
Abstract
In recent years, colorectal cancer incidence and mortality have increased significantly due to poor lifestyle choices. Despite the development of various treatments, their effectiveness against advanced/metastatic colorectal cancer remains unsatisfactory due to drug resistance. However, ferroptosis, a novel iron-dependent cell death process induced by lipid peroxidation and elevated reactive oxygen species (ROS) levels along with reduced activity of the glutathione peroxidase 4 (GPX4) antioxidant enzyme system, shows promise as a therapeutic target for colorectal cancer. This review aims to delve into the regulatory mechanisms of ferroptosis in colorectal cancer, providing valuable insights into potential therapeutic approaches. By targeting ferroptosis, new avenues can be explored for innovative therapies to combat colorectal cancer more effectively. In addition, understanding the molecular pathways involved in ferroptosis may help identify biomarkers for prognosis and treatment response, paving the way for personalized medicine approaches. Furthermore, exploring the interplay between ferroptosis and other cellular processes can uncover combination therapies that enhance treatment efficacy. Investigating the tumor microenvironment’s role in regulating ferroptosis may offer strategies to sensitize cancer cells to cell death induction, leading to improved outcomes. Overall, ferroptosis presents a promising avenue for advancing the treatment of colorectal cancer and improving patient outcomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) is one of the most common gastrointestinal malignancies. According to the latest data from the World Health Organization’s International Agency for Research on Cancer (IARC), CRC is ranked the third most common morbidity (10.0%) and second greatest mortality (9.4%) among all cancers worldwide [1]. Up to date, various therapeutics have achieved great progress for CRC, such as surgery, chemotherapy, radiotherapy, targeted therapy and immunotherapy [2]. Radical surgery and adjuvant chemoradiotherapy has exhibited a promising efficacy and prognosis in the treatment of early-stage CRC [2]. But for patients with advanced or invasive/metastatic CRC, no standard and efficient therapeutic regimen is available [3]. Although targeted therapy for CRC shows a promising specific and potent efficacy, drug resistance or off-target effect still exist [4]. The immunotherapy mainly benefits CRC patients with high microsatellite instability or defective DNA mismatch repair system (MSI-H/dMMR), which account for about 5-15% of the overall CRC patients [5, 6]. Thus, there is a clear urgent to develop more efficient and broad-spectrum therapeutic strategies for CRC.
Studies have shown that individuals who intake daily substantial heme iron and patients with iron overload disease have an increased risk of developing CRC [7]. The survival of CRC cells, incredibly depends on intracellular iron level, and cells can obtain iron from blood supply and intestinal lumen, making them the only malignancy with abundant iron uptake sources [8]. Compared with normal cells, CRC cells had a high level of iron ions so that they may be more vulnerable ferroptosis [8]. Therefore, the induction of ferroptosis is a promising treatment for CRC.
Ferroptosis is a novel programmed cell death (PCD) that is dependent upon intracellular iron level and different from other cell deaths including autophagy, apoptosis, necrosis and pyroptosis [9, 10]. Ferroptotic cells present with increased mitochondrial membrane density, shrunken mitochondria, reduced mitochondrial cristae, swollen organelle and cytoplasmic, formed vesicle, ruptured plasma membrane rupture, and condensed chromatin [9]. Extensive accumulation of iron ions in cells triggers the Fenton reaction (Fe2+ provides an electron to H2O2), which induces the generation of free radicals, raises the level of ROS, damages lipids, proteins and DNA, and may lead to ferroptosis [11].
In this review, we summarize the key mechanisms of ferroptosis and elaborate on the regulation, function, and application of ferroptosis in CRC, which may provide a platform for a better understanding of the complex role of ferroptosis in CRC.
The core mechanism and characteristics of ferroptosis
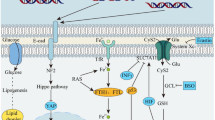
The accumulation of lipid peroxides, decreased glutathione (GSH) activity, and iron overload are proposed as core mechanisms and features of ferroptosis (Fig. 1) [12].

Mechanism of ferroptosis. Ferroptosis is induced by iron-dependent lipid peroxidation. PUFAs synthesized by acetyl-CoA carboxylase alpha (ACACA) and fatty acid synthase (FASN) or released by lipophagy are peroxidized via a series of reactions mainly catalyzed by ACSL4, LPCAT3 and ALOXs. Glycolysis and glutaminolysis cooperatively fuel the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) in mitochondria to produce reactive oxygen species (ROS). Tf binds to TfR1 to facilitate Fe3+ uptake, followed by metalloreductase STEAP3-mediated reduction of Fe3+. Excessively high level of labile iron pool (LIP) triggers Fenton reaction and then promotes ALOXs activity and generation of ROS. Extracellular cystine is imported into cells by the cystine/glutamate antiporter system xc− and oxidized to cysteine, which is essential for GSH synthesis catalyzed by glutamate–cysteine ligase (GCL) and glutathione synthetase (GSS). The transsulfuration pathway act as another source of cysteine besides system xc−. The mevalonate pathway is involved in the synthesis of selenoproteins (including GPX4). GPX4 plays a central role in resisting ferroptosis. In addition, GSH-independent antioxidant systems, including FSP1/CoQ10H2, GCH1/BH4/DHFR and DHODH/CoQ10H2, are newly discovered ferroptosis inhibitors in recent years. FSP1 inhibits ferroptosis by promoting the production of CoQ10H2. GCH1/BH4 promotes the formation of CoQ10 to resist ferroptosis. GLUTs, glucose transporters; DMT1, divalent metal transporter 1; α-KG, α-ketoglutarate; GSSG, glutathione oxidized; IPP, isopentenyl pyrophosphate; FPP, farnesyl pyrophosphate
Lipid peroxidation
Under oxidative stress, large quantities of polyunsaturated fatty acids (PUFAs) in cells are oxidized to lipid bilayer peroxides (PLOOH). These peroxides can damage cell membranes, leading to plasma membrane rupture and ferroptosis [13]. Arachidonic acid (AA) and adrenal acid (AdA) are the most easily oxidized lipids in cells, which is mediated by two necessary enzymes, acyl-CoA synthetase long-chain family member 4 (ACSL4) and human lysophosphatidic transferase 3 (LPCAT3) [14]. PUFAs are esterified to acyl-CoA by ACSL4, which primarily catalyzes the binding of free AA (or AdA) to CoA to produce AA-CoA or AdA-CoA [14]. After that, the derivatives are catalyzed by LPCAT3 and embedded into membrane phospholipids (phospholipidation), which yields AA-PE/AdA-PE [14, 15]. In contrast to LPCAT3, MBOAT1/2 enzymes reduce the content of PE-PUFAs by incorporating MUFAs into phospholipids, inhibiting ferroptosis [16]. These PUFAs in the form of phospholipids (PUFA-PL) are directly oxidized by the lipoxygenases (ALOXs) to the phospholipid hydroperoxide PLOOH [17, 18].
Moreover, NADPH–cytochrome P450 reductase (POR) and NADH-cytochrome b5 reductase 1 (CYB5R1) also mediate the lipid peroxidation of membrane phospholipids by facilitating the passage of electrons from NAD(P)H to oxygen to promote the production of H2O2, which will activate ferroptosis [19].
Antioxidant defense
(1) GPX4-dependent pathway
Cystine/glutamate antiporter (system xc−)-reduced GSH-glutathione peroxidase 4 (GPX4) (System xc−/GSH/GPX4 axis) is a classical pathway that plays an important role in regulating ferroptosis. System xc− is a heterodimer formed through the linkage of SLC7A11 (xCT) and SLC3A2 (4F2hc) by disulfide bonds, which results in the exchange of extracellular cystine with intracellular glutamate and promotes GSH synthesis [20]. Cysteine is a rate-limiting factor in GSH synthesis because of its low content (to avoid cytotoxicity) and high content of other amino acids [21]. GPX4 is a critical antioxidant protein in cells by oxidizing GSH to reduce lipid peroxides and protect the cell membrane from being damaged by hydroperoxides, allowing it to be a core inhibitor of ferroptosis [22, 23]. GPX4 is synthesized by the mevalonate pathway [24] and degraded by heat shock protein 90 (HSP90) [25, 26]. Otherwise, Wang et al. [27] revealed that serine/arginine-rich splicing factor 9 (SFRS9) could specifically bind to GPX4 mRNA to enhance its expression, which suppressed lipid peroxidation in CRC cells.
The transsulfuration pathway is another important source of cysteine generation to compensate for the lack of cysteine when the uptake of cysteine by system xc− is inhibited [28]. Methionine is converted to honocysteine and then transformed into cysteine by cystathionine-β-synthase (CBS) which act as a ferroptosis suppressor [23, 28]. In CRC cells, the byproduct H2S generated by the enzymatic reaction mediated by cystathionine-γ-lyase (CTH) promotes the sulfhydrylation of OTU domain-containing ubiquitin aldehyde-binding protein 1 (OTUB1) at the cysteine 91 site to enhance the stability of SLC7A11 [29]. Moreover, the CTH inhibitor AOAA can suppress the sulfhydrylation of OTUB1 to foster ferroptosis in CRC cells [29].
(2) FSP1-CoQ10 pathway
Ferroptosis suppressor protein 1 (FSP1), also known as apoptosis-inducing factor mitochondrial 2 (AIFM2), is a newly identified suppressor of ferroptosis [30]. FSP1 is an oxidoreductase localized on the cytosolic membrane, and it reduces CoQ10 to CoQ10H2 with NADPH as an electron donor [31]. CoQ10H2 can trap lipophilic radicals that mediate lipid peroxidation reactions to inhibit ferroptosis [31, 32]. In CRC cells, FSP1 inhibits the activation ferroptosis through via suppressing CoQ10 generation and promoting ESCRT-III-mediated repair of impaired cell membranes [31, 33]. In addition, N-acetyltransferase 10 (NAT10) improve the stability of FSP1 through N4-acetylcytidine (ac4C) modification of FSP1 mRNA, which resists ferroptosis in CRC cells [34].
(3) GCH1-BH4 axis
GCH1-BH4-phospholipids is another anti-ferroptosis pathway [35]. GTP cyclohydrolase-1 (GCH1) is the rate-limiting enzyme for the synthesis of potent antioxidant tetrahydrobiopterin (BH4) [35]. BH4 is a coenzyme of phenylalanine hydroxylase, which suppresses the activity of enzyme and lipid peroxidation to inhibit cell ferroptosis [35]. Moreover, GCH1-BH4 can promote the synthesis of CoQ10 to exert antioxidant activity, which simultaneously inhibit cell ferroptosis [35]. In addition, the GCH1-BH4 axis selectively blocks the degradation of phospholipids that contain two PUFA tails and maintain the structure of cell membrane [35]. GCH1 mRNA level is significantly correlated with the susceptibility of tumor cells to ferroptosis, hence, it is a potential biomarker to indicate ferroptosis sensitivity. In CRC cells, the inhibition of GCH1-BH4 axis activates NCOA4-mediated autophagy of ferritin (FTH) and subsequently promote erastin-induced ferroptosis, indicating that the combination of GCH1 inhibitor and erastin is a potential strategy for CRC treatment [36].
(4) DHODH
Dihydroorotate dehydrogenase (DHODH) is an iron-containing flavin-dependent enzyme on the outer side of the inner mitochondrial membrane [37]. It plays a vital role in the de novo synthesis of pyrimidines [37]. DHODH can oxidize dihydroorotate (DHO) to orotate (OA) and reduce CoQ10 level through transferring electrons to it, thereby inhibiting ferroptosis in the inner mitochondrial membrane [38]. DHODH and GPX4 are the two critical anti-ferroptosis molecules in the inner mitochondrial membrane. In tumor with low GPX4 expression, DHODH inhibitor brequinar can induce ferroptosis to suppress tumor progression, whereas in tumor with high GPX4 expression, the combination of brequinar and sulfasalazine (SAS) exhibits potent anti-tumor effects, indicating that their combination for inducing ferroptosis is a potential strategy for cancer treatment [38].
Iron toxicity
Iron is a main component of hemoglobin, myoglobin and cytochrome enzymes [39]. Iron primarily exists as Fe2+ or is stored in FTH as Fe3+ in cells [40]. When the level of Fe2+ is overloaded in cells, Fe2+ and H2O2 will undergo the Fenton reaction to generate hydroxyl radicals (HO•), which oxidizes PUFAs and leads to cell membrane damage and rupture [41]. Transferrin (Tf) is responsible for iron transportation via forming the Tf-Fe3+ complex in plasma [42]. The binding of Tf to the transferrin receptor (TfR) on the cell surface will induce endocytosis [43]. Modulating iron content of cancer cells via alternating iron absorption, export, or storage can affect the susceptibility of cells to ferroptosis [14]. In CRC cells, iron-responsive element-binding protein 2 (IREB2) is shown to enhance the stability of TFRC mRNA (encodes TfR1) and promotes cellular uptake of iron, thereby increasing the sensitivity of CRC cells to ferroptosis [44]. The export of iron mainly relies on ferroportin (FPN, also known as SLC40A1), which inhibits ferroptosis by reducing intracellular Fe2+ concentration [45]. It is reported that the binding of FPN with Hepcidin will promote the degradation of FPN and subsequently regulate iron metabolism [46]. Otherwise, Ye et al. [47] found that HMGB1 could upregulate the expression of transferrin receptor protein 1(TfR1) through the RAS-JNK/p38 pathway, which increased the content of intracellular Fe2+ and activated ferroptosis.
Autophagy and ferroptosis
Autophagy is a biological process that provides energy to cells under stress by degrading cytoplasm or organelles through lysosomes [48]. Studies have indicated that autophagy has a crosstalk with ferroptosis [49]. Here we will summarize the role of autophagy-related molecules in ferroptosis.
(1) NCOA4 mediates FTH autophagy
Nuclear receptor coactivator 4 (NCOA4) is a carrier receptor that mediates the transport of FTH to lysosomes for degradation, which allows it to increase the content of free iron in cells and activate ferroptosis [14, 50]. NCOA4 expression is regulated by E3 ubiquitin-protein ligase HERC2 [51]. When the intracellular iron content is overloaded, HERC2 can bind to NCOA4 and promote its ubiquitination and subsequent degradation to restore the level of Fe2+ in cells [51].
(2) Lipophagy promotes ferroptosis
Lipids are stored in lipid droplets (LDs) [52]. LDs are selectively transported by autophagosomes to lysosomes for degradation, which provide free lipids to cells [26]. The increased lipids will lead to lipid peroxidation and induce ferroptosis [53]. During lipophagy, Ras-related protein Rab-7a (RAB7A) plays a vital role to mediate lysosomal recruitment of LDs, and the knockdown of RAB7A markedly restrains RSL3-induced ferroptosis [26, 53]. In contrast to RAB7A, tumor protein D52 (TPD52) is able to promote the formation of LDs to inhibit cell ferroptosis [26].
(3) Clockophagy
Clockophagy is a newly discovered form of autophagy that selectively degradate aryl hydrocarbon receptor nuclear translocator-like protein 1 (ARNTL, also known as BMAL1), which is a core component of the biological clock [54]. ARNTL can inhibit ferroptosis by repressing the transcription of EGLN2 [54]. Sequestosome 1 (SQSTM1) is a receptor for ARNTL that can promote ferroptosis by activating ARNTL autophagy [54]. Although studies have indicated a correlation between clockophagy and ferroptosis, the effect of the circadian clock on ferroptosis still requires further investigation.
(4) BECN1 regulates ferroptosis through a dual pathway
BECN1 is an autophagy-related gene and is able to regulate ferroptosis by boosting FTH autophagy [55, 56]. The expression of BECN1 is promoted by ELAV-like RNA binding protein 1 (ELAVL1), which increases BECN1 mRNA stability by binding the adenylate uridylate-rich elements (AREs) in the 3ʹ-UTR region of BECN1, which expedites the autophagic degradation of FTH [56]. Moreover, after the phosphorylation by AMPK, the BECN1 protein can bind to SLC7A11, block the activity of system xc−, and lead to ferroptosis in CRC cells [57]. The above studies show that BECN1 regulates ferroptosis either dependent or independent of cell autophagy [26].
Signaling pathways and molecules regulating ferroptosis in CRC
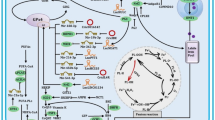
Recent studies revealed several signaling pathways and proteins that could regulate ferroptosis in CRC cells. They may be targets for the treatment of CRC (Fig. 2).

Signaling pathways and molecules regulating ferroptosis in CRC. MiR-539 inhibits GPX4 expression by targeting the SAPK/JNK signaling pathway in CRC cells. miR-545 targets Tf to inhibit Fe3+ uptake. In CRC cells, circ_0007142 resisted ferroptosis by removing the suppression of miR-874-3p on GDPD5. CircABCB10 resisted ferroptosis through reducing the inhibition of miR-326 on CCL5. p53 controls ferroptosis in CRC cells bidirectionally. On the one hand, p53 resists ferroptosis by up-regulating the expression of SLC7A11 and inhibiting the binding of DPP4 and NADPH oxidase 1 (NOX1). On the other hand, p53 promotes ferroptosis in the CYGB/p53-YAP axis. The intercellular and intracellular E-Cad/NF2/Hippo signaling pathway negatively regulates ferroptosis. OTUD1 directly binds to IREB2 to prevent it from ubiquitination and degradation, thus enhancing TfR1-mediated iron uptake. AMPK stimulates CRC cells ferroptosis through three pathways, including promoting BECN1 phosphorylation and inhibiting the activity of mTOR complex 1 (mTORC1) and SCD1. HIF-2α facilitates ferroptosis by controlling the expression of ferroptosis-related genes, such as PLIN2 and HILPDA
(1) p53
p53 is a well-studied tumor suppressor that promotes cell cycle arrest, cell senescence or apoptosis [58]. Previous studies have shown that the activation of p53 is required for inducing ferroptosis by suppressing SLC7A11 transcription in osteosarcoma and breast cancer cells [59]. However, Xie et al. [60] found that p53 could increase the expression of SLC7A11 by expediting the nuclear localization of dipeptidyl-peptidase-4 (DPP4) and suppressing its activity in CRC cells. Consequently, p53 exerted diverse effects on the expression of SLC7A11 in CRC and non-CRC cells. DPP4 is a serine protease located on cell membrane and regulates blood glucose levels. In TP53−/− CRC cells, the nuclear localization process of DPP4 is inhibited, which promotes cellular lipid peroxidation and leads to ferroptosis [60]. In TP53−/− CRC cells, the knockdown of DPP4 can partially reverse erastin-induced ferroptosis, indicating that DPP4 is involved in cell ferroptosis [60]. Furthermore, the sensitivity to ferroptosis is increased in TP53-mutant HCT116 cells compared to wild-type ones [60]. In conclusion, p53 is able to regulate cell ferroptosis through various mechanisms, which differs in different cancers. These findings highlight the critical role of p53 and DPP4 in regulating ferroptosis [60].
(2) YAP
Yes-associated protein (YAP) is the downstream effector of Hippo signaling pathway and it can promote ferroptosis via elevating the expression of ferroptosis regulators such as ACSL4 and TfR1 in CRC cells [61,62,63]. Some signaling molecules such as cytoglobin (CYGB), NF2 and E-cadherin (E-Cad) can regulate the expression and activity of YAP [62, 64]. CYGB, also known as stellate cell activation-associated protein, is a ROS regulator found in hepatic stellate cells [64]. Ye et al. [64] found that CYGB could accelerate lipid peroxidation and increase the sensitivity of CRC cells to ferroptosis by activating p53-YAP axis.
NF2 (also known as Merlin) is a tumor suppressor that can activate the Hippo pathway and restrain the activity of YAP, thereby suppressing the ferroptosis of CRC cells [62]. A study found that the interaction between epithelial cells could regulate ferroptosis, and a greater cell density indicated a lower sensitivity to ferroptosis [62]. E-Cad is responsible for this effect since it can inhibit the activity of YAP by activating the NF2 or Hippo pathway and thereby regulating ferroptosis [62]. The reduced CRC cell density or knock out of E-Cad/NF2 can enhance the activity of YAP and trigger ferroptosis in CRC cells [62]. Hence, the E-Cad/NF2/Hippo/YAP signaling pathway is a way for neighboring cells to regulate ferroptosis [62].
(3) OTUD1
Song et al. [44] found that OTU domain-containing protein 1 (OTUD1) could directly bind to IREB2 and promote its deubiquitination, thereby reducing the degradation of IREB2 and increase the expression of its downstream gene TFRC. Increased expression of TFRC promotes the accumulation of intracellular iron and ROS and sensitize cells to ferroptosis [44]. Moreover, OTUD1-induced ferroptosis can recruit immune cells and provoke immune responses against tumors [44]. Therefore, OTUD1 has the ability to promote cell ferroptosis and anti-tumor immune response. It is a promising target for CRC treatment [44].
(4) HIF-2ɑ
Hypoxia-inducible factor 2ɑ (HIF-2ɑ) is a regulator of gene transcription in response to decreased oxygen concentration [65]. HIF-2ɑ is overexpressed in CRC and can promote the progression of CRC [66]. The activated HIF-2ɑ upregulates the expression of lipid and iron regulatory genes, elevates intracellular iron levels, and enhances the susceptibility of CRC cells to ferroptosis [67]. Moreover, the activated HIF-2ɑ can boost irreversible oxidation cysteine to increase ROS level, thereby promoting CRC ferroptosis [67]. Thus, oxidative stress agonists selectively targeting activated HIF-2ɑ such as ferroptosis agonists (erastin and RSL3) can be used for CRC treatment [67].
(5) ROS/AMPK/SCD1 axis
TP53-induced glycolysis and apoptosis regulator (TIGAR) is a downstream of p53 and exerts an antioxidant activity in cells [68, 69]. Liu et al. [68] found that the silence of TIGAR increases the sensitivity of CRC cells (SW620 and HCT116 cells) to ferroptosis and expedite the accumulation of malondialdehyde (MDA). In addition, the knockout of TIGAR can promote ROS level and AMPK phosphorylation but reduce the expression of SCD1 in CRC cells [68]. Stearoyl-CoA desaturase 1 (SCD1) is a ferroptosis suppressor since it can regulate lipid metabolism and promote coenzyme Q10 (CoQ10) synthesis [70]. The knockdown of TIGAR is shown to reduce SCD1 expression in a redox- and AMPK-dependent manner and results in promoted ferroptosis [68]. Altogether, TIGAR can suppress ferroptosis through the ROS/AMPK/SCD1 pathway in CRC cells, indicating that targeting TIGAR may be a therapeutic approach for CRC via activating ferroptosis.
(6) ELOVL6-ACSL4
Elongation of very long chain fatty acids protein 6 (ELOVL6) is a momentous enzyme that catalyzes the elongation of saturated and monounsaturated fatty acids, and it is highly expressed in CRC specimens [71, 72]. Tian et al. [71] found that ELOVL6 was a potential target of apatinib. They demonstrated that apatinib has the ability to decrease cell viability in CRC cells and stimulate ferroptosis through the regulation of the ELOVL6/ACSL4 signaling pathway. According to their findings, ELOVL6 exerted an inhibitory activity of ferroptosis by binding and inhibiting the activity of ACSL4; apatinib indirectly promoted the expression of ACSL4 by reducing the expression of ELOVL6, thereby inducing ferroptosis in CRC cells [71].
(7) Non-coding RNA
Non-coding RNA mainly includes microRNA (miRNA), long non-coding RNA (lncRNA) and circular RNA (circRNA), which are essential for cellular biology and can regulate ferroptosis in CRC. The length of miRNA is 17–25 nucleotides, and it is able to degradate target genes by binding to their 3’-UTRs [73, 74]. Different miRNAs exert different regulatory effects on ferroptosis, which depends on their target genes. For example, miR-539 activates the SAPK/JNK pathway by targeting tumor necrosis factor alpha-induced protein 8 (TNFAIP8), thereby downregulating GPX4 expression and inducing ferroptosis in CRC cells, whereas miR-545 inhibits the accumulation of Fe2+ by targeting Tf, thereby suppressing ferroptosis of CRC cells [73, 75].
LncRNA has a length of more than 200 nucleotides, which is closely involved in cancer development [76, 77]. Han et al. [78] found that LINC00239 inhibits NF-E2-related factor 2 (Nrf2) ubiquitination by binding to Kelch-like ECH-associated protein 1 (Keap1), thereby suppresses ferroptosis in CRC cells. However, LINC01606 inhibits ferroptosis in colon cancer cells by up-regulating SCD1 expression and activating Wnt/β-catenin signaling pathway [79]. In addition, LINC01606 can interact with miR-423-5p to enhance the stemness of colon cancer cells [79]. In conclusion, LINC01606 can promote the progression of colon cancer, which may be a biomarker for the treatment of colon cancer [79].
Recent studies have revealed that circRNAs can regulate ferroptosis in tumor cells. CircRNAs are enriched with miRNA binding sites and can function as miRNA sponges [80]. For example, circ_0007142 can sponge miR-874-3p and inhibit its binding to glycerophosphodiester phosphodiesterase domain-containing protein 5 (GDPD5), thereby blocking ferroptosis in CRC cells. Hence, the knockdown of circ_0007142 can induce ferroptosis in CRC cells [80]. Also, circABCB10 diminishes the degradation of CCL5 that is resistant to ferroptosis by sponging miR-326 to induce ferroptosis [81]. Consequently, the circABCB10/miR-326/CCL5 axis is a target to modulate ferroptosis in CRC [81].
Drug targeting ferroptosis in CRC
By targeting the pathways involved in ferroptosis, small molecule drugs have shown great potential in suppressing CRC growth and improving patient outcomes. In order to provide a comprehensive overview of the ongoing research in this area, we have compiled a table summarizing the previous in vivo studies using animal models to investigate the efficacy of small molecules in targeting ferroptosis as a strategy for CRC treatment (Table 1, at the end of the text). Here, we have listed several representative drugs.
(1) IMCA
2-imino-6-methoxy-2 H-chromene-3-carbothioamide (IMCA) is a benzopyran derivative with various biological activities [82, 83]. Zhang et al. [83] found that IMCA could reduce the phosphorylation of mTOR (a pharmacological target of rapamycin) and p70S6 kinase (a downstream target protein of mTOR) by promoting AMPK phosphorylation at Thr172, thereby suppressing the activity of SLC7A11 and activating ferroptosis in CRC cells. Consequently, IMCA not only reduced the viability of CRC cell lines but also inhibited tumor growth in vivo with minimal organ toxicity. [83].
(2) Gallic acid
Gallic acid is a natural polyphenolic compound widely found in tea, oak bark and other plants. Additionally, gallic acid demonstrates anti-neoplastic activities by regulating multiple oncogenic targets, highlighting its potential as a dietary component [84, 85]. Gallic acid can inhibit CRC cell proliferation and this effect can be reversed by ferroptosis inhibitors [86]. The application of gallic acid significantly increased the contents of ROS, MDA and Fe2+ whereas that of GSH was decreased in HCT116 cells and Caco-2 cells, indicating that gallic acid can induce ferroptosis in CRC cells [86]. Also, gallic acid can reduce the expression of GPX4 and SLC7A11 and elevate that of TfR1 to promote ferroptosis in CRC [86].
(3) TalaA
Talaroconvolutin A (TalaA) is a natural product isolated from the endophytic fungus Talaromyces purpureogenus [87]. Xia et al. [87] found that TalaA induces ferroptosis in CRC cells through down-regulating the expression of SLC7A11 and GSS (glutathione synthetase) and upregulating that of ALOXE3 and HMOX1 [87]. Besides, TalaA has a stronger ability to induce ferroptosis in CRC cells than the erastin [87]. As a result, in vivo experiments demonstrated that TalaA effectively suppressed the growth of xenograft tumors. Hence, TalaA may become a candidate drug for treating CRC.
(4) Aspirin
Aspirin (acetylsalicylic acid) is a non-steroidal anti-inflammatory drug (NSAID) commonly used for pain relief and anti-thrombosis [88, 89]. A previous study demonstrated that the use of aspirin was associated with improved survival in patients with PIK3CA-mutant CRC, but not in patients with PIK3CA-wild type cancer [90]. Chen et al. further investigated the effects of aspirin on CRC with an oncogenic PIK3CA mutation and found that it increased the sensitivity of these cells to ferroptosis inducers [91]. Mechanistically, aspirin inhibited the PI3K/AKT/mTOR pathway, leading to down-regulation of its downstream protein, SREBP-1/SCD1. This down-regulation promoted ferroptosis in PIK3CA-mutant CRC cells. SCD1 is responsible for the conversion of saturated fatty acids (SFAs) into monounsaturated fatty acids (MUFAs) [92]. By inhibiting the expression of SCD1, aspirin reduced the production of MUFAs, thus enhancing RSL3-induced ferroptosis in PIK3CA-mutant CRC cells. Furthermore, the combination of aspirin and RSL3 showed significant anti-tumor efficacy in a human CRC xenograft model [91].
(5) Vitamin C
Vitamin C (VitC) is an antioxidant that can activate oxidative stress [93]. It is able to delay cetuximab resistance in CRC patients, restricts CRC organoid growth and destroys organoid structure, which are associated with VitC-induced ferroptosis [93]. The combination of VitC and cetuximab upraises labile iron pool (LIP) levels in CRC cells, and this effect is abolished by an iron chelator deferoxamine that can reduce iron accumulation and deposition [93]. The inclusion of VitC in combination with cetuximab impedes the emergence of drug persisters, curbs the growth of CRC organoids, and significantly delays the progression of acquired resistance in CRC patient-derived xenografts. This discovery holds immense importance in the treatment of CRC [93].
(6) β-elemene
β-elemene, a bioactive compound isolated from Chinese herbal medicine turmeric, exhibits anticancer activity on various types of cancer including CRC [94, 95]. Chen et al. [95] reported that combining β-elemene with cetuximab could increase the level of ROS, lipid peroxidation and Tf. It could also promote GSH consumption and decrease the expression of ferroptosis negative regulator proteins including GPX4, SLC7A11, and FTH1, thereby inducing ferroptosis in RAS-mutant CRC cells [95]. Moreover, β-elemene and cetuximab synergistically decreased the migration of CRC cells via inhibiting epithelial-mesenchymal transition (EMT) [95]. Hence, the combination of β-elemene and cetuximab may be an effective treatment for patients with RAS-mutant CRC by inducing ferroptosis and inhibiting cancer growth and migration [95].
Nanotechnology-enabled ferroptosis therapy
With the rapid development of nanotechnology and biomaterials, the rational design of nanomedicines can greatly improve the accumulation and release of ferroptosis inducers at tumor sites, thereby enhancing their therapeutic effects [96].
Using zinc oxide-coated virus-like silica nanoparticles (VZnO), Pan et al. were able to directly scavenge H2S, thereby diminishing GSH content and suppressing GPX4 activity in CRC cells. As a consequence, the powerful removal of lipid peroxides is compromised, eventually leading to the induction of ferroptosis in CRC cells [97]. Resveratrol (RSV) has been shown to promote ferroptosis by downregulating the expression of GPX4 and SLC7A11 [98]. To improve the delivery efficiency of RSV, Zhang et al. developed RSV-NPs@RBCm, in which RSV-loaded poly (ε-caprolactone) -poly (ethylene glycol) (PCL-PEG) nanoparticles are coated with erythrocyte membrane. This innovative nanosystem allows RSV-NPs@RBCm to evade macrophage phagocytosis and exhibits strong tumor specificity, resulting in a significant enhancement of the anti-tumor effect of RSV. Li et al. developed a biocompatible nanosystem known as fusiform iron oxide-hydroxide nanospindles (FeOOH NSs) for nuclear magnetic resonance (NMR) co-imaging. These FeOOH NSs have the unique capability to induce ferroptosis by scavenging endogenous H2S. As a result, FeOOH NSs not only enable tumor imaging through magnetic resonance imaging (MRI), but also effectively initiate ferroptosis in CRC tumors, synergistically combining these two therapeutic approaches to enhance treatment efficacy [99]. Li et al. developed glycyrrhetinic acid-based nanoplatforms (GCMNPs) that induce ferroptosis in CRC cells by suppressing GPX4 expression. Ferumoxytol, an iron-based nanomaterial used in ferrotherapy, also induces ferroptosis and immunogenic cell death (ICD) [100]. The combined use of GCMNPs and ferumoxytol synergistically delays CRC tumor progression and enhances immune cell infiltration. The feasibility and characteristics of nanoplatforms and nanomaterials for CRC therapy are underscored by these results, demonstrating their potential in triggering ferroptosis.
Ferroptosis and chemotherapy
The resistance of advanced CRC to oxaliplatin-based chemotherapy drugs is the primary cause of treatment failure [101]. However, studies have showed that ferroptosis reversal can overcome this resistance in CRC [102]. For instance, the KIF20A/NUAK1/PP1β/GPX4 signaling pathway has been identified as a mediators of oxaliplatin resistance in CRC cells by inhibiting ferroptosis [103]. Additionally, Zhang et al. [104] discovered that microsomal triglyceride transfer protein (MTTP) upregulates the expression of GPX4 and SLC7A11, leading to the inhibition of ferroptosis and reduced sensitivity of CRC to oxaliplatin. Another contributor to oxaliplatin resistance is cyclin-dependent kinase 1 (CDK1), which promotes the polyubiquitination and degradation of ACSL4 protein, effectively inhibiting ferroptosis [105].On the other hand, oxaliplatin itself can induce ferroptosis in CRC cells by inhibiting Nrf2 signaling pathway [106]. Furthermore, when combined with erastin, oxaliplatin exhibits a significant synergistic anti-tumor effect by inducing ferroptosis in CRC cells [106].
Moreover, approximately 50-60% of CRC patients develop resistance to 5-fluorouracil (5-FU)-based chemotherapy drugs [107]. Lipocalin 2 (LCN2), through its ability to reduce intracellular iron levels and promote the expression of GPX4 and SLC7A11, inhibits ferroptosis and enhance resistance to 5-FU [108]. Therefore, targeting LCN2 function with monoclonal antibodies may serve a potential therapeutic strategy for CRC patients resistant to 5-FU [108]. Additionally, pyrroline-5-carboxylate reductase 1 (PYCR1) has been found to promote tumor growth in CRC and reduce the sensitivity of CRC to 5-FU by inhibiting ferroptosis [109]. In summary, the reversal of chemoresistance in advanced CRC through targeting ferroptosis holds promise.
Ferroptosis and immunotherapy
In recent years, some studies have found that immunotherapy is also intrinsically related to ferroptosis. For example, Wang et al. [110] show that immunotherapy-activated CD8+ T cells promote lipid peroxidation and ferroptosis in cancer cells, such as B16 melanoma cells and HT-1080 fibrosarcoma cells, which contributes to the anti-tumor efficacy of immunotherapy. Mechanistically, interferon gamma (IFNγ) released by CD8+ T cells reduces the expression of SLC3A2 and SLC7A11 in tumor cells, promoting lipid peroxidation and ferroptosis [110]. Furthermore, the combination of cysteine endonuclease, an engineered enzyme that degrades cystine or cysteine, with PD-L1 synergistically enhance T cell-mediated anti-tumor immunity and induce cysteine endonuclease-induced ferroptosis in tumor cells [110]. Therefore, ferroptosis of tumor cells is a mechanism by which CD8+ T cells mediate the removal of tumors in vivo, and targeting ferroptosis may improve the efficacy of immunotherapy for cancer [110].
Lv et al. reported that apolipoprotein L3 (APOL3) increased IFNγ and decreased lactic acid concentration by binding and degrading L-lactate dehydrogenase A (LDHA), promoting ferroptosis of CRC cells and enhancing the cytotoxicity of CD8+ T cells [111]. Therefore, APOL3-LDHA may be a potential molecular target for simultaneous targeting ferroptosis and immunotherapy. Additionally, Chen et al. found that 20-hydroxyeicosatetraenoic acid (20-HETE), derived from cytochrome P450 1B1 (CYP1B1), can promote the expression of F-box only protein 10 (FBXO10) by activating the protein kinase C (PKC) signaling pathway. This leads to the ubiquitination and degradation of ACSL4, inducing the resistance of CRC cells to ferroptosis. Conversely, inhibiting CYP1B1 sensitize tumor cells to PD-1 antibodies [112]. Therefore, CYP1B1 may be a potential target for targeted CRC immunotherapy, but we still need to further determine the mechanism by which CYP1B1 improves the efficacy of CRC immunotherapy (Fig. 3).

The interaction between ferroptosis and immunotherapy in CRC. OTUD1 increases the sensitivity of CRC cells to ferroptosis by upregulating TfR1. Overexpression of OTUD1 causes CRC cells to release DAMPs (ATP, HSP70, HSP90 and HMGB1) and promotes tumor ICD. APOL3-LDHA promotes CD8+ T cells infiltration and induced ferroptosis of CRC cells by increasing IFNγ content and reducing lactic acid concentration. CYP1B1 inhibition can make CRC tumor sensitive to PD-1 antibody and reduce ACSL4 ubiquitination to promote ferroptosis in CRC cells. ZnP@DHA/Pyro-Fe induces ICD by mediating ferroptosis in CRC cells, which promotes antigen presentation (promote CRC cells to release DAMPs such as HMGB1). GCMNPs down-regulates GPX4 expression to stimulate ferroptosis in CRC cells and produce a large number of free radicals, which lead to ICD and enhance the sensitivity of CRC to immunotherapy. AA, arachidonic acid; DAMPs, damage-associated molecular patterns; ICD, immunogenic cell death; CTL, cytotoxic T lymphocytes; iDCs, immature dendritic cells; mDCs, myeloid dendritic cells
Immunogenic cell death (ICD) is a rare form of cell death characterized by changes in cell surface composition and the release of soluble mediators [113]. In the presence of tumor cells and antigen, ICD inducers accelerate dendritic cell maturation by activating the danger signal pathway, activating antitumor activities of CD4+/CD8+ T cells and improving the susceptibility of MSI-L CRCs to PD-1/PD-L1 inhibitors [100, 114].
Discussion
Ferroptosis is a newly discovered nonapoptotic regulated cell death and is a potential way to inhibit CRC. The mechanism of ferroptosis mainly includes three classic pathways: lipid peroxidation, reduced activity of GPX4 and iron overload [12]. Numerous studies have identified several ferroptosis modulators, such as GSH-independent antioxidant molecules (including FSP1 [31], DHODH [38], and GCH1-BH4 axis [35]) and autophagy-related molecules (including NCOA4 [50] and BECN1 [57]). Although progress has been made in comprehending the occurrence and mechanisms of ferroptosis, there remains a lack of knowledge regarding the specific execution molecules [115]. Therefore, further investigation into the mechanisms of ferroptosis is necessary to facilitate its translation into clinical treatments for CRC and other cancers.
While ferroptosis is characterized by lipid peroxidation, GPX4 degradation, and increased levels of ROS, studies have indicated that lipid peroxidation and elevated ROS can also occur in other forms of cell death, making it challenging to differentiate ferroptosis from other types of cell death. Consequently, there is a need for molecular markers with high specificity and sensitivity to distinguish ferroptosis in the future [115]. Additionally, the signaling molecules and pathways that regulate ferroptosis can vary among different tumor types. Moreover, the same molecule may exert opposite effects on ferroptosis in different tumor cells. Therefore, investigations should be conducted specifically in CRC to explore the signaling molecules or pathways that regulate ferroptosis, which could potentially unveil novel therapeutic targets for CRC [115].
It is worth noting that extensive ferroptosis has been observed in patients with inflammatory bowel disease (IBD) [116, 117]. IBD refers to a group of chronic, relapsing, and incurable gastrointestinal disorders characterized by an unclear etiology, prolonged course, and a high risk of developing colorectal cancer [116]. IBD includes Crohn’s disease (CD) and Ulcerative colitis (UC) [118]. Studies have shown that blocking ferroptosis can alleviate UC symptoms and promote intestinal repair by inhibiting endoplasmic reticulum (ER) stress-mediated cell death [119]. Ferroptosis inhibitors have also demonstrated effectiveness in improving colitis [120, 121]. Similarly, dysregulation of iron and lipid peroxidation contributes to CD. Xue-Jie-San (XJS), a Chinese herbal medicine formula, has shown promise in ameliorating CD colitis by inhibiting the FGL1/NF-κB/STAT3 pathway and enhancing the GPX4 antioxidant system in intestinal epithelial cells (IECs), thereby counteracting ferroptosis [122].
In summary, the role of ferroptosis in colorectal diseases is complex. While it negatively regulates IBD, it contributes to the treatment of CRC. Understanding the interplay between ferroptosis and these diseases is crucial for developing targeted therapies. Although some progress has been made in studying ferroptosis in CRC, there are still many unknowns that need to be explored. Further research is needed to develop more effective treatment options and expand the application of ferroptosis in CRC treatment.
Conclusions and perspectives
CRC is a severe disease with limited treatment options. To enhance the efficacy of current treatments for CRC, our study proposes combining ferroptosis with chemotherapy, immunotherapy, and targeted therapy. However, there is a need for further investigation to fully understand the molecular mechanisms involved in translating ferroptosis into clinical applications. In our future research, we will employ advanced genomic, proteomic, and metabolomic techniques to identify key regulators, signaling cascades, and critical metabolic alterations associated with ferroptosis in CRC. This will enable us to develop high-specificity and low-toxicity ferroptosis-targeted drugs through methods such as high-throughput screening and innovative drug design approaches. Additionally, comprehensive preclinical studies and well-designed clinical trials will be crucial in evaluating the therapeutic potential, pharmacokinetics, toxicity, and optimal treatment strategies of ferroptosis-targeted approaches in CRC. By undertaking these investigations, we aim to validate the benefits and safety profile of targeting ferroptosis in CRC patients and contribute to the development of effective and safe therapies for this devastating disease.
Data availability
All data relevant to this review is included in the text, references, and figures.
Abbreviations
- ROS:
-
Reactive oxygen species
- GPX4:
-
Glutathione peroxidase 4
- CRC:
-
Colorectal cancer
- IECs:
-
Intestinal epithelial cells
- PCD:
-
Programmed cell death
- GSH:
-
Glutathione
- PUFAs:
-
Polyunsaturated fatty acids
- AA:
-
Arachidonic acid
- AdA:
-
Adrenal acid
- ACSL4:
-
Acyl-CoA synthetase long-chain family member 4
- LPCAT3:
-
Lysophosphatidic transferase 3
- CYB5R1:
-
Cytochrome b5 reductase 1
- CBS:
-
Cystathionine-β-synthase
- CTH:
-
Cystathionine-γ-lyase
- OTUB1:
-
OTU domain-containing ubiquitin aldehyde-binding protein 1
- FSP1:
-
Ferroptosis suppressor protein 1
- NAT10:
-
N-acetyltransferase 10
- GCH1:
-
GTP cyclohydrolase-1
- DHODH:
-
Dihydroorotate dehydrogenase
- Tf:
-
Transferrin
- IREB2:
-
Iron-responsive element-binding protein 2
- FPN:
-
Ferroportin
- TfR1:
-
Transferrin receptor protein 1
- LDs:
-
Lipid droplets
- ARNTL:
-
Aryl hydrocarbon receptor nuclear translocator-like protein 1
- SQSTM1:
-
Sequestosome 1
- ELAVL1:
-
ELAV-like RNA binding protein 1
- AREs:
-
Adenylate uridylate-rich elements
- DPP4:
-
Dipeptidyl-peptidase-4
- YAP:
-
Yes-associated protein
- CYGB:
-
Cytoglobin
- OTUD1:
-
OTU domain-containing protein 1
- TIGAR:
-
TP53-induced glycolysis and apoptosis regulator
- MDA:
-
Malondialdehyde
- SCD1:
-
Stearoyl-CoA desaturase1
- ELOVL6:
-
Elongation of very long chain fatty acids family member 6
- TNFAIP8:
-
Tumor necrosis factor alpha-induced protein 8
- Nrf2:
-
NF-E2-related factor 2
- EMT:
-
Epithelial-mesenchymal transition
- LIP:
-
Labile iron pool
- MTTP:
-
Microsomal triglyceride transfer protein
- 5-FU:
-
5-fluorouracil
- PYCR1:
-
Pyrroline-5-carboxylate reductase 1
- IFNγ:
-
Interferon gamma
- APOL3:
-
Apolipoprotein L3
- CYP1B1:
-
Cytochrome P450 1B1
- FBXO10:
-
F-box only protein 10
- PKC:
-
Protein kinase C
- ICD:
-
Immunogenic cell death
- IBD:
-
Inflammatory bowel disease
- CD:
-
Crohn’s disease
- UC:
-
Ulcerative colitis
- IECs:
-
Intestinal epithelial cells
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68:394–424. https://doi.org/10.3322/caac.21492
Dekker E, Tanis PJ, Vleugels JLA, Kasi PM (2019) Wallace. Colorectal cancer. Lancet 394:1467–1480. https://doi.org/10.1016/s0140-6736(19)32319-0
Sorbarikor P, V A P (2019) Targeted therapy for colorectal cancer metastases: a review of current methods of molecularly targeted therapy and the use of tumor biomarkers in the treatment of metastatic colorectal cancer. Cancer 125:4139–4147. https://doi.org/10.1002/cncr.32163
Clarke PA, Roe T, Swabey K, Hobbs SM, McAndrew C, Tomlin K, Westwood I, Burke R, van Montfort R, Workman P (2019) Dissecting mechanisms of resistance to targeted drug combination therapy in human colorectal cancer. Oncogene 38:5076–5090. https://doi.org/10.1038/s41388-019-0780-z
Zhang J, Shen L, Li X, Song W, Liu Y, Huang L (2019) Nanoformulated Codelivery of Quercetin and Alantolactone promotes an Antitumor response through synergistic immunogenic cell death for microsatellite-stable colorectal Cancer. ACS Nano 13:12511–12524. https://doi.org/10.1021/acsnano.9b02875
Biller LH (2021) Diagnosis and treatment of metastatic colorectal Cancer: a review. JAMA 325:669–685. https://doi.org/10.1001/jama.2021.0106
Lewandowska A, Rudzki G, Lewandowski T, Stryjkowska-Gora A, Rudzki S (2022) Title: risk factors for the diagnosis of Colorectal Cancer. Cancer Control 29:10732748211056692. https://doi.org/10.1177/10732748211056692
Schwartz AJ, Goyert JW, Solanki S, Kerk SA, Chen B, Castillo C, Hsu PP, Do BT, Singhal R, Dame MK, Lee HJ, Spence JR, Lakhal-Littleton S, Vander Heiden MG, Lyssiotis CA, Xue X, Shah YM (2021) Hepcidin sequesters iron to sustain nucleotide metabolism and mitochondrial function in colorectal cancer epithelial cells. Nat Metab 3:969–982. https://doi.org/10.1038/s42255-021-00406-7
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149:1060–1072. https://doi.org/10.1016/j.cell.2012.03.042
Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G (2019) The molecular machinery of regulated cell death. Cell Res 29:347–364. https://doi.org/10.1038/s41422-019-0164-5
Torti SV, Torti FM (2013) Iron and cancer: more ore to be mined. Nat Rev Cancer 13:342–355. https://doi.org/10.1038/nrc3495
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR (2016) Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 113:E4966–E4975. https://doi.org/10.1073/pnas.1603244113
Wang Y, Wei Z, Pan K, Li J, Chen Q (2020) The function and mechanism of ferroptosis in cancer. Apoptosis 25:786–798. https://doi.org/10.1007/s10495-020-01638-w
Chen X, Kang R, Kroemer G, Tang D (2021) Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol 18:280–296. https://doi.org/10.1038/s41571-020-00462-0
Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, Superti-Furga G (2015) Stockwell. Human haploid cell Genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol 10:1604–1609. https://doi.org/10.1021/acschembio.5b00245
Liang D, Feng Y, Zandkarimi F, Wang H, Zhang Z, Kim J, Cai Y, Gu W, Stockwell BR, Jiang X (2023) Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones. Cell 00522–00526. https://doi.org/10.1016/j.cell.2023.05.003
Feimei K, Jiao L, Daolin T, Rui K (2020) Oxidative damage and antioxidant defense in ferroptosis. Front Cell Dev Biol 8:586578. https://doi.org/10.3389/fcell.2020.586578
Seok YW, K K J, G M M P, Milesh SMS (2016) S B R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 113:E4966–E4975. https://doi.org/10.1073/pnas.1603244113
Yan B, Ai Y, Sun Q, Ma Y, Cao Y, Wang J, Zhang Z, Wang X (2021) Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the Oxidoreductases POR and CYB5R1. Mol Cell 81:355–369. e10
Cao JY, Dixon SJ (2016) Mechanisms of ferroptosis. Cell Mol Life Sci 73:2195–2209. https://doi.org/10.1007/s00018-016-2194-1
Lee J, Kang ES, Kobayashi S, Homma T, Sato H, Seo HG, Fujii J (2017) The viability of primary hepatocytes is maintained under a low cysteine-glutathione redox state with a marked elevation in ophthalmic acid production. Exp Cell Res 361:178–191. https://doi.org/10.1016/j.yexcr.2017.10.017
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, Schreiber SL, Stockwell BR (2014) Regulation of ferroptotic cancer cell death by GPX4. Cell 156:317–331. https://doi.org/10.1016/j.cell.2013.12.010
Friedmann Angeli JP, Krysko DV, Conrad M (2019) Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer 19:405–414. https://doi.org/10.1038/s41568-019-0149-1
Yang WS, Stockwell BR (2016) Ferroptosis: death by lipid peroxidation. Trends Cell Biol 26:165–176. https://doi.org/10.1016/j.tcb.2015.10.014
Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M, Shan B, Pan H, Yuan J (2019) Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci U S A 116:2996–3005. https://doi.org/10.1073/pnas.1819728116
Liu J, Kuang F, Kroemer G, Klionsky DJ, Kang R, Tang D (2020) Autophagy-dependent ferroptosis: Machinery and Regulation. Cell Chem Biol 27:420–435. https://doi.org/10.1016/j.chembiol.2020.02.005
Wang R, Xing R, Su Q, Yin H, Wu D, Lv C, Yan Z (2021) Knockdown of SFRS9 inhibits progression of Colorectal Cancer through triggering ferroptosis mediated by GPX4 reduction. Front Oncol 11:683589. https://doi.org/10.3389/fonc.2021.683589
Wang L, Cai H, Hu Y, Liu F, Huang S, Zhou Y, Yu J, Xu J, Wu F (2018) A pharmacological probe identifies cystathionine beta-synthase as a new negative regulator for ferroptosis. Cell Death Dis 9:1005. https://doi.org/10.1038/s41419-018-1063-2
Chen S, Bu D, Zhu J, Yue T, Guo S, Wang X, Pan Y, Liu Y, Wang P (2021) Endogenous hydrogen sulfide regulates xCT stability through persulfidation of OTUB1 at cysteine 91 in colon cancer cells. Neoplasia 23:461–472. https://doi.org/10.1016/j.neo.2021.03.009
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, Bassik MC, Nomura DK, Dixon SJ, Olzmann JA (2019) The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575:688–692. https://doi.org/10.1038/s41586-019-1705-2
Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Goya Grocin A, Xavier da TN, Silva E, Panzilius CH, Scheel A, Mourao K, Buday M, Sato J, Wanninger T, Vignane V, Mohana M, Rehberg A, Flatley A, Schepers A, Kurz D, White M, Sauer M, Sattler EW, Tate W, Schmitz A, Schulze VO, Proneth GM, Popowicz DA, Pratt JPF, Angeli M, Conrad (2019) FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575:693–698. https://doi.org/10.1038/s41586-019-1707-0
Stefely JA, Pagliarini DJ (2017) Biochemistry of mitochondrial coenzyme Q biosynthesis. Trends Biochem Sci 42:824–843. https://doi.org/10.1016/j.tibs.2017.06.008
Dai E, Zhang W, Cong D, Kang R, Wang J, Tang D (2020) AIFM2 blocks ferroptosis independent of ubiquinol metabolism. Biochem Biophys Res Commun 523:966–971. https://doi.org/10.1016/j.bbrc.2020.01.066
Zheng X, Wang Q, Zhou Y, Zhang D, Geng Y, Hu W, Wu C, Shi Y, Jiang J (2022) N-acetyltransferase 10 promotes colon cancer progression by inhibiting ferroptosis through N4-acetylation and stabilization of ferroptosis suppressor protein 1 (FSP1) mRNA. Cancer Commun (Lond) 42:1347–1366. https://doi.org/10.1002/cac2.12363
Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Muller C, Zandkarimi F, Merl-Pham J, Bao X, Anastasov N, Kossl J, Brandner S, Daniels JD, Schmitt-Kopplin P, Hauck SM, Stockwell BR, Hadian K, Schick JA (2020) GTP cyclohydrolase 1/Tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci 6:41–53. https://doi.org/10.1021/acscentsci.9b01063
Hu Q, Wei W, Wu D, Huang F, Li M, Li W, Yin J, Peng Y, Lu Y, Zhao Q, Liu L (2022) Blockade of GCH1/BH4 Axis activates Ferritinophagy to mitigate the resistance of Colorectal Cancer to Erastin-Induced ferroptosis. Front Cell Dev Biol 10:810327. https://doi.org/10.3389/fcell.2022.810327
Wang F, Min J (2021) DHODH tangoing with GPX4 on the ferroptotic stage. Signal Transduct Target Ther 6:244. https://doi.org/10.1038/s41392-021-00656-7
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, Koppula P, Wu S, Zhuang L, Fang B, Poyurovsky MV, Olszewski K, Gan B (2021) DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593:586–590. https://doi.org/10.1038/s41586-021-03539-7
Konstantin S (2021) Role of Iron in Cancer. Semin Cancer Biol 76:189–194. https://doi.org/10.1016/j.semcancer.2021.04.001
Xuexian F, Zhaoxian C, Hao W, Dan H, Qi C, Pan Z, Feng G, Yingying Y, Zijun S, Qian W, Peng A, Sicong H, Jianwei P, Hou-Zao C, Jinghai C, Andreas L, Junxia M, Fudi W (2020) Loss of Cardiac Ferritin H facilitates Cardiomyopathy via Slc7a11-Mediated ferroptosis. Circul Res 127:486–501. https://doi.org/10.1161/CIRCRESAHA.120.316509
Bebber CM, Muller F, Clemente LP, von Weber J (2020) Karstedt. Ferroptosis in Cancer Cell Biology. Cancers (Basel) 12:164. https://doi.org/10.3390/cancers12010164
Gao M, Monian P, Quadri N, Ramasamy R, Jiang X (2015) Glutaminolysis and transferrin regulate ferroptosis. Mol Cell 59:298–308. https://doi.org/10.1016/j.molcel.2015.06.011
Grignano E, Birsen R, Chapuis N (2020) From Iron Chelation to overload as a therapeutic strategy to induce ferroptosis in leukemic cells. Front Oncol 10:586530. https://doi.org/10.3389/fonc.2020.586530
Song J, Liu T, Yin Y, Zhao W, Lin Z, Yin Y, Lu D, You F (2021) The deubiquitinase OTUD1 enhances iron transport and potentiates host antitumor immunity. EMBO Rep 22:e51162. https://doi.org/10.15252/embr.202051162
Geng N, Shi BJ, Li SL, Zhong ZY, Li YC, Xua WL, Zhou H, Cai JH (2018) Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur Rev Med Pharmacol Sci 22:3826–3836. https://doi.org/10.26355/eurrev_201806_15267
Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J (2004) Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306:2090–2093. https://doi.org/10.1126/science.1104742
Fanghua Y, Wenwen C, Min X, Minghua Y, Yan Y, Lizhi C, Liangchun Y (2019) HMGB1 regulates erastin-induced ferroptosis via RAS-JNK/p38 signaling in HL-60/NRASQ61L cells. Am J Cancer Res 9:730–739
Mizushima N, Komatsu M, Autophagy (2011) Renovation of cells and tissues. Cell 147:728–741. https://doi.org/10.1016/j.cell.2011.10.026
Kang R, Tang D (2017) Autophagy and ferroptosis - what’s the connection? Curr Pathobiol Rep 5:153–159. https://doi.org/10.1007/s40139-017-0139-5
Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC (2014) Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509:105–109. https://doi.org/10.1038/nature13148
Mancias JD, Pontano Vaites L, Nissim S, Biancur DE, Kim AJ, Wang X, Liu Y, Goessling W, Kimmelman AC (2015) Harper. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife 4:e10308. https://doi.org/10.7554/eLife.10308
Liu K, Czaja MJ (2013) Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ 20:3–11. https://doi.org/10.1038/cdd.2012.63
Bai Y, Meng L, Han L, Jia Y, Zhao Y, Gao H, Kang R, Wang X, Tang D, Dai E (2019) Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun 508:997–1003. https://doi.org/10.1016/j.bbrc.2018.12.039
Minghua Y, Pan C, Jiao L, Shan Z, Guido K, K D J, L M T, Z H J K, Rui T, Daolin (2019) Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv 5:eaaw2238. https://doi.org/10.1126/sciadv.aaw2238
H L X JS (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402:672–676. https://doi.org/10.1038/45257
Zili Z, Zhen Y, Ling W, Hai D, Jiangjuan S, Anping C, Feng Z, Shizhong Z (2018) Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy 14:2083–2103. https://doi.org/10.1080/15548627.2018.1503146
Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, Xie Y, Liu J, Klionsky DJ, Kroemer G, Lotze MT, Zeh HJ, Kang R, Tang D (2018) AMPK-Mediated BECN1 phosphorylation promotes ferroptosis by directly blocking System Xc(-) activity. Curr Biol 28:2388–99e5. https://doi.org/10.1016/j.cub.2018.05.094
Berkers CR, O D Maddocks EC, Cheung I, Mor KH, Vousden (2013) Metabolic regulation by p53 family members. Cell Metab 18:617–633. https://doi.org/10.1016/j.cmet.2013.06.019
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W (2015) Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520:57–62. https://doi.org/10.1038/nature14344
Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, Zhong M, Yuan H, Zhang L, Billiar TR, Lotze MT, Zeh HJ 3rd, Kang R, Kroemer G, Tang D (2017) The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep 20:1692–1704. https://doi.org/10.1016/j.celrep.2017.07.055
Fu V, Plouffe SW, Guan KL (2017) The Hippo pathway in organ development, homeostasis, and regeneration. Curr Opin Cell Biol 49:99–107. https://doi.org/10.1016/j.ceb.2017.12.012
Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, Chen ZN, Jiang X (2019) Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 572:402–406. https://doi.org/10.1038/s41586-019-1426-6
Sharma U, Tuli HS, Uttam V, Choudhary R, Sharma B, Sharma U, Prakash H, Jain A (2022) Role of hedgehog and Hippo signaling pathways in cancer: a special focus on non-coding RNAs. Pharmacol Res 186:106523. https://doi.org/10.1016/j.phrs.2022.106523
Ye S, Xu M, Zhu T, Chen J, Shi S, Jiang H, Zheng Q, Liao Q, Ding X, Xi Y (2021) Cytoglobin promotes sensitivity to ferroptosis by regulating p53-YAP1 axis in colon cancer cells. J Cell Mol Med 25:3300–3311. https://doi.org/10.1111/jcmm.16400
Imamura T, Kikuchi H, Herraiz MT, Park DY, Mizukami Y, Mino-Kenduson M, Lynch MP, Rueda BR, Benita Y, Xavier RJ, Chung DC (2009) HIF-1alpha and HIF-2alpha have divergent roles in colon cancer. Int J Cancer 124:763–771. https://doi.org/10.1002/ijc.24032
Xue X, Taylor M, Anderson E, Hao C, Qu A, Greenson JK, Zimmermann EM, Gonzalez FJ, Shah YM (2012) Hypoxia-inducible factor-2alpha activation promotes colorectal cancer progression by dysregulating iron homeostasis. Cancer Res 72:2285–2293. https://doi.org/10.1158/0008-5472.CAN-11-3836
Singhal R, Mitta SR, Das NK, Kerk SA, Sajjakulnukit P, Solanki S, Andren A, Kumar R, Olive KP, Banerjee R, Lyssiotis CA, Shah YM (2021) HIF-2alpha activation potentiates oxidative cell death in colorectal cancers by increasing cellular iron. J Clin Invest 131:e143691. https://doi.org/10.1172/JCI143691
Liu MY, Li HM, Wang XY, Xia R, Li X, Ma YJ, Wang M, Zhang HS (2022) TIGAR drives colorectal cancer ferroptosis resistance through ROS/AMPK/SCD1 pathway. Free Radic Biol Med 182:219–231. https://doi.org/10.1016/j.freeradbiomed.2022.03.002
Bensaad K, Tsuruta A, Selak MA, Vidal M, Vousden KH (2006) TIGAR, a p53-Inducible Regulator of Glycolysis and apoptosis. Cell 126:107–120. https://doi.org/10.1016/j.cell.2006.05.036
Tesfay L, Paul BT, Konstorum A, Deng Z, Cox AO, Lee J, Furdui CM, Hegde P, Torti FM (2019) Torti. Stearoyl-CoA desaturase 1 protects ovarian Cancer cells from ferroptotic cell death. Cancer Res 79:5355–5366. https://doi.org/10.1158/0008-5472.CAN-19-0369
Tian X, Li S, Ge G (2021) Apatinib promotes ferroptosis in Colorectal Cancer cells by targeting ELOVL6/ACSL4 signaling. Cancer Manag Res 13:1333–1342. https://doi.org/10.2147/CMAR.S274631
Su YC, Feng YH, Wu HT, Huang YS, Tung CL, Wu P, Chang CJ, Shiau AL, Wu CL (2018) Elovl6 is a negative clinical predictor for liver cancer and knockdown of Elovl6 reduces murine liver cancer progression. Sci Rep 8:6586. https://doi.org/10.1038/s41598-018-24633-3
Yang Y, Lin Z, Han Z, Wu Z, Hua J, Zhong R, Zhao R, Ran H, Qu K, Huang H, Tang H, Huang J, Liu Z, Hong X, Peng Z, Zhuang G (2021) miR-539 activates the SAPK/JNK signaling pathway to promote ferropotosis in colorectal cancer by directly targeting TIPE. Cell Death Discov 7:272. https://doi.org/10.1038/s41420-021-00659-x
Kashyap D, Tuli HS, Garg VK, Goel N (2018) Oncogenic and tumor-suppressive roles of MicroRNAs with special reference to apoptosis: molecular mechanisms and therapeutic potential. Mol Diagn Ther 22:179–201. https://doi.org/10.1007/s40291-018-0316-1
Zheng S, Hu L, Song Q, Shan Y, Yin G, Zhu H, Kong W, Zhou C (2021) miR-545 promotes colorectal cancer by inhibiting transferring in the non-normal ferroptosis signaling. Aging-Us 13:26137–26147. https://doi.org/10.18632/aging.203801
Lu SR, Li Q, Lu JL, Liu C, Xu X, Li JZ (2018) Long non-coding RNA LINC01503 promotes colorectal cancer cell proliferation and invasion by regulating miR-4492/FOXK1 signaling. Exp Ther Med 16:4879–4885. https://doi.org/10.3892/etm.2018.6775
Cai HJ, Zhuang ZC, Wu Y, Zhang YY, Liu X, Zhuang JF, Yang YF, Gao Y, Chen B, Guan GX (2021) Development and validation of a ferroptosis-related lncRNAs prognosis signature in colon cancer. Bosn J Basic Med Sci 21:569–576. https://doi.org/10.17305/bjbms.2020.5617
Han Y, Gao X, Wu N, Jin Y, Zhou H, Wang W, Liu H, Chu Y, Cao J, Jiang M, Yang S, Shi Y, Xie X, Chen F, Han Y, Qin W, Xu B, Liang J (2022) Long noncoding RNA LINC00239 inhibits ferroptosis in colorectal cancer by binding to Keap1 to stabilize Nrf2. Cell Death Dis 13:742. https://doi.org/10.1038/s41419-022-05192-y
Luo Y, Huang S, Wei J, Zhou H, Wang W, Yang J, Deng Q, Wang H, Fu Z (2022) Long noncoding RNA LINC01606 protects colon cancer cells from ferroptotic cell death and promotes stemness by SCD1-Wnt/beta-catenin-TFE3 feedback loop signalling. Clin Transl Med 12:e752. https://doi.org/10.1002/ctm2.752
Wang Y, Chen H, Wei X (2021) Circ_0007142 downregulates miR-874-3p-mediated GDPD5 on colorectal cancer cells. Eur J Clin Invest 51:e13541. https://doi.org/10.1111/eci.13541
Xian ZY, Hu B, Wang T, Cai JL, Zeng JY, Zou Q, Zhu PX (2020) CircABCB10 silencing inhibits the cell ferroptosis and apoptosis by regulating the miR-326/CCL5 axis in rectal cancer. Neoplasma 67:1063–1073. https://doi.org/10.4149/neo_2020_191024N1084
Xiu C, Hua Z, Xiao BS, Tang WJ, Zhou HP, Liu XH (2017) Novel benzopyran derivatives and their therapeutic applications: a patent review (2009–2016). Expert Opin Ther Pat 27:1031–1045. https://doi.org/10.1080/13543776.2017.1338687
Zhang L, Liu W, Liu F, Wang Q, Song M, Yu Q, Tang K, Teng T, Wu D, Wang X, Han W, Li Y (2020) IMCA induces ferroptosis mediated by SLC7A11 through the AMPK/mTOR pathway in Colorectal Cancer. Oxid Med Cell Longev 2020:1675613. https://doi.org/10.1155/2020/1675613
Khan BA, Mahmood T, Menaa F, Shahzad Y, Yousaf AM, Hussain T, Ray SD (2018) New Perspectives on the efficacy of gallic acid in Cosmetics & Nanocosmeceuticals. Curr Pharm Des 24:5181–5187. https://doi.org/10.2174/1381612825666190118150614
Aggarwal D, Kaur G, Tuli HS, Mistry H, Sak K, Khan MA, Yerer MB, Mittal S, Garg VK (2022) Gallic acid: a Dietary Polyphenol that exhibits anti-neoplastic activities by modulating multiple oncogenic targets. Anti-cancer Agents Med Chem 22:499–514. https://doi.org/10.2174/1871520621666211119085834
Hong Z, Tang P, Liu B, Ran C, Yuan C, Zhang Y, Lu Y, Duan X, Yang Y, Wu H (2021) Ferroptosis-related genes for overall survival prediction in patients with Colorectal Cancer can be inhibited by gallic acid. Int J Biol Sci 17:942–956. https://doi.org/10.7150/ijbs.57164
Xia Y, Liu S, Li C, Ai Z, Shen W, Ren W, Yang X (2020) Discovery of a novel ferroptosis inducer-talaroconvolutin A-killing colorectal cancer cells in vitro and in vivo. Cell Death Dis 11:988. https://doi.org/10.1038/s41419-020-03194-2
Argentina O, Niki Z-M, D G M, D J S L, Lenard H, David H, Ernest V, Eduardo B, Pratip M, Steven (2017) Beyond COX-1: the effects of aspirin on platelet biology and potential mechanisms of chemoprevention. Cancer Metastasis Rev 36. https://doi.org/10.1007/s10555-017-9675-z
Christopher E, C T E F, Emma A, Brian WN, Mr (2017) Non-steroidal anti-inflammatory drugs (NSAIDs) for chronic non-cancer pain in children and adolescents. Cochrane Database Syst Rev 8. https://doi.org/10.1002/14651858.CD012537.pub2
Xiaoyun L, Paul L, Reiko N, Teppei M, Aya K, Mai Y, Yu I, Rong QZ, Yoshifumi B, Kaori S, Ruifang S, Katsuhiko N, Edward MJAG (2012) C A T, O shuji. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med 367. https://doi.org/10.1056/NEJMoa1207756
Chen H, Qi Q, Wu N, Wang Y, Feng Q, Jin R, Jiang L (2022) Aspirin promotes RSL3-induced ferroptosis by suppressing mTOR/SREBP-1/SCD1-mediated lipogenesis in PIK3CA-mutatnt colorectal cancer. Redox Biol 55:102426. https://doi.org/10.1016/j.redox.2022.102426
Rita M, Alessia N, Elena PM, Claudia DV, Marcello M-S, Gennaro C (2018) Metabolic features of cancer stem cells: the emerging role of lipid metabolism. Oncogene 37. https://doi.org/10.1038/s41388-018-0141-3
Lorenzato A, Magri A, Matafora V, Audrito V, Arcella P, Lazzari L, Montone M, Lamba S, Deaglio S, Siena S, Bertotti A, Trusolino L, Bachi A, Di Nicolantonio F, Bardelli A, Arena S (2020) Vitamin C restricts the emergence of Acquired Resistance to EGFR-Targeted Therapies in Colorectal Cancer. Cancers (Basel) 12:685. https://doi.org/10.3390/cancers12030685
Zhai B, Zhang N, Han X, Li Q, Zhang M, Chen X, Li G, Zhang R, Chen P, Wang W, Li C, Xiang Y, Liu S, Duan T, Lou J, Xie T, Sui X (2019) Molecular targets of β-elemene, a herbal extract used in traditional chinese medicine, and its potential role in cancer therapy: a review. Biomed Pharmacother 114:108812. https://doi.org/10.1016/j.biopha.2019.108812
Chen P, Li X, Zhang R, Liu S, Xiang Y, Zhang M, Chen X, Pan T, Yan L, Feng J, Duan T, Wang D, Chen B, Jin T, Wang W, Chen L, Huang X, Zhang W, Sun Y, Li G, Kong L, Chen X, Li Y, Yang Z, Zhang Q, Zhuo L, Sui X, Xie T (2020) Combinative treatment of beta-elemene and cetuximab is sensitive to KRAS mutant colorectal cancer cells by inducing ferroptosis and inhibiting epithelial-mesenchymal transformation. Theranostics 10:5107–5119. https://doi.org/10.7150/thno.44705
Liu M, Liu B, Liu Q, Du K, Wang Z, He N (2019) Nanomaterial-induced ferroptosis for cancer specific therapy. Coord Chem Rev 382:160–180. https://doi.org/10.1016/j.ccr.2018.12.015
Pan X, Qi Y, Du Z, He J, Yao S, Lu W, Ding K, Zhou M (2021) Zinc oxide nanosphere for hydrogen sulfide scavenging and ferroptosis of colorectal cancer. J Nanobiotechnol 19:392. https://doi.org/10.1186/s12951-021-01069-y
Zhang Z, Ji Y, Hu N, Yu Q, Zhang X, Li J, Wu F, Xu H, Tang Q, Li X (2022) Ferroptosis-induced anticancer effect of resveratrol with a biomimetic nano-delivery system in colorectal cancer treatment. Asian J Pharm Sci 17:751–766. https://doi.org/10.1016/j.ajps.2022.07.006
Li Y, Chen W, Qi Y, Wang S, Li L, Li W, Xie T, Zhu H, Tang Z, Zhou M (2020) H2 S-Scavenged and activated Iron oxide-hydroxide nanospindles for MRI-Guided photothermal therapy and ferroptosis in Colon cancer. Small 16:e2001356. https://doi.org/10.1002/smll.202001356
Li Q, Su R, Bao X, Cao K, Du Y, Wang N, Wang J, Xing F, Yan F, Huang K, Feng S (2022) Glycyrrhetinic acid nanoparticles combined with ferrotherapy for Improved Cancer Immunotherapy. Acta Biomater 109–120. https://doi.org/10.1016/j.actbio.2022.03.030
Li L, Shang J, Zhang Y, Liu S, Peng Y, Zhou Z, Pan H, Wang X, Chen L, Zhao Q (2017) MEG3 is a prognostic factor for CRC and promotes chemosensitivity by enhancing oxaliplatin-induced cell apoptosis. Oncol Rep 38:1383–1392. https://doi.org/10.3892/or.2017.5828
Wang G, Wang J-J, Zhi-Min Z, Xu X-N, Shi F, Fu X-L (2023) Targeting critical pathways in ferroptosis and enhancing antitumor therapy of platinum drugs for colorectal cancer. Sci Prog 106. https://doi.org/10.1177/00368504221147173
Changshun Y, Yu Z, Shengtao L, Yi L, Weihua L (2021) Suppressing the KIF20A/NUAK1/Nrf2/GPX4 signaling pathway induces ferroptosis and enhances the sensitivity of colorectal cancer to oxaliplatin. Aging 13:13515–13534. https://doi.org/10.18632/aging.202774
Zhang Q, Deng T, Zhang H, Zuo D, Zhu Q, Bai M, Liu R, Ning T, Zhang L, Yu Z, Zhang H, Ba Y (2022) Adipocyte-derived exosomal MTTP suppresses ferroptosis and promotes Chemoresistance in Colorectal Cancer. Adv Sci 9:e2203357. https://doi.org/10.1002/advs.202203357
Kaixuan Z, Weihao L, Yue W, Zifei Z, Linjie Z, Weili Z, Yue X, Chi Z (2023) Inhibition of CDK1 Overcomes Oxaliplatin Resistance by Regulating ACSL4-mediated Ferroptosis in Colorectal Cancer. Advanced science (Weinheim, Baden-Wurttemberg, Germany): e2301088. https://doi.org/10.1002/advs.202301088
Liu B, Wang H (2022) Oxaliplatin induces ferroptosis and oxidative stress in HT29 colorectal cancer cells by inhibiting the Nrf2 signaling pathway. Exp Ther Med 23:394. https://doi.org/10.3892/etm.2022.11321
Sharma P, Shimura T, Banwait JK, Goel A (2020) Andrographis-mediated chemosensitization through activation of ferroptosis and suppression of beta-catenin/Wnt-signaling pathways in colorectal cancer. Carcinogenesis 41:1385–1394. https://doi.org/10.1093/carcin/bgaa090
Chaudhary N, Choudhary BS, Shah SG, Khapare N, Dwivedi N, Gaikwad A, Joshi N, Raichanna J, Basu S, Gurjar M, Saklani SPKA, Gera P, Ramadwar M, Patil P, Thorat R, Gota V, Dhar SK, Gupta S, Das M, Dalal SN (2021) Lipocalin 2 expression promotes tumor progression and therapy resistance by inhibiting ferroptosis in colorectal cancer. Int J Cancer 149:1495–1511. https://doi.org/10.1002/ijc.33711
Borong Z, Zhongchao M, Ying Y, Yanan S, Miao Z, Xinlin Y, Wei X, Xiaofeng Q (2022) The role of PYCR1 in inhibiting 5-fluorouracil-induced ferroptosis and apoptosis through SLC25A10 in colorectal cancer. Hum Cell 35:1900–1911. https://doi.org/10.1007/s13577-022-00775-5
Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, Xia H, Zhou J, Li G, Li J, Li W, Wei S, Vatan L, Zhang H, Szeliga W, Gu W, Liu R, Lawrence TS, Lamb C, Tanno Y, Cieslik M, Stone E, Georgiou G, Chan TA, Chinnaiyan A, Zou W (2019) CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569:270–274. https://doi.org/10.1038/s41586-019-1170-y
Lv Y, Tang W, Xu Y, Chang W, Zhang Z, Lin Q, Ji M, Feng Q, He G, Xu J (2023) Apolipoprotein L3 enhances CD8 + T cell antitumor immunity of colorectal cancer by promoting LDHA-mediated ferroptosis. Int J Biol Sci 19:1284–1298. https://doi.org/10.7150/ijbs.74985
Chen C, Yang Y, Guo Y, He J, Chen Z, Qiu S, Zhang Y, Ding H, Pan J, Pan Y (2023) CYP1B1 inhibits ferroptosis and induces anti-PD-1 resistance by degrading ACSL4 in colorectal cancer. Cell Death Dis 14:271. https://doi.org/10.1038/s41419-023-05803-2
Kroemer G, Galluzzi L, Kepp O (2013) Immunogenic cell death in Cancer Therapy. Annu Rev Immunol 31:51–72. https://doi.org/10.1146/annurev-immunol-032712-100008
Song W, Shen L, Wang Y, Liu Q, Goodwin TJ, Li J, Dorosheva O, Liu T, Liu R, Huang L (2018) Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap. Nat Commun 9:2237. https://doi.org/10.1038/s41467-018-04605-x
Chen X, Comish PB, Tang D (2021) Characteristics and biomarkers of ferroptosis. Front Cell Dev Biol 9:637162. https://doi.org/10.3389/fcell.2021.637162
Tang B, Zhu J, Fang S, Wang Y, Vinothkumar R, Li M, Weng Q, Yang LY, Qiu R, Xu M, Zhao Z, Ji J (2021) Pharmacological inhibition of MELK restricts ferroptosis and the inflammatory response in colitis and colitis-propelled carcinogenesis. Free Radic Biol Med 172:312–329. https://doi.org/10.1016/j.freeradbiomed.2021.06.012
Huang J, Zhang J, Ma J, Ma J, Liu J, Wang F, Tang X, Moreira H (2022) Inhibiting ferroptosis: a Novel Approach for Ulcerative Colitis therapeutics. Oxidative Med Cell Longev 2022:9678625. https://doi.org/10.1155/2022/9678625
Maloy KJ, Powrie F (2011) Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 474:298–306. https://doi.org/10.1038/nature10208
Xu M, Tao J, Yang Y, Tan S, Liu H, Jiang J, Zheng F, Wu B (2020) Ferroptosis involves in intestinal epithelial cell death in ulcerative colitis. Cell Death Dis 11:86. https://doi.org/10.1038/s41419-020-2299-1
Chen Y, Zhang P, Chen W, Chen G (2020) Ferroptosis mediated DSS-induced ulcerative colitis associated with Nrf2/HO-1 signaling pathway. Immunol Lett 225:9–15. https://doi.org/10.1016/j.imlet.2020.06.005
Xu J, Liu S, Cui Z, Wang X, Ning T, Wang T, Zhang N, Xie S, Min L, Zhang S, Liang C, Zhu S (2021) Ferrostatin-1 alleviated TNBS induced colitis via the inhibition of ferroptosis. Biochem Biophys Res Commun 573:48–54. https://doi.org/10.1016/j.bbrc.2021.08.018
Gao Y, Zhang Z, Du J, Yang X, Wang X, Wen K, Sun X (2023) Xue-Jie-San restricts ferroptosis in Crohn’s disease via inhibiting FGL1/NF-κB/STAT3 positive feedback loop. Front Pharmacol 14:1148770. https://doi.org/10.3389/fphar.2023.1148770
Zhu J-F, Liu Y, Li W-T, Li M-H, Zhen C-H, Sun P-W, Chen J-X, Wu W-H, Zeng W (2023) Ibrutinib facilitates the sensitivity of colorectal cancer cells to ferroptosis through BTK/NRF2 pathway. Cell Death Dis 14:151. https://doi.org/10.1038/s41419-023-05664-9
Guo S, Zhao W, Zhang W, Li S, Teng G, Liu L (2023) Vitamin D promotes ferroptosis in Colorectal Cancer Stem cells via SLC7A11 downregulation. Oxid Med Cell Longev 2023:4772134. https://doi.org/10.1155/2023/4772134
Gao Z, Jiang J, Hou L, Ji F (2022) Lysionotin induces ferroptosis to Suppress Development of Colorectal Cancer via promoting Nrf2 degradation. Oxid Med Cell Longev 2022:1366957. https://doi.org/10.1155/2022/1366957
Miao Q, Deng WQ, Lyu WY, Sun ZT, Fan SR, Qi M, Qiu SH, Zhu YR, Lin JP, Chen MF, Deng LJ (2023) Erianin inhibits the growth and metastasis through autophagy-dependent ferroptosis in KRAS(G13D) colorectal cancer. Free Radic Biol Med 204:301–312. https://doi.org/10.1016/j.freeradbiomed.2023.05.008
Lin H, Yamei Y, Meiyang F, Shanfeng G, Lingyu Z, Xiaofan X, Rufeng L, Xuan X, Xiaofei W, Lei N, Dongdong T, Chen H, Youlong C, Juan Y (2022) Pt3R5G inhibits colon cancer cell proliferation through inducing ferroptosis by down-regulating SLC7A11. Life Sci 306:120859. https://doi.org/10.1016/j.lfs.2022.120859
Kuang H, Sun X, Liu Y, Tang M, Wei Y, Shi Y, Li R, Xiao G, Kang J, Wang F, Peng J, Xu H, Zhou F (2023) Palmitic acid-induced ferroptosis via CD36 activates ER stress to break calcium‐iron balance in colon cancer cells. FEBS J 290:3664–3687. https://doi.org/10.1111/febs.16772
Acknowledgements
Not applicable.
Funding
This work was supported by the grants from National Natural Science Foundation of China (82100194 to J.J., 82273050 to M.Y.), Hubei Provincial Natural Science Foundation of China (2021CFB035 to J.J., 2022CFB304 to M. Y.).
Author information
Authors and Affiliations
Contributions
Siyue Liu and Ming Yue drafted the initial manuscript; Jue Jiang, Siyue Liu and Xiaoliu Liu conceived the structure of the manuscript; Siyue Liu, Jue Jiang, Yukang Lu, Ying Wang and Shiwen Luo revised the manuscript. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
All authors approve the submission of the manuscript.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liu, S., Yue, M., Lu, Y. et al. Advancing the frontiers of colorectal cancer treatment: harnessing ferroptosis regulation. Apoptosis 29, 86–102 (2024). https://doi.org/10.1007/s10495-023-01891-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-023-01891-9