on previous mapping studies (Henderson et al. 2004; Turri et al. 2001a, b; Turri et al. 2001a, b). It is possible that non-coding regulatory variants within those windows influence distal genes not included in our gene list. In addition, while GeneWeaver is the most extensive database currently available, such user-curated databases remain limited by current existing data, which could be biased by over-representation of certain rat or mouse strains. Most historic mouse genetic mapping studies involved C57BL/6 mice or their derivatives, crossed to strains that are now appreciated to be quite closely related to this strain. Thus, we are identifying genes that are most represented among these crosses, but may miss other anxiety-related genes that are only found in certain specific crosses. As such databases grow as new studies are included, the power of these analyses will continue to improve, and additional target genes may yet be identified.
In summary, this study reports the first examination of whole-genome sequence similarity across replicate strains of inbred mice differentially selected for high and low OFA. The results highlight the strong effects of selection and the high polygenicity of the trait, as demonstrated by the low ϕ coefficient and relatively high proportion, approximately 10%, of associated variants, respectively. In light of this polygenicity, where individual loci are expected to be of small effect in both large sample human GWAS and in mouse linkage mapping studies, the combination of new WGS data and GeneWeaver analyses of legacy allowed us to prioritize genes for future study. This cross-species analysis successfully and substantially reduced the number of priority genes within QTL regions of High and Low strains identified over twenty years ago, from 2428 genes to two likely candidates, illustrating the value and power of applying novel integrative bioinformatics approaches to legacy datasets. Using RNA sequencing, DNA methyl-sequencing, or other additional cutting-edge genomics approaches, these animals have potential for further discovery of genetic pathways and molecular mechanisms contributing to anxiety-related behaviors.
Data availability
Raw reads from the 8 individuals sequenced have been deposited in the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) with accession PRJNA605645.
References
Antoine MW, Vijayakumar S, McKeehan N, Jones SM, Hébert JM (2017) The severity of vestibular dysfunction in deafness as a determinant of comorbid hyperactivity or anxiety. J Neurosci Off J Soc Neurosci 37(20):5144–5154. https://doi.org/10.1523/JNEUROSCI.3545-16.2017
Anxiety and Depression Association of America, ADAA. Available at https://adaa.org/about-adaa/press-room/facts-statistics. Accessed 25 Sept 2019
Ashbrook DG, Williams RW, Lu L, Hager R (2015) A cross-species genetic analysis identifies candidate genes for mouse anxiety and human bipolar disorder. Front Behav Neurosci 9:171. https://doi.org/10.3389/fnbeh.2015.00171
Baker EJ, Jay JJ, Bubier JA, Langston MA, Chesler EJ (2011) GeneWeaver: a web-based system for integrative functional genomics. Nucleic Acids Res 40(D1):D1067–D1076. https://doi.org/10.1093/nar/gkr968
Bandelow B, Michaelis S, Wedekind D (2017) Treatment of anxiety disorders. Dialogues Clin Neurosci 19(2):93–107
Bloch-Zupan A, Sedano H, Scully C (2012) Dento/oro/craniofacial anomalies and genetics. Elsevier, Amsterdam. https://doi.org/10.1016/C2011-0-06093-3
Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, Tobin J, Lieberer E, Sterner C, Landoure G, Arora R, Sirimanna T, Thompson D, Cross JH, Van’t Hoff W, Al Masri O, Tullus K, Yeung S, Anikster Y, Klootwijk E, Hubank M, Dillon MJ, Heitzmann D, Arcos-Burgos M, Knepper MA, Dobbie A, Gahl W, Warth R, Sheridan E, Kleta R (2009) Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. New Engl J Med 360(19):1960–1970. https://doi.org/10.1056/NEJMoa0810276
Bolivar V, Flaherty L (2003) A region on chromosome 15 controls intersession habituation in mice. J Neurosci 23(28):9435–9438. https://doi.org/10.1523/JNEUROSCI.23-28-09435.2003
Bubier JA, Wilcox TD, Jay JJ, Langston MA, Baker EJ, Chesler EJ (2016) Cross-species integrative functional genomics in GeneWeaver reveals a role for Pafah1b1 in altered response to alcohol. Front Behav Neurosci 10:1. https://doi.org/10.3389/fnbeh.2016.00001
Chapman EJ, Knowles MA (2009) Necdin: A multi functional protein with potential tumor suppressor role? Working Hypothesis 48(11). https://doi.org/10.1002/mc.20567
Chen Y, Zhou H, An Y, Jia X, Sun T, Guan X, Zhang J (2019) Combined effects of olfactory dysfunction and chronic stress on anxiety- and depressive- like behaviors in mice. Neurosci Lett 692:143–149. https://doi.org/10.1016/j.neulet.2018.11.010
Danecek P, Schiffels S, Durbin R (2014) Multiallelic calling model in bcftools (-m). 2
DeFries JC, Gervais MC, Thomas EA (1978) Response to 30 generations of selection for open-field activity in laboratory mice. Behav Genet 8(1):3–13. https://doi.org/10.1007/BF01067700
Devane CL, Chiao E, Franklin M, Kruep EJ (2005) Anxiety disorders in the 21st century: status, challenges, opportunities, and comorbidity with depression. Am J Manag Care 11(12 Suppl):S344–353
Duval ER, Javanbakht A, Liberzon I (2015) Neural circuits in anxiety and stress disorders: a focused review. Ther Clin Risk Manag 11:115–126. https://doi.org/10.2147/TCRM.S48528
Eser D, Leicht G, Lutz J, Wenninger S, Kirsch V, Schüle C, Karch S, Baghai T, Pogarell O, Born C, Rupprecht R, Mulert C (2009) Functional neuroanatomy of CCK-4-induced panic attacks in healthy volunteers. Hum Brain Mapp 30(2):511–522. https://doi.org/10.1002/hbm.20522
Fan K, Wu X, Fan B, Li N, Lin Y, Yao Y, Ma J (2012) Up-regulation of microglial cathepsin C expression and activity in lipopolysaccharide-induced neuroinflammation. J Neuroinflamm 9:96. https://doi.org/10.1186/1742-2094-9-96
Flint J, Corley R, DeFries JC, Fulker DW, Gray JA, Miller S, Collins AC (1995) A simple genetic basis for a complex psychological trait in laboratory mice. Science (New York, N.Y.) 269(5229):1432–1435. https://doi.org/10.1126/science.7660127
Gonzales NM, Seo J, Hernandez Cordero AI, St Pierre CL, Gregory JS, Distler MG, Abney M, Canzar S, Lionikas A, Palmer AA (2018) Genome wide association analysis in a mouse advanced intercross line. Nat Commun 9(1):5162. https://doi.org/10.1038/s41467-018-07642-8
Gottschalk MG, Domschke K (2017) Genetics of generalized anxiety disorder and related traits. Dialogues Clin Neurosci 19(2):159–168
Hashioka S, Inoue K, Miyaoka T, Hayashida M, Wake R, Oh-Nishi A, Inagaki M (2019) The possible causal link of periodontitis to neuropsychiatric disorders: more than psychosocial mechanisms. Int J Mol Sci 20(15):3723. https://doi.org/10.3390/ijms20153723
Henderson ND, Turri MG, DeFries JC, Flint J (2004) QTL analysis of multiple behavioral measures of anxiety in mice. Behav Genet 34(3):267–293. https://doi.org/10.1023/B:BEGE.0000017872.25069.44
Jiao H, Hong W, Nevo E, Li K, Zhao H (2019) Convergent reduction of V1R genes in subterranean rodents. BMC Evol Biol 19(1):176. https://doi.org/10.1186/s12862-019-1502-4
Le-Niculescu H, Balaraman Y, Patel SD, Ayalew M, Gupta J, Kuczenski R, Shekhar A, Schork N, Geyer MA, Niculescu AB (2011) Convergent functional genomics of anxiety disorders: translational identification of genes, biomarkers, pathways and mechanisms. Transl Psychiatry 1(5):e9–e9. https://doi.org/10.1038/tp.2011.9
Levey DF, Gelernter J, Polimanti R, Zhou H, Cheng Z, Aslan M, Quaden R, Concato J, Radhakrishnan K, Bryois J, Sullivan PF, Stein MB (2020) Reproducible genetic risk loci for anxiety: results from ∼200,000 participants in the Million Veteran Program. Am J Psychiatry. https://doi.org/10.1176/appi.ajp.2019.19030256
Li H (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics (Oxford, England) 27(21):2987–2993. https://doi.org/10.1093/bioinformatics/btr509
Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv:1303.3997 [q-Bio]. https://arxiv.org/abs/1303.3997
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 25(14):1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009) The sequence alignment/map format and SAMtools. Bioinformatics (Oxford, England) 25(16):2078–2079. https://doi.org/10.1093/bioinformatics/btp352
Lo C-L, Lossie AC, Liang T, Liu Y, Xuei X, Lumeng L, Zhou FC, Muir WM (2016) High resolution genomic scans reveal genetic architecture controlling alcohol preference in bidirectionally selected rat model. PLoS Genet 12(8):e1006178. https://doi.org/10.1371/journal.pgen.1006178
Meier SM, Deckert J (2019) Genetics of anxiety disorders. Curr Psychiatry Rep 21(3):16. https://doi.org/10.1007/s11920-019-1002-7
Meier SM, Trontti K, Purves KL, Als TD, Grove J, Laine M, Pedersen MG, Bybjerg-Grauholm J, Bækved-Hansen M, Sokolowska E, Mortensen PB, Hougaard DM, Werge T, Nordentoft M, Breen G, Børglum AD, Eley TC, Hovatta I, Mattheisen M, Mors O (2019) Genetic variants associated with anxiety and stress-related disorders: a Genome-Wide Association Study and Mouse-Model Study. JAMA Psychiatry. https://doi.org/10.1001/jamapsychiatry.2019.1119
Mouse Map Converter. (n.d.). Available at https://churchill-lab.jax.org/mousemapconverter/. Accessed 9 Oct 2019
Mouse Genomes Project. Available at https://www.sanger.ac.uk/science/data/mouse-genomes-project. Accessed 1 Mar 2020
Mi H, Dong Q, Muruganujan A, Gaudet P, Lewis S, Thomas PD (2009) PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res 38:D204–D210. https://doi.org/10.1093/nar/gkp1019
Miller AH, Raison CL (2016) The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol 16(1):22–34. https://doi.org/10.1038/nri.2015.5
Mozhui K, Ciobanu DC, Schikorski T, Wang X, Lu L, Williams RW (2008) Dissection of a QTL hotspot on mouse distal chromosome 1 that modulates neurobehavioral phenotypes and gene expression. PLoS Genet 4(11):e1000260. https://doi.org/10.1371/journal.pgen.1000260
NIMH » Any Anxiety Disorder. (n.d.). Available at https://www.nimh.nih.gov/health/statistics/any-anxiety-disorder.shtml. Accessed 25 Sept 2019
Parker CC, Carbonetto P, Sokoloff G, Park YJ, Abney M, Palmer AA (2014) High-resolution genetic mapping of complex traits from a combined analysis of F2 and advanced intercross mice. Genetics 198(1):103–116. https://doi.org/10.1534/genetics.114.167056
Peterlik D, Flor PJ, Uschold-Schmidt N (2016) The emerging role of metabotropic glutamate receptors in the pathophysiology of chronic stress-related disorders. Curr Neuropharmacol 14(5):514–539. https://doi.org/10.2174/1570159X13666150515234920
Philip VM, Duvvuru S, Gomero B, Ansah TA, Blaha CD, Cook MN, Hamre KM, Lariviere WR, Matthews DB, Mittleman G, Goldowitz D, Chesler EJ (2010) High-throughput behavioral phenotyping in the expanded panel of BXD recombinant inbred strains. Genes, Brain, Behav 9(2):129–159. https://doi.org/10.1111/j.1601-183X.2009.00540.x
Purves KL, Coleman JRI, Meier SM, Rayner C, Davis KAS, Cheesman R, Bækvad-Hansen M, Børglum AD, Wan Cho S, Jürgen Deckert J, Gaspar HA, Bybjerg-Grauholm J, Hettema JM, Hotopf M, Hougaard D, Hübel C, Kan C, McIntosh AM, Mors O et al (2019) A major role for common genetic variation in anxiety disorders. Mol Psychiatry. https://doi.org/10.1038/s41380-019-0559-1
Reference GH (n.d.). CTSC gene. Genetics Home Reference. Available at https://ghr.nlm.nih.gov/gene/CTSC. Accessed 5 Feb 2020
Reich D, Thangaraj K, Patterson N, Price AL, Singh L (2009) Reconstructing Indian population history. Nature 461(7263):489–494. https://doi.org/10.1038/nature08365
Rieseberg LH, Raymond O, Rosenthal DM, Lai Z, Livingstone K, Nakazato T, Durphy JL, Schwarzbach AE, Donovan LA, Lexer C (2003) Major ecological transitions in wild sunflowers facilitated by hybridization. Science 301(5637):1211–1216. https://doi.org/10.1126/science.1086949
Seibenhener ML, Wooten MC (2015) Use of the open field maze to measure locomotor and anxiety-like behavior in mice. J Vis Exp JoVE. https://doi.org/10.3791/52434
Shimada-Sugimoto M, Otowa T, Hettema JM (2015) Genetics of anxiety disorders: genetic epidemiological and molecular studies in humans. Psychiatry Clin Neurosci 69(7):388–401. https://doi.org/10.1111/pcn.12291
Sokal RR, Rohlf FJ (1995) Biometry: The Principles and Practice of Statistics in Biological Research, 3rd edn. W.H. Freeman and Co., New York
Turri MG, Datta SR, DeFries J, Henderson ND, Flint J (2001a) QTL analysis identifies multiple behavioral dimensions in ethological tests of anxiety in laboratory mice. Curr Biol 11(10):725–734. https://doi.org/10.1016/s0960-9822(01)00206-8
Turri MG, Henderson ND, DeFries JC, Flint J (2001b) Quantitative trait locus mapping in laboratory mice derived from a replicated selection experiment for open-field activity. Genetics 158(3):1217–1226
Wadhawan A, Reynolds MA, Makkar H, Scott AJ, Potocki E, Hoisington AJ, Brenner LA, Dagdag A, Lowry CA, Dwivedi Y, Postolache TT (2020) Periodontal pathogens and neuropsychiatric health. Curr Top Med Chem. https://doi.org/10.2174/1568026620666200110161105
Ward J, Tunbridge EM, Sandor C, Lyall LM, Ferguson A, Strawbridge RJ, Lyall DM, Cullen B, Graham N, Johnston KJA, Webber C, Escott-Price V, O’Donovan M, Pell JP, Bailey MES, Harrison PJ, Smith DJ (2019) The genomic basis of mood instability: identification of 46 loci in 363,705 UK Biobank participants, genetic correlation with psychiatric disorders, and association with gene expression and function. Mol Psychiatry. https://doi.org/10.1038/s41380-019-0439-8
Waters RP, Pringle RB, Forster GL, Renner KJ, Malisch JL, Garland T Jr, Swallow JG (2013) Selection for increased voluntary wheel-running affects behavior and brain monoamines in mice. Brain Res 1508:9–22. https://doi.org/10.1016/j.brainres.2013.01.033
Zhou Z, Blandino P, Yuan Q, Shen P-H, Hodgkinson CA, Virkkunen M, Watson SJ, Akil H, Goldman D (2019) Exploratory locomotion, a predictor of addiction vulnerability, is oligogenic in rats selected for this phenotype. Proc Natl Acad Sci 116(26):13107–13115. https://doi.org/10.1073/pnas.1820410116
Acknowledgements
We would like to thank Dr. Erich J Baker for his assistance with GeneWeaver updates and functions.
Funding
This study was funded by NIDA and NIAAA (Grant No. R01 AA019776) and NIMH (Grant No. R01 MH100141-06). R01 AA018776 (NIH NIDA and NIAAA) to EJC. R01 MH100141-06 (NIMH to Matthew C. Keller) supports LME. R21 MH116263A (NIMH) supports CAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Aimee L. Thomas, Luke M. Evans, Michaela D. Nelsen, Elissa J. Chesler, Matthew S. Powers, Winona C. Booher, Christopher A. Lowry, John C. DeFries, Marissa A. Ehringer have no conflict of interest to declare.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Human and animal rights and informed consent
The local Institutional Animal Care and Use Committee (IACUC) approved all protocols using the guidelines set forth by the Office of Laboratory Animal Welfare. All animal protocols were approved by the IACUC and follow all international regulations for human treatment. No human subjects were involved in the study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Thomas, A.L., Evans, L.M., Nelsen, M.D. et al. Whole-Genome Sequencing of Inbred Mouse Strains Selected for High and Low Open-Field Activity. Behav Genet 51, 68–81 (2021). https://doi.org/10.1007/s10519-020-10014-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10519-020-10014-y