
Abstract
Cyprinid herpesvirus 2 (CyHV-2) is the etiological agent of herpesviral hematopoietic necrosis disease (HVHND), which causes severe mortality in ornamental goldfish (Carassius auratus), crucian carp (Carassius auratus), and gibel/prussian carp (Carassius gibelio). Quick and hassle-free point-of-care detection of CyHV-2 is vital for the maintenance of ornamental fish health. In this manuscript, we describe the development of a rapid and sensitive RPA (recombinase polymerase amplification) assay, coupled with lateral flow dipsticks (LFD), that can achieve sensitive diagnosis of CyHV-2 in goldfish within 20 min at 36 °C with the satisfactory detection limit of 102 gene copies per reaction. This is the first report wherein major capsid protein (MCP) of CyHV-2 was targeted for RPA-LFD assay development. The assay did not show any cross-reactivity with other viral pathogens like cyprinid herpesvirus 3 (CyHV-3), spring viremia of carp virus (SVCV), infectious spleen and kidney necrosis virus (ISKNV), and viral nervous necrosis virus (VNNV). Furthermore, screening of CyHV-2 infection in CyHV-2-infected goldfish did not yield any false positive/negative results. In short, the RPA-LFD assay developed in this study presents a simple, rapid, and sensitive method for point-of-care diagnosis of CyHV-2, especially under resource-limited conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cyprinid herpesvirus 2 (CyHV-2), the etiological agent of herpesviral hematopoietic necrosis disease (HVHND), is an infectious pathogen of gold fish (Carassius auratus), crucian carp (Carassius auratus), and various hybrids of carp and goldfish that causes devastating economic losses to the aquaculture industry (Fichi et al. 2016; Li et al. 2019). This linear double-stranded DNA virus in the genus Cyprinivirus of family Alloherpesviridae causes very high mortality (50–100%) among the infected fish (Kong et al. 2017; Thangaraj et al. 2020). Owing to the lack of appropriate drugs/vaccines for CyHV-2, timely and accurate diagnosis of an infection is quintessential for the maintenance of fish health in ornamental aquaculture.
A plethora of nucleic acid–based, cell culture, and immunological diagnostic methods have been developed for the diagnosis of CyHV-2 infections (Kong et al. 2017). Nevertheless, PCR is the most recommended method on account of its diagnostic specificity and sensitivity. The requirement of specialized equipment like thermocycler, gel imaging system, long reaction time, and trained personnel restricts its on-site application, particularly in resource-limited conditions. The isothermal recombinase polymerase amplification (RPA) developed by TwistDx in 2006 is a rapid, simple, and cost-effective alternative for the on-site identification of pathogens in a sample (Chen et al. 2020). The RPA permits amplification of target DNA at low reaction temperature (37–42 °C) within as short as 20 min (Chen et al. 2020). Furthermore, the strand displacing polymerase used in RPA exhibits more tolerance to inhibitors in samples compared to Taq polymerase used in conventional PCR (Moore et al. 2017). Among the various visualization methods for RPA amplicons, lateral flow dipstick (LFD) assay is better suited for point-of-care (POC) testing as it facilitates analysis of results with the naked eye (Li et al. 2018). The present study was focused to develop RPA primers and probes targeting the highly conserved major capsid protein (MCP) gene (ORF 92) of CyHV-2 for rapid diagnosis of HVHND. Major capsid protein is one of the most widely used biomarkers of viral infection in fish and has often been used as a capture antigen in immunological studies and for vaccine development (Shen et al. 2018). To date, there are very few studies on the major capsid protein of CyHV-2, and therefore, this study is the first of its kind focusing the development of an easy and reliable RPA-LFD assay prototype for the POC detection of CyHV-2.
Materials and methods
Propagation and purification of CyHV-2 from infected gold fish
Diseased gold fish exhibiting clinical signs of HVHND were collected from an ornamental fish farm. Gills, kidney, and spleen of the infected fish were excised aseptically and homogenized with sterile Dulbecco’s phosphate buffered saline (DPBS). The tissue homogenate was freeze-thawed thrice and was centrifuged at 3000 × g for 20 min at 4 °C. The supernatant was collected and filtered through 0.22-µm filter (Millipore) and was subjected to CyHV-2 isolation using sucrose gradient centrifugation. The purified virus was stored at − 80 °C for further use. The filtrate was also inoculated on to the confluent monolayer of fantail goldfish fin (FtGF) cell line for the observation of cytopathic effects.
Genomic DNA extraction and PCR amplification
The stored purified viral samples were subjected to DNA extraction using salting out method following Miller et al. (1988). Individual as well as pooled infected tissues such as gills, liver, and spleen were taken for DNA extraction. CyHV-2 infection was confirmed by PCR using the primers targeting DNA polymerase (Engelsma et al. 2013).
Design of RPA primers and probes
Nucleotide sequence data for MCP gene of CyHV-2 was retrieved from GenBank (Accession KU527546), and the conserved regions were identified after multiple sequence alignment using BioEdit. Different combinations of primers and probes targeting the conserved region of MCP were designed according to the guidelines in the TwistAmp Assay Design manual (TwistAmp Basic kit, TwistDX, Cambridge, UK). The TwistAmp kit formulations allow rapid and sensitive amplification of short target sequences (80–500 bp) within the DNA. As per the kit guidelines, the recommended amplicon length should not exceed 500 bp for an ultra-rapid assay. As Jaroenram and Owens (2014) suggested, around 35 bp could only be possible for primer construction, and as the primer length decreases, the amplification efficiency increases as well. However, the length of TwistAmp primers should be longer than the normal PCR primers according to the instructions of TwistAmp manual. In the case of LFD RPA primers, biotin labels were added at 5′-end to detect the cellular nucleotide. The probes designed for LFD assay were based on the nucleotides of RPA primers and which mainly comprise fluorophores (FAM) at 5′-end and a polymerase extension blocking group, C3-spacer at 3′-end. A tetrahydrofuran residue (Z) was added as an internal basic nucleotide analogue. This is the site where enzyme nfo cleaves and generates a new 3′-OH group, thereby promoting polymerase extension and further modifying probe in to primer. The RPA primers including LFD primers and probes were selected by testing the combination that yielded the highest sensitivity. Table 1 lists the RPA and LFD primers and probes (Sigma Aldrich, USA) used in the current study.
Recombinase polymerase amplification (RPA) and Lateral flow dipstick (LFD) assay
Normal PCR was carried out using RPA 1F/1R and RPA 4F/4R primers specific for MCP gene of CyHV-2 with product sizes of 335 bp and 230 bp, respectively. The reaction conditions consisted of an initial denaturation at 95 °C/2 min, followed by 28 cycles of denaturation at 95 °C/1 min, annealing at 55 °C/1 min, extension at 72 °C/1 min with a final extension of 72 °C/10 min. The RPA reaction was performed after confirmation of the positive amplification of MCP gene. The RPA reaction mixture for 335 bp amplicon comprised of forward primer (LFD-RPA 1F, 10 µM) (4 µl), LFD probe 335 (10 µM), 1X rehydration buffer, DNase-free water, and template DNA in a total volume of 47.5 µL. The amplification of 230 bp product was performed by the addition of reverse primer (LFD-RPA 4F) (10 µM) and the corresponding LFD probe 230 (10 µM). Furthermore, the total volume of reaction mixture was transferred into the lyophilized TwistAmp reaction pellets containing the enzyme nfo (Endonuclease 1 V) (TwistAmp nfo new formulation, TwistDX, Cambridge, UK) and 2.5 µl of magnesium acetate (280 mM) was placed on the cap of the reaction tube to commence the reaction. Triplicates were kept for all reactions along with positive (provided in the kit) and negative (without template) controls. After proper mixing by centrifugation, the reaction tubes were immediately incubated at temperatures ranging from 30 to 40 °C for 40 min. Optimal reaction time was also determined by performing RPA reaction at intervals ranging from 5 to 40 min at the optimal temperature. For the detection of products using LFD assay, the RPA product (10µL) was mixed with HybriDetect Assay Buffer (90µL) and the LFD strip (Milenia HybiDetect1, Milenia Biotec, Giessen, Germany) was incubated in the solution at room temperature and the observations were recorded immediately after 5 min.
Diagnostic sensitivity of RPA-LFD assay
To test the sensitivity of RPA assay, the MCP amplicons were cloned into pGEM®-T Easy Vector (Promega, Madison, WI, USA) and different copies of the recombinant plasmid were used as templates for the RPA reaction. Based on the size of plasmid (3015 bp) and the insert (335 bp), the copy number was calculated following Whelan et al. (2003) and was subjected to tenfold dilutions from 1 X 109 to 1 X 101 copies µL−1 to determine the exact sensitivity of the RPA-LFD assay.
Diagnostic specificity and applicability of RPA-LFD assay
Specificity of the newly designed RPA primers and probes was determined by performing RPA-LFD assay with the positive DNA of other fish viruses such as, cyprinid herpesvirus 3 (CyHV-3), spring viremia of carp virus (SVCV), infectious spleen and kidney necrosis virus (ISKNV), and viral nervous necrosis virus (VNNV) under the specified conditions.
The applicability of this assay was validated by performing the RPA-LFD assay for gold fish (n = 14) collected from Ernakulam fish farm, with or without CyHV-2 and comparison of the results with routine PCR assays.
Results
Isolation and identification of CyHV-2
The virus was successfully purified and genomic DNA (> 10 Kb) was extracted from the isolated virus. Besides, DNA was also extracted from the pooled organs as such of the infected fish. This obviates the need for virus isolation using sophisticated protocols, thereby simplifying the process and can be taken as a major advantage in field diagnosis. PCR amplification of the DNA polymerase gene generated amplicons of 362 bp, confirming the CyHV-2 infection in the tested gold fish samples. CyHV-2 infection was also confirmed by typical cytopathic effects in FtGF cell lines, such as cytoplasmic vacuolization, followed by the detachment of cell monolayer.
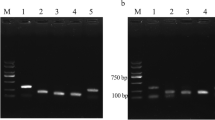
In the screening of CyHV-2 RPA primers, the amplification efficiency was found to be highest for the primer sets RPA 1F/1R and RPA 4F/4R with product sizes of 335 bp and 230 bp, respectively (Fig. 1). However, the primer set LFD- RPA 1F yielded better results on RPA-LFD assay when compared to the LFD-RPA 4R primer set. Hence, the former set was used for subsequent studies. The MCP gene sequence (335 bp) obtained in this study was submitted to GenBank (MZ399719), and it showed 100% similarity to CyHV-2 major capsid protein gene (KU527546, MT643194) on BLAST analysis.

Gel picture showing the positive amplification of RPA-MCP gene of CyHV-2 using two different RPA primer sets; 1F/1R and 4F/4R with 100bp Ladder
Diagnostic sensitivity of RPA and LFD assay
Upon optimization of RPA reaction, significant amplification was observed in the temperature range of 35–38 °C and hence, 36 °C was selected as the standard temperature for the assay. The assay successfully detected the positive samples after 15 min of RPA reaction, and hence, 20 min was determined as the optimal reaction time for RPA assay, by considering the incubation of 5 min for the LFD assay. After RPA reaction, the positive and negative amplifications of different copies of recombinant plasmids (109 to 101) were confirmed by observing the amplified products in 1% agarose gel (Fig. 2). Thereafter, on LFD assay two bands were formed in LFD strips for CyHV-2 positive samples, one in the control line and another in the test line, unlike the negative control and CyHV-2 negative samples wherein, only a single band appeared in the control line. A flow-control signal is only produced in the negative control in contrast to positive signals in test samples. The positive and negative signals in the LFD strips upon testing different copies (109 to 101) of the recombinant plasmids are shown in Fig. 3. On performing the RPA-LFD assay with different dilution series of pGEM®-T Easy Vector, harboring the MCP gene, the detection limit was found to be 102 gene copies per reaction.

Gel picture showing the recombinase polymerase amplification of different copies (109 to 101) of pGEM-TMCP with controls (P: positive control, N: negative control)

LFD signals shown by different copies (109 to 101) of pGEM-T-MCP with controls (P: positive control, N: negative control)
Diagnostic specificity and applicability of RPA and LFD assay
The analysis of diagnostic specificity by investigation of the cross-reactivity of RPA assay against other viral pathogens revealed strong specificity of the RPA primers for CyHV-2 and hardly exhibited positive results for other tested viruses. During the RPA-LFD analysis of 14 gold fish samples for CyHV-2, 5 tested positive in concordance with the PCR test results of the same samples. This suggests that the primers and probes developed in our study are accurate, efficient, and reliable for on-site detection of CyHV-2.
Discussion
Development of a rapid detection tool for subduing the disease outbreaks is the need of the hour in aquaculture. RPA-LFD is a simple, rapid, and cost-effective isothermal amplification method for the on-site detection of major viral pathogens in aquaculture such as hemocyte iridescent virus, carp edema virus, cyprinid herpes virus, and nodavirus (Jamalpure et al. 2021; Chen et al. 2020; Soliman et al. 2018; Prescott et al. 2016). Amongst these, cyprinid herpes virus 2 is a significant viral pathogen affecting ornamental fish aquaculture (Li et al. 2019). Therefore, the current study was designed to develop an RPA-LFD assay for point-of-care diagnosis of CyHV-2 infections. As major capsid protein, pORF92 is the most abundant protein in CyHV-2 virions that plays a vital role during CyHV-2 infection process (Gao et al. 2020); ORF92 was employed as a marker gene for the development of RPA-LFA. Previously, an MCP-based RPA assay was developed for CyHV-3 in latently infected koi carp (Prescott et al. 2016). Despite the highly conserved nature of MCP, no study has reported the development of MCP-based RPA assay for CyHV-2 infections.
The RPA-LFD assay demonstrated excellent sensitivity by facilitating the detection of low copy numbers of the MCP-harboring plasmids, corresponding to a low viral load in fish samples. Our RPA-LFD assay showed a comparable limit of detection (below 100 copies), exhibited by most of the RPA assays developed for fish viruses (Chen et al. 2020; Mai et al. 2021). Previously, Wang et al. (2018) developed an RPA-LFA assay targeting ORF72, encoding the capsid triplex subunit 2 for rapid detection for CyHV-2. However, the entire assay took 30 min for completion, and furthermore, the specificity of the assay was expressed in terms of DNA concentration, which is not an accurate representation of sensitivity of the reaction, while the assay developed in our study permitted successful detection of exact copy number of CyHV-2 in as little as 20 min. The absence of cross-reactivity of our RPA primers with other viruses confirmed superior performance of this method over other conventional diagnostic techniques that generate several false positive/negative results. This highlighted the specificity of designed primers and probes, which is in close agreement with the specificity shown by the RPA primers developed in recent studies for the detection of CyHV-2, CyHV-3 and other fish pathogens (Chen et al. 2020; Mai et al. 2021; Wang et al. 2018). The RPA-based assay developed in this study permitted successful identification of 5 positive cases of CyHV-2, without giving any false positives/negatives for the remaining samples, verified using PCR. Due to the high sensitivity, specificity, rapidity, reproducibility, and applicability, the developed assay is a quantum leap in comparison to conventional PCR and other gold standard diagnostic techniques like real-time PCR. This RPA-LFD assay is therefore, a promising alternative for POC diagnosis of CyHV-2, in the event of limited access to expensive equipment or trained personnel to perform complicated assays.
Conclusion
A simple and reliable RPA-LFD assay targeting the MCP gene of CyHV-2 was developed for the first time which could accomplish successful detection of 100 copies of the viral gene within 20 min at 36 °C. This presents a rapid and sensitive point-of-care diagnosis of CyHV-2 under resource- limited conditions.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Code availability
Not applicable.
References
Chen Z, Huang J, Zhang F, Zhou Y, Huang H (2020) Detection of shrimp hemocyte iridescent virus by recombinase polymerase amplification assay. Mol Cell Probes 49:101475
Engelsma MY, Way K, Dodge MJ, Voorbergen-Laarman M et al (2013) Detection of novel strains of cyprinid herpesvirus closely related to koi herpesvirus. Dis Aquat Organ 107:113–120
Fichi G, Susini F, Cocumelli C et al (2016) New detection of Cyprinid herpesvirus 2 in mass mortality event of Carassius carassius (L.), in Italy. J Fish Dis 39:1523–1527
Gao W, Wen H, Wang H, Lu J, Lu L, Jiang Y (2020) Identification of structure proteins of cyprinid herpesvirus 2. Aquaculture 523:735184
Jamalpure S, Vimal S, Ahmed AN, Hameed ASS, Paknikar KM, Rajwade JM (2021) On-site detection of nodavirus in post larval (PL) stage of the giant prawn, Macrobrachium rosenbergii: a test to nip the problem in the bud. Aquaculture 534:736292
Jaroenram W, Owens L (2014) Recombinase polymerase amplification combined with a lateral flow dipstick for discriminating between infectious Penaeus stylirostris densovirus and virus-related sequences in shrimp genome. J Virol Methods 208:144–151. https://doi.org/10.1016/j.jviromet.2014.08.006
Kong SY, Jiang Y, Wang Q, Lu JF, Xu D, Lu LQ (2017) Detection methods of Cyprinid herpesvirus 2 infection in silver crucian carp (Carassius auratus gibelio) via a pORF72 monoclonal antibody. J Fish Dis 40:1791–1798
Li K, Rui Y, Mingtian Z, Tingting Z, Yuchao G, Yang Z et al (2019) Recombinant baculovirus BacCarassius-D4ORFs has potential as a live vector vaccine against CyHV-2. Fish Shellfish Immun 92:101–110
Li J, Macdonald J, Stetten F (2018) Review: a comprehensive summary of a decade development of the recombinase polymerase amplification. Analyst 144:31–67. https://doi.org/10.1039/c8an01621f
Mai HN, Caro LFA, Cruz-Flores R, Dhar AK (2021) Development of a recombinase polymerase amplification (RPA) assay for acute hepatopancreatic necrosis disease (AHPND) detection in Pacific white shrimp (Penaeus vannamei). Mol Cell Probes 57:101710
Miller SA, Dyke DD, Polesk HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acid Res 16:12–15
Moore M, Jaykus LY (2017) Development of a recombinase polymerase amplification assay for detection of epidemic human noroviruses. Sci Rep 7:40244
Prescott MA, Reed AN, Jin L (2016) Rapid detection of Cyprinid herpesvirus 3 in latently infected koi by recombinase polymerase amplification. J Aquat Anim Health 28:173–180. https://doi.org/10.1080/08997659.2016.1185048
Shen Z, Yousheng J, Jianfei L, Motohiko S, Dan X, Liqun L (2018) Application of a monoclonal antibody specific for the ORF92 capsid protein of Cyprinid herpesvirus 2. J Virol Methods 261:22–27
Soliman H, El-Matbouli M (2018) Rapid detection and differentiation of carp oedema virus and cyprinid herpes virus-3 in koi and common carp. J Fish Dis 41:761–772. https://doi.org/10.1186/s13071-018-2825-5
Thangaraj RS, Nithianantham SR, Dharmaratnam A, Kumar R, Pradhan PK, Gopakumar ST, Sood N (2020) Cyprinid herpesvirus-2 (CyHV-2): a comprehensive review. Rev Aquacult 13:796–821
Wang H, Sun M, Xu D, Podok P, Xie J, Jiang et al (2018) Rapid visual detection of cyprinid herpesvirus 2 by recombinase polymerase amplification combined with a lateral flow dipstick. J Fish Dis 41:1201–1206
Whelan JA, Russel NB, Whelan MA (2003) A method for the absolute quantification of cDNA using real time PCR. J Immunol Meth 278:261–269
Acknowledgements
The authors state their gratitude to Director, ICAR National Bureau of Fish Genetic Resources, Lucknow, and Director General and Deputy Director General (Fy. Sc.), Indian Council of Agricultural Research (ICAR), New Delhi. The authors thank ICAR National Fellow Scheme of ICAR (ICAR letter no F. No Agri.edn.27/2/2015 HRD dated 13.02.2017), Department of Agricultural Research and Education, Ministry of Agriculture and Farmer’s Welfare, Government of India, for funding.
Author information
Authors and Affiliations
Contributions
The following contributions had been done by the authors:
Conceived or designed the study: Thangaraj Raja Swaminathan.
Performed research: Prasannan Geetha Preena, Thaliyil Veetil Arun Kumar, Arathi Dharmaratnam, and Tina Kollannoor Johny.
Data analysis and interpretation: Thaliyil Veetil Arun Kumar, Prasannan Geetha Preena, Arathi Dharmaratnam and Thangaraj Raja Swaminathan.
Manuscript draft: Prasannan Geetha Preena, Thaliyil Veetil Arun Kumar, Tina Kollannoor Johny, Arathi Dharmaratnam, and Thangaraj Raja Swaminathan.
All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
All the experimental challenge procedures in this study (Proposal number: NBFGR/IAEC/2019/0014) were evaluated and approved by the Institute Animal ethics Committee (IAEC) of ICAR National Bureau of Fish Genetic Resources (NBFGR) (CPCESA Registration No: 909/GO/Re/S/05/CPCSEA dated 09.09.2005 and CPCSEA Ref file No. 25/111/2014-CPCESA dated 05th December 2018) vide approval Number G/IAEC/2019/1 dated 04 October 2019.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Handling Editor: Gavin Burnell
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Prasannan Geetha Preena and Thaliyil Veetil Arun Kumar contributed equally to this work
Rights and permissions
About this article

Cite this article
Preena, P.G., Kumar, T.V.A., Johny, T.K. et al. Quick hassle-free detection of cyprinid herpesvirus 2 (CyHV-2) in goldfish using recombinase polymerase amplification-lateral flow dipstick (RPA-LFD) assay. Aquacult Int 30, 1211–1220 (2022). https://doi.org/10.1007/s10499-021-00806-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10499-021-00806-2