
Abstract
Autoimmune diseases are pathological conditions that result from the misidentification of self-antigens in immune system, leading to host tissue damage and destruction. These diseases can affect different organs and systems, including the blood, joints, skin, and muscles. Despite the significant progress made in comprehending the underlying pathogenesis, the complete mechanism of autoimmune disease is still not entirely understood. In autoimmune diseases, the innate immunocytes are not functioning properly: they are either abnormally activated or physically disabled. As a vital member of innate immunocyte, neutrophils and their modes of death are influenced by the microenvironment of different autoimmune diseases due to their short lifespan and diverse death modes. Related to neutrophil death pathways, apoptosis is the most frequent cell death form of neutrophil non-lytic morphology, delayed or aberrant apoptosis may contribute to the development anti-neutrophil cytoplasmic antibodies (ANCA)-associated vasculitis (AAV). In addition, NETosis, necroptosis and pyroptosis which are parts of lytic morphology exacerbate disease progression through various mechanisms in autoimmune diseases. This review aims to summarize recent advancements in understanding neutrophil death modes in various autoimmune diseases and provide insights into the development of novel therapeutic approaches for autoimmune diseases.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Autoimmune disease is a complex group of conditions marked by the breakdown of self-tolerance to various autoantigens. These multifaceted pathologies arise from a complicated interplay between genetic and environmental factors. In cases of pathological autoimmunity, the intrinsic immune system of the organism is mobilized against self-tissues, leading to the recruitment of autoantibodies, self-reactive lymphocytes, and other innate immune cells. This immune response culminates in a cascade of inflammatory processes, resulting in the infliction of tissue damage and the development of chronic inflammation [1]. Autoimmune disease is a multifaceted and heterogeneous illness with a wide array of clinical manifestations, characterized by the immune system of body attacking its own tissues and organs. This disease can be classified into two major categories: systemic and organ-specific autoimmune diseases [2, 3]. The systemic autoimmune diseases, including rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), Sjogren's syndrome (SS) and systemic sclerosis (SSC) are distinguished by their pathologic impact on multiple organs and tissues. Conversely, organ-specific immune diseases, such as psoriasis, ulcerative colitis (UC), Crohn's disease (CD), and multiple sclerosis (MS), display a localized manifestation of autoantibodies accumulation in specific systems and organs. The pathogenesis of autoimmune diseases is intricate, involving a complex interplay between genetic, environmental, and psychological factors, leading to the activation of both innate and adaptive immune responses.
Within the immune system, macrophages, dendritic cells, and neutrophils are all considered to be integral players in the development and progression of autoimmune diseases [4, 5]. Specifically, neutrophils have garnered considerable attention in recent years due to their ability to impact the autoimmune process through both direct and indirect mechanisms. For example, in response to pathogenic invasion, such as bacterial infections, neutrophils detect and recognize the pathogens through specific receptors. Upon recognition, neutrophils utilize their toxic products, such as myeloperoxidase (MPO), neutrophil elastase (NE), reactive oxygen species (ROS), and cytokines, to perform their phagocytic function to eradicate the invading pathogens directly [6]. Nevertheless, excessive infiltration of neutrophils in sites of tissue injury can lead to chronic inflammation by releasing cytokines and proteases, thereby causing tissue damage. Furthermore, the infiltration of neutrophils can regulate the activity of adaptive immune responses by activating B cells and T cells, thereby leading to immune system imbalance indirectly [7]. Therefore, the relationship between neutrophil death and inflammation and autoimmunity deserves further exploration.
As a type of white blood cell, neutrophils are derived from hematopoietic stem cells and undergo maturation within the confines of the bone marrow. This process of granulopoiesis is marked by a series of distinct stages, each of which is characterized by a unique morphology and granular content. From promyelocytes to myelocytes to metamyelocytes, the neutrophilic precursors gradually adopt the classic segmented appearance and develop the characteristic intracellular granules that confer their potent antimicrobial properties. This complex process of neutrophil differentiation and maturation is crucial for the maintenance of immune homeostasis [8]. The intricate regulation of neutrophil development, production, and release is orchestrated by a multifaceted network of signaling molecules, but perhaps none is more central than the granulocyte colony-stimulating factor (GM-CSF). Acting through a cascade of downstream effectors, GM-CSF finely tunes the delicate balance of neutrophil homeostasis by suppressing the expression of CXR4, a receptor that promotes retention of immature neutrophils in the bone marrow, and simultaneously enhancing the activity of CXCR2, a receptor that stimulates the release of mature neutrophils into the bloodstream. This exquisite modulation of neutrophil trafficking and differentiation is essential for the proper functioning of the immune system and serves as a prime target for pharmacological intervention in a host of hematological disorders and inflammatory diseases [9]. Actually, neutrophils are the sentinel guardians of the innate immune system which are characterized by their remarkable ability to rapidly migrate to sites of infection or injury and unleash a potent arsenal of antimicrobial effector molecules. However, this prodigious performance comes at a cost, as these cells are inherently short-lived and have a limited lifespan in the circulation, with a half-life of a mere 8 h in humans. The physiological range of neutrophil count in a healthy individual is estimated to be within the range of 1500–8000 per microlitre of blood, which contributes to the remarkable surge of millions of these cells each day. The high turnover rate of neutrophils reflects the tremendous metabolic demands required for their effector function, as well as the need to prevent the accumulation of damaged or dysfunctional cells that could cause tissue damage or autoimmunity. While this tight regulation of neutrophil turnover is critical for immune homeostasis, it also poses a major challenge for therapeutic targeting, as any intervention that alters neutrophil survival or turnover could have far-reaching consequences for immune function and host defense [10].
Recent research has revealed that neutrophils possess multiple programmed cell death pathways, which can be categorized into lytic and non-lytic morphologies based on the final cell death morphology [11]. Apoptosis, the most common non-lytic morphology, is characterized by ‘a dance of death’ and is typically immunologically silent [12]. Conversely, lytic morphology pathways such as necroptosis, pyroptosis, neutrophil extracellular traps (NETs) and ferroptosis release cellular molecules by rupturing the plasma membrane, resulting in the formation of damage-associated molecular patterns (DAMPs) that promote inflammatory reactions. Consequently, autoimmune diseases and chronic inflammation can develop [13, 14]. The importance of studying neutrophil death in autoimmune disease mechanisms and treatment modalities is emphasized by these findings.
However, the significance of survival mechanisms cannot be overlooked even in short-lived cells such as neutrophil. Autophagy plays a crucial role in maintaining the normal state and functionality of neutrophil. Unlike the diverse modes of cell death, autophagy is activated in response to accumulated cellular damage, oxidative stress, and nutritional starvation. Its primary function is to degrade damaged organelles, unwanted proteins, and other substances, thereby preserving overall cellular homeostasis. Recent studies have revealed that autophagy not only acts as a conserved and self-protective mechanism in neutrophil but also exhibits intricate interactions with various cell death pathways, including apoptosis, NETosis, pyroptosis, and the release of inflammatory factors. Consequently, autophagy becomes interdependent and plays a crucial role in the development of numerous autoimmune diseases. Therefore, the relationship between autophagy and the mode of neutrophil death in autoimmune diseases has also attracted interest.
Here, we reviewed recent progress in understanding the pathogenesis, abnormalities of different neutrophil death pathways and relationship between survival and death mechanism in autoimmune diseases, with the goal of developing targeted therapeutic agents to modulate neutrophil death.
Apoptosis: the king of non-lytic neutrophil cell death
Apoptosis is a regulated morphology that widely exists in the neutrophil cell death kingdom. During the apoptosis, no intracellular components are released outside the cell, the cell shape becomes round and the volume decreases. What’s more, the nucleus shrinks and breaks, chromatin condenses along the nucleus, typical cytoplasmic organelles do not change in ultrastructure. However, the plasma membrane does not break. Eventually, the cell fragments were swallowed by phagocytes, and were degraded by proteases and nucleases in lysosomes after being transported to lysosomes, which prevented the inflammatory contents of cells from being released to the surrounding areas and was beneficial to tissue renewal [15]. Therefore, normal apoptosis as a non-lytic morphology of the neutrophil cell death can maintain the stability of the organism.
Currently there are two known pathways of apoptosis, endogenous pathway and exogenous pathway. Endogenous apoptosis refers to the imbalance of intracellular homeostasis caused by the changes of microenvironment such as stimulation of toxic substances, DNA damage and secretion of growth factors, which leads to the increase of permeability of mitochondrial outer membrane and entrance of Cytochrome C (Cyt-C) to the cytoplasm to activate the caspase cascade of apoptosis [16]. The endogenous pathway is usually controlled by Bcl-2 protein family. Some proteins (such as Bax and Bid) promote the occurrence of apoptosis while some proteins (such as Bcl-2) have the opposite effect. It is the balance between these pro-apoptotic proteins and anti-apoptotic proteins that ensures the normal progress of apoptosis [17]. Exogenous pathway refers to the pathway used by cytotoxic T lymphocytes (CTL) and natural killer cells (NK) to attack their target cells, which is initiated by the activation of tumor necrosis factor-α (TNF-α), Fas ligand and TNF-related apoptosis-inducing ligand (TRAIL) receptors DR4 and DR5 located on the cell surface.
As a non-lytic neutrophil cell death pathway, normal apoptosis is considered to be an effective strategy to avoid tissue damage which can eliminate neutrophil and prevent neutrophil from secreting large amounts of inflammatory cytokines [13, 18]. Nevertheless, recent studies show that abnormal apoptosis are associated with autoimmune diseases.
Some autoimmune diseases are associated with the untimely clearance of apoptotic neutrophil, where the phagocytic clearance of apoptotic neutrophil is impaired, thus triggering the autoantigenic clusters that maintain autoimmune disease. However, it has been also shown that some autoimmune diseases exhibit delayed apoptosis of neutrophil, resulting in the accumulation of neutrophil that can lead to the release of inflammatory factors and cytokines and the recruitment to other immune cells, which leads to inflammation eventually. In conclusion, abnormal neutrophil apoptosis is likely to be the underlying cause of some autoimmune diseases.
Disrupted clearance mechanisms of apoptotic neutrophils in autoimmune pathology
In recent years, many targets involved in neutrophil death have been extensively studied, which have been certified to be related to the development of autoimmune diseases, especially SLE. This section will explore the relationship between apoptotic neutrophil clearance and autoimmune diseases, focusing on the ‘key players’ in apoptotic neutrophil clearance.
Autoantigen protease 3 (PR3), also known as myeloblastin, is a kind of serine protease existed in neutrophil azurophilic granules, which can regulate granulocyte differentiation [19]. It degrades a variety of extracellular matrices such as elastin, haemoglobin, collagen type IV and many other tissue components. Generally speaking, the expression of PR3 on cell surface is increased in apoptotic neutrophil, which limits their phagocytic clearance by macrophages and promotes a pro-inflammatory microenvironment [20,21,22]. In recent research, PR3 is considered as the target for ANCA in granulomatosis with polyangiitis (GPA) [23, 24]. Studies have shown that PR3 enhances the degree of the occurrence of certain autoimmune diseases such as GPA and RA [19].
The efferocytosis of macrophages prevent the release of intracellular substances and the autoimmune response of body to its own antigens. If efferocytosis is impaired, it can lead to autoimmune diseases or tissue damage [25]. During apoptotic neutrophil death in GPA patients, increased expression of PR3 on the membrane induces abnormal phagocytosis of apoptotic neutrophils by macrophages thereby inducing an inflammatory response [19]. It has been shown that PR3 causes abnormal macrophage efferocytosis associated with calreticulin (CRT), an endoplasmic reticulum chaperone protein that releases a ‘eat me signal’ in the membrane of apoptotic neutrophils, thereby inducing macrophages to take up the apoptotic neutrophils. This promotes the regression of inflammation. During apoptotic neutrophil death in GPA patients, PR3 may block the binding of CRT to one of its major receptors, the low-density lipoprotein receptor-related protein (LRP), and the complex of PR3 and CRT may also impair the function of LRP and thereby disrupt the phagocytic function of macrophages [26], preventing the timely clearance of apoptotic neutrophils and ultimately amplify inflammation. Apoptotic neutrophils may also bind to cytokines at the site of inflammation, leading to autoimmune [23]. In patients with vasculitis, the combination between ANCA and PR3 also modulates neutrophil apoptosis and thus has an effect on phagocytosis. However, PR3-ANCA can lead to increased uptake by macrophages, resulting in TNF-α production to induce inflammation and autoimmunity.
Inflammation and autoimmunity through autoantibody modulation of apoptotic cells may be important not only in vasculitis but also in other autoimmune diseases [27]. Antiphospholipid syndrome (APS), SLE, may lead to inflammation through the same mechanism [28, 29]. Therefore, PR3 and its conjugates with other substances can not only modulate neutrophil apoptosis, but also have an impact on the clearance of neutrophils by macrophages and can be an important therapeutic target for certain autoimmune diseases (Table 1).
In fact, it can be seen from the relationship between PR3 and apoptotic neutrophil clearance that phagocytosis of macrophages plays an important role in the process of apoptotic cells clearance. Phagocytosis removes altered self-cells, especially apoptotic cells, and complement protein C1q is one of the mediators of phagocytosis which plays an important role in preventing the formation of antigen–antibody complexes and in the activation of the complement pathway [30].
C1q binds tightly to CD91 and CRT on the surface of apoptotic neutrophils, recognising apoptotic neutrophils and enabling phagocytes to perform their phagocytosis. In the presence of CRT, the addition of extracellular C1q accelerates the uptake of cellular debris. However, in patients with SLE, C1q and the bound proteins and receptors are absent or blocked, and anti-CRT and anti-C1q antibodies have been found in the serum [31]. The recognition of apoptotic cells by C1q is also affected by the aforementioned PR3, which is involved in the clearance of apoptotic neutrophils and the regulation of the immune response, and shares ligands such as CRT. It has been found that PR3 binds to C1q, thereby impairing C1q recognition of apoptotic neutrophils and resulting in defective clearance of apoptotic neutrophils [32]. In conclusion, C1q acts by promoting the clearance of apoptotic neutrophils, and when C1q is deficient or genetically mutated, it can lead to the development of autoimmune diseases such as SLE and RA [33]. The abnormal interaction between C1q, PR3 and CRT leads to the development of autoimmune diseases by delaying the clearance of apoptotic neutrophils, providing a new idea and therapeutic target for the treatment of autoimmune diseases characterized by defective clearance of apoptotic neutrophils.
In addition, CD44 is an adhesion molecule involved in extracellular cell matrix (ECM) interactions [34] and is widely expressed in a variety of tissues and involved in cell adhesion and migration, lymphocyte activation and proliferation [35]. The interaction of CD44 with hyaluronic acid (HA) has been extensively studied [36] and this action is thought to be closely related to the regulation of the immune response [37,38,39].In SLE, reduced expression of CD44 on the surface of neutrophils leads to failure of macrophages to clear apoptotic neutrophils, resulting in secondary necrosis and ultimately the release of autoantigenic clusters that can trigger autoimmunity [40, 41]. The ability of CD44 to promote cell clearance may be due to its association with cytoskeletal molecules, whereas in SLE, changes in the cytokine milieu such as the release of IL-10 lead to the downregulation of CD44, which results in impaired clearance of apoptotic neutrophils and promotes the development of autoimmune disease. It is easy to see that although CD44 plays an important role in immune regulation, the mechanisms that promote inflammation in different autoimmune diseases vary [35]. Therefore, understanding the role of CD44 in the development of inflammation and autoimmune diseases is essential for the use of this molecule as a drug target.
Overlived neutrophils make contribution to autoimmune disease
Over-activation or delayed apoptosis of neutrophils can lead to continued inflammation and amplification of the inflammatory response, which can lead to tissue damage [42, 43]. These processes occur in a variety of autoimmune diseases, of which RA is the most representative.
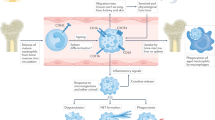
Programmed cell death-ligand 1 (PD-L1) is a ligand for programmed death 1 (PD-1) and is thought to be one of the key factors that inhibits T cell signaling, mediate tolerance mechanisms and provide immune homeostasis [44, 45]. Recent studies have revealed that PD-L1 can regulate neutrophil apoptosis and thus influence inflammation [46]. PD-L1 upregulates and interacts with the p85 subunit of PI3K to phosphorylate downstream Akt and delay neutrophil apoptosis, which in turn causes inflammatory responses and tissue damage [46]. As in RA, upregulation of PD-L1 causes delayed apoptosis of neutrophils, which results in massive infiltration of neutrophils in the synovium and contributes to inflammation. Monocyte chemotactic protein-inducible protein-1 (MCPIP-1), also known as Regnase-1, is a potent inhibitor of the inflammatory response [29], which appears to play an important role in neutrophil death. It can limit tissue damage by specifically targeting genes in different cell types through its ribonuclease activity and thereby reducing inflammatory factor storms [47]. MCPIP-1 controls the balance between anti- and pro-apoptotic proteins in neutrophils, thereby regulating their lifespan. MCPIP-1 is upregulated in dead neutrophils, but this effect is counteracted by GM-CSF, which causing a delay in neutrophil apoptosis, which leads to an excessive accumulation of neutrophils, resulting in inflammation and tissue damage, and even the development of autoimmune diseases [48]. In SLE, elevated type I interferon (IFN-I) expression promotes the expression of MCPIP-1, which in turn promotes reduced production of miR-146a, which has a suppressive autoimmune function [49]. However, therapeutic approaches targeting MCPIP-1 need to be cautious, as its complete inhibition or over-activation may produce detrimental immunopathological outcomes, and the search for therapeutic strategies with modulatory effects on MCPIP-1 could lead to promising results in autoimmune diseases. In addition, the chemokine-like inflammatory cytokine macrophage migration inhibitory factor (MIF) has been shown to be associated with neutrophil apoptosis, which contributes to the development of RA, MIF binds to its receptor CXCR2 and releases the survival factor CXCL8 from peripheral blood mononuclear cell (PBMC), thereby causing delayed neutrophil apoptosis and resulting in neutrophil infiltration, leading to inflammation [50]. It has also been shown that high intracellular K+ concentration or inhibition of K+ efflux inhibits neutrophil apoptosis by suppressing cytochrome c-dependent apoptotic vesicle production [51]. In patients with RA, neutrophils are exposed to pro-inflammatory mediators in the synovial fluid, thereby delaying apoptosis and contributing to the amplification and continued production of inflammation [52]. It follows that in some autoimmune diseases such as RA, inhibition of delayed neutrophil apoptosis and thus inflammation may be the key to reducing the symptoms of autoimmune disease (Fig. 1).

Yin-yang balance of neutrophil apoptosis. In healthy individuals, the processes of normal neutrophil apoptosis and clearance of apoptotic neutrophil debris are balanced, and this equilibrium is crucial for maintaining homeostasis. Disruption of this balance can lead to autoimmune diseases. There are two known apoptotic pathways, the endogenous and exogenous pathways, which regulate neutrophil function. Abnormal clearance of apoptotic neutrophils can trigger autoimmune diseases, such as SLE. In SLE patients, binding of C1q to CRT on the surface of apoptotic neutrophils is disrupted by binding of PR3 to CRT, preventing macrophage recruitment and resulting in accumulation of apoptotic neutrophils. Delayed neutrophil apoptosis is also a trigger of autoimmune diseases, with RA being a typical example. Inhibition of neutrophil apoptosis can result in inflammation due to infiltration and activation of inflammatory cells. Factors such as high intracellular potassium concentration, activation of TNFR1 and GM-CSF receptor, and inhibition of potassium efflux can lead to inhibition of apoptosis and inflammation
NETosis: neutrophil-specific way of death
Neutrophils as the most abundant innate immune effector cells in the human immune system are equipped with a wide range of effective antibacterial agents and are mainly stored in specialized granules. Neutrophils normally protect the host against the spread of bacteria and fungi, but if they over defend, they can damage the host tissue. Therefore, its deployment is regulated by three strategies: phagocytosis, degranulation and NETs release. NETs are fibrous assembly networks of nuclei and particles that are released in activated neutrophil-cell membranes and can inhibit and kill bacteria outside the cell. NETs consist of citrulline group proteins and highly decompressed chromatin fibers released in a process known as NETosis [53]. Most of the DNA in NETs comes from the nucleus and also contains mitochondrial DNA. NETs appear to be faceless: normally, they can capture and kill pathogens, but beneath this mask may be the evil face of autoimmune disease.
There are many important roles in the formation of NETs. The DNA that makes up NETs does not have active transcription levels but condenses into heterochromatin in the nucleus. Heterochromatin decompression is mediated by peptidylarginine deiminase 4 (PAD4), which catalyzes the conversion of histone arginine to citrullines, reducing the strong positive charge of histones and consequently weakening histone-DNA binding. This attenuated interaction then opens up the nucleosome and constitutes a prerequisite for NETs formation [54]. In addition to PAD4, NE is an essential member of NETosis, which cleaves histones during the formation of NETs. In addition, MPO can accept the stimulation of ROS and trigger the activation and transport of NE [53, 55]. Therefore, the search for treatments for NETs-related diseases can start from these closely related roles and explore new and effective treatments with NETs as the entry point.
As shown above, NETs contain histone and chromatin as well as important autoantigens, which increased NETs production, unique low density granulocyte populations and impaired NETs degradation leading to persistent inflammation and tissue damage related to autoimmune diseases. NETs can also activate other immune cells, and their components may amplify the inflammatory response by activating the complement pathway and inflammasome. NETs may also promote autoantibody formation in diseases such as RA, AAV and SLE by providing a constant source of autoantigens. Thus, NETs can be used as biomarkers to provide insights into disease diagnosis and treatment [56]. NETs are indeed strongly associated with a variety of autoimmune diseases, but the roles they play in the pathogenesis of various autoimmune diseases are changeable. We can understand that the same person plays a different role in different circumstances. Roles are different, but they can lead to seemingly the same result. In this review, we will focus on the role of NETs in several major autoimmune diseases. In addition, new drugs for the treatment of autoimmune diseases targeting NETs are summarized (Table 1).
NETs in SLE: chief culprit
SLE is a systemic autoimmune disease whose early pathological manifestations are mostly manifested in the skin and joints and the kidney, cardiovascular and other important organ systems are also affected. SLE patients will produce antinuclear antibodies (ANA) targeting a variety of nuclear components, such as anti-dsDNA antibodies, anti-ribosome antibodies, etc. Anti-dsDNA antibodies are highly specific and can form immune complexes with exposed chromatin to cause inflammation and promote disease [57]. Under normal conditions, a variety of dsDNA clearance mechanisms exist in cells. For example, dsDNA is rapidly degraded by DNA enzymes in the cytoplasm. Therefore, the failure of these mechanisms, such as inefficient clearance of apoptotic cell debris and NETs that are essentially DNA, will lead to the accumulation of dsDNA, inducing the production of autoantibodies and becoming the trigger of disease [58]. The relationship between NETs and SLE is not unfounded but traceable. Some researchers explored the effect of NETs on SLE using MRL/lpr mice which is a classic SLE mouse model and found that PAD inhibitors could reduce albuminuria and the deposition of immune complex in the kidney, which reduce the severity of skin diseases at the same time [59]. Immunocomplexes containing ribonucleoproteins (RNP IC) in lupus patients have also been shown to induce NETs. Simultaneously low-density granulocytes (LDG), a subset of proinflammatory neutrophils characteristic of SLE patients exhibit enhanced idiopathic NETs on biopsy [60].
Further investigation reveals that the role of NETs in SLE is closely related to IFN-I production. Researches show that SLE serum contains immune complexes of its own DNA, antimicrobial cationic peptide LL37 and human neutrophil peptide (HNP), which are protected from nuclease degradation and interact with the FC-γ receptor IIa on pDC to activate plasmacytoid dendritic cells (pDC) to release IFN-I [61]. However, NETs in SLE contain DNA and a large amount of LL37, which contribute to pDC production of high levels of IFN-α in a manner which is dependent on DNA and toll-like receptor 9 (TLR9) [62]. At the same time, there is a positive feedback regulatory mechanism for NETs in SLE, that is, mature SLE neutrophils are activated by IFN-I and die after exposure to SLE derived anti-ribonucleoprotein antibodies and release NETs [63]. The released NETs then activate pDC to produce IFN-I, which aggravates the progression of SLE. Meanwhile, the increased content of histone acetylation and methylation residues in NETs in SLE patients may indirectly increase the stimulative capacity of NETs by enhancing the binding of antimicrobial peptides in NETs [64]. In recent years, researchers have found that the mechanism by which NETs aggravate SLE is not limited to IFN-I. In SLE patients with endothelium dependent impaired vasodilation, matrix metalloproteinase-9 (MMP-9) and anti-MMP-9 immune complexes (IC) enhance NETs in LDG. Meanwhile, LDG-derived NETs are the source of MMP-9 activation in endothelial cells (EC), triggering endothelial dysfunction [65]. Although mitochondrial DNA and chromatin are present in NETs, other components such as RNA, especially siRNAs, may play a role in immune disorders and tissue damage. Studies have shown that extracellular LL37 can bind to extracellular endogenous RNA and transfer it to the DC endosomes to activate the IFN-I response via TLR7/TLR8 [66]. Meanwhile, abnormal downregulation of miR-4512 in monocytes and macrophages has been confirmed to promote the formation of NETs by targeting CXCL2 and TLR4 in SLE [67].
Delayed apoptosis of neutrophils and impaired clearance of NETs play similar roles in SLE on a macro level, both of which lead to the increase of anti-dsDNA antibodies and thus the occurrence of disease. This also reflects that although the mechanisms are different, different ways of dying of neutrophils may lead to the same outcome, and that there may be multiple ways of dying of neutrophils in the same autoimmune disease. And the same way of cell death will lead to different outcomes in different diseases through different pathways, perhaps this is also the significance of further research on the death of neutrophils.
NETs in AAV: mouth-watering prey
AAV is the general term for a class of multi-system autoimmune small vasculitis, including microscopic poly-vasculitis (MPA), eosinophilic granuloma with poly-vasculitis (EGPA) and GPA [68]. It is characterized by granuloma formation, arterioles and venules formation, and inflammation of capillaries. When blood vessels are in an inflammatory state, they may rupture or block causing systemic inflammation such as purpura rash and segmental glomerular infarction [69]. ANCA is mainly IgG autoantibodies against cytoplasmic components of neutrophils, which can bind to MPO and PR3 from neutrophils, and is considered to be a key marker to trigger the reaction of this disease [70]. Studies have shown that neutrophils and their interaction with ANCAs play a key role in the pathogenesis of AAV. Firstly, ANCAs activates neutrophils, which bind to antigens and attach to the antigen and Fcγ receptors on neutrophils activated by proinflammatory cytokines to promote respiratory burst and degranulation of neutrophils, aggravating AAV [71, 72]. NETs are released by ANCA-stimulated neutrophils and contain targeted autoantigens PR3 and MPO. The deposition of NETs and MPO-DNA complex in the inflammatory kidney after ANCA binding with PR3, MPO and other autoantigens suggests that the formation of NETs induces vasculitis and promotes the autoimmune response in patients with vasculitis [73]. Fragments of NETs were also found in the microvascular system of patients with small vasculitis, suggesting that NETs are pathogenic in AAV [74]. In MRL/lpr mice, myeloid dendritic cells (mDCs) loaded with NETs-derived neutrophil-extracellular DNA led to the production of MPO-ANCA and PR3-ANCA with vasculitis-like kidney injury. The pathogenicity of NETs is also demonstrated by NETs-mediated damage to the vascular endothelium and its surrounding tissues [75]. Meanwhile, citrullinated histone H3 (Cit-H3) is also a key component of NETs, and the presence of Cit-H3 positive neutrophils is a specific marker for NETs formation in AAV [76]. Further study revealed that high mobility group protein (HMGB1) was strongly associated with the NETs in AAV. HMBG1 increases ANCA translocation to regulate ANCA-mediated respiratory burst and neutrophil degranulation. In neutrophils treated with HMGB1 and ANCA positive IgG, HMBG1 can interact with TLR2 and TLR4 to mediate increased release of NETs in a NOX-dependent manner [77]. From the above description, it can be found that ANCA may promote the formation of NETs, which in turn aggravate the progression of AAV.
In fact, the specific mechanisms by which NETs facilitate the progress of AAV are not yet fully defined and comprehensive. It has been shown that ANCA-induced NETs activate alternative complement pathways to initiate inflammatory amplification cascades. Neutrophil cell membrane and neutrophil-derived particles can activate the alternative complement pathway, leading to C5a production. C5a can interact with C5a receptors of neutrophils to attract and activate neutrophils, forming an amplifying loop of ANCA activation of neutrophils [78]. At the same time, studies have shown that patients with AAV have a high incidence of venous thromboembolism events and are in a hypercoagulable state. C5a activates the coagulation system, and more importantly, both MPO-ANCA IgG and PR3-ANCA IgG stimulate C5A-modified neutrophils to produce NETs expressing tissue factor (TF), thus activating the coagulation system and aggravating AAV [79]. At the same time, multiple studies have shown that cellular immunoglobulin can affect NETs and thus aggravate the progression of AAV. For example, T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3) have been reported to be important regulatory factors of T cells. Studies have found that the expression of TIM-3 in DCs of active MPO-AAV patients is significantly decreased, while the expression of TLR4 is significantly increased. Blocking TIM-3 can further enhance the expression of NETs mediated DC cytokines [80]. It has also been shown that platelets stimulated by the TLR pathway can induce the formation of NETs. TLR9-stimulated platelets release CXCL4 significantly in AAV and subsequently increase the formation of NETs. In addition, neutralizing anti-CXCl4 antibodies significantly inhibited AAV platelet-enhanced NETs formation. TLR9 signal transduction and CXCL4 release underlie the critical role of platelets in NETs formation in the pathogenesis of AAV [81].
Therefore, if the role of NETs in SLE is the ‘culprit’, the role of NETs in AAV is a prey precisely targeted by autoantibodies. PR3, which constitutes NETs, binds to MPO and ANCA to activate neutrophilic degranulation and promote the production of NETs. At the same time, NETs directly cause vascular inflammation by destroying endothelial cells and activating the complement system, and indirectly act as a link between the innate and adaptive immune systems by producing PR3-ANCA and MPO-ANCA, creating a vicious cycle [82].
NETs in RA: danger maker
RA is an autoimmune disease and a chronic joint disease characterized by synovial inflammation, destruction of articular cartilage and production of anti-citrullinated peptide antibodies (ACPAs). The specific mechanism leading to the development of RA is not fully understood, but the formation of ACPAs is considered to be the key pathogenic factor. PAD2 and PAD4 may produce these citrulline antigens and are detected in the synovium of RA, which is closely associated with neutrophil infiltration [83]. Neutrophils as a member of innate immune cells infiltrate extensively in the joint cavity of patients with RA, in which neutrophils occupy a position that can not be ignored. NETs as an inflammatory mode of neutrophil death undoubtedly exacerbate the progression of RA. The histone citrullination catalyzed by PAD4 is a key step in NETs, in which citrullinated histones are neutralized.
It has been demonstrated that neutrophils externalize citrulline autoantigens associated with RA pathogenesis during the development of NETs which strongly induce the formation of NETs. In fact, NETs are in a positive feedback mechanism in RA. The inflammatory cytokines interleukin-17A (IL-17A) and TNF-αinduce the production of NETs in RA, and NETs induce the production of IL-6, IL-8, chemokines, and adhesion molecules, which aggravate the inflammatory response through the externalization of citrulline autoantigens and immune-stimulating molecules, which may promote abnormal joint and peripheral adaptations and innate immune responses and perpetuate the pathogenesis of the disease. In summary, the primary role of NETs in RA is that of the primary provider of autoantigens, in other words, NETs is a danger maker. Not only does PAD4 promote NETs participation in positive feedback regulatory mechanisms in RA, but other NETs-related proteases also show their skills. Neutrophils in blood were abnormally activated in RA, which increased the production of ROS and cytokines. At the same time, the expression levels of MPO, NE and lactoferrin related to NETs increased in synovial fluid (SF). These granule proteins can divide and activate cytokines and chemokines, who cause the degradation of cartilage and promote the production of RA [84]. Joanna Clarke et al. studied the direct impact of NETs on RA. They believe that NE can not only degrade cartilage, but stimulate the formation of downstream inflammation mediated by fibroblast like synovial cells (FLS), and promote the release of PAD2 in FLS. Autoantibodies such as anti-citrulline protein antibodies in RA will activate macrophages to release proinflammatory cytokines and activate neutrophils in synovium, so as to release more NE and aggravate joints inflammation. The NE of NETs aggravates the joint inflammation, and the joint inflammation in turn promotes the production of NE, the two promote and influence each other [85].
In recent years, researchers have made a detailed analysis of the types and proteomics of NETs in RA, which provides a basis for the pathological diagnosis of RA. Elinor A. Chapman and others found that ROS required for the production of NETs come from two ways, one from the activation of NOX2 and the other dependent on mitochondria. The occurrence of RA is NOX2 independent NETs. At the same time, it was found that many NETs proteins increased in RA synovial fluid, including cathepsin, MPO, MMP8, MMP9, Lcn2 and PADI2. These proteins contribute to the cellular infiltration and degradation of collagen in synovial joint. It provides a basis for predicting the severity and cure of RA patients [86].
NETs emerge as multi-faceted adversaries: unleashing a cascade of autoimmune disorders
Focusing on the skin, psoriasis is a chronic systemic inflammatory disease, which affects not only the skin, but various internal organs. One of the important signs of psoriasis is the infiltration of epidermal neutrophils, which will lead to the occurrence of NETs [87]. There have been many studies on the occurrence and pro-inflammatory mechanism of NETs in psoriasis. Neutrophils amplify skin inflammation by releasing NETs and activating IL-36 and TLR4 signals, and further increase the infiltration of neutrophils into the skin [88]. Other studies have shown that neutrophils and NETs can promote inflammation through several different mechanisms, such as the activation of inflammasome, triggering TLR7 and TLR9 through self-antigen complexes (such as LL-37-DNA), promoting macrophage apoptosis, or processing and activating IL-36 cytokines. At the same time, NETs also contain RNA. In previous studies, DNA research in NETs is more extensive. However, it is ignored that RNA is also a rich part of mammalian NETs. The RNA associated with NETs may be the effective component of RNA-LL37 and PMA triggering NETs [89]. In addition, for Stevens Johnson Syndrome (SJS) and toxic epidermal necrolysis (TEN), Manao Kinoshita et al. found that CD8+ T cells that trigger skin infiltration in the early stage of inflammation produce lipocalin-2, which triggers the formation of NETs in the early damaged skin, and then releases LL-37, which induces keratinocytes to express formyl peptide receptor 1 (FPR1), resulting in cell necrosis [90].
With a specific focus on the digestive system, IBD represents an autoimmune condition characterized by inflammation and damage to the gastrointestinal tract, where NETs also play a role. A recent investigation demonstrated elevated levels of NETs in both the plasma and colon tissue of individuals diagnosed with IBD, thereby indicating the potential involvement of NETs in the onset and progression of this disease. Notably, IBD is associated with an increased risk of thrombosis and heightened hypercoagulability. To mitigate disease progression, the administration of DNase has been shown to impede the formation of NETs, reduce the release of thrombogenic cytokine mediators by macrophages, and directly inhibit platelet activation [91]. Histones, constituents of NETs, possess the ability to directly impair the permeability and structural integrity of the intestinal epithelium. Experimental evidence using monolayers of Caco-2 intestinal cells demonstrates that PAD4 mediates the disruption of epithelial permeability by causing the degradation of zonula occludens protein 1 (ZO-1) and adhesion junction protein E-cadherin. This process ultimately triggers apoptosis and contributes to the progression of IBD. Nevertheless, the precise underlying mechanism of this phenomenon remains to be fully elucidated [92]. In the context of UC and CD, the involvement and specific functionality of NETs exhibit subtle distinctions. Investigations have revealed significantly elevated expression levels of NETs-associated proteins, including Cit-H3, PAD4, MPO, and NE in the colonic tissue of UC patients when compared to both healthy individuals and CD patients. Notably, the production of NETs in UC patients is stimulated by the presence of TNF-α that activates circulating neutrophils. Moreover, the activation of extracellular signal-regulated kinase 1/2 (ERK1/2) in lamina propria mononuclear cells (LPMC) in UC patients ultimately amplifies the production of TNF-α and IL-1β, thereby exacerbating the disease process [93]. The use of PAD4 inhibitors can reduce the production of NETs and improve DSS induced acute enteritis [94]. Intriguingly, emerging evidence suggests that achieving a balanced state rather than complete elimination of NETs may be beneficial in patients with UC. Rectal bleeding has long been recognized as a prominent clinical manifestation in UC patients; however, the formation of fibrin and neutrophil-generated immune clots provides a protective mechanism by stabilizing the compromised mucosal tissue and effectively halting bleeding. In this intricate process, PAD4 facilitates the condensation of neutrophil chromatin, whereby the outer chromatin of NETs impedes blood vessel patency and further aggregates to fulfill a hemostatic function. Hence, NETs play a vital protective role in the context of UC-related bleeding [95]. While numerous proteins associated with NETs are more commonly observed in UC than in CD, there is evidence indicating a specific correlation between NETs and CD. Essential constituents of NETs, including Cit-H3, NE, and MPO, can indeed be detected in stool samples from CD patients, suggesting their potential involvement in the pathogenesis of CD. Furthermore, additional studies have demonstrated enhanced expression of Cit-H3 and PAD4 in mice with 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis, further supporting the role of NETs in CD pathogenesis. Notably, the administration of PAD4 inhibitor Cl-amidine effectively mitigated the clinical colitis index and reduced the expression of pro-inflammatory factors in mice, indicating its therapeutic potential in CD [96]. The production of NETs is positively correlated with the severity of CD, which may be caused by the excessive production of bactericidal oxidant hypochlorous acid (HOCl) in the extracellular region mediated by MPO, which leads to host tissue damage and intensification of CD. Perianal fistulising Crohn's disease (pfCD) is a disabling phenotype of CD. Studies have shown the presence of NETs in perianal fistulas, increased production of NETs is associated with enhanced TNF-α production and adverse fistula remission [97]. In conclusion, the excessive generation of NETs worsens the onset and progression of CD and hampers its recovery. Furthermore, while MPO and NE have been identified as serum biomarkers for diagnosis and prognosis in patients with CD and UC, ANCAs targeting PR3 are more commonly detected in UC compared to CD, making them a reliable means of distinguishing between the two diseases.
Regarding the nervous system, emerging evidence indicates the involvement of neutrophils in the pathogenesis of MS. However, unlike in other diseases, neutrophils do not prominently appear in central nervous system tissue biopsies of MS patients. This discrepancy may be attributed to the transient nature of neutrophils as short-lived cells, which primarily respond to specific disease stages such as disease onset or relapse [98]. Due to this characteristic, the investigation of NETs in MS remains inadequate. Nevertheless, several studies have identified correlations between neutrophil-associated factors, such as NE, CXCL1, and CXCL5 in plasma, and the severity of the disease. Additionally, elevated levels of MPO have been significantly observed in the serum of individuals with MS [99]. Moreover, studies have shown that the serum NETs level in relapsing–remitting (RR)-MS (RRMS) patients is higher than that in other patients with MS pressure type, and the frequency in males belonging to this subtype is significantly higher than that in females, suggesting that NETs may be related to some sex differences in this disease. At the same time, NETs may affect the process of RRMS through T cell immune regulation [100].
Irrespective of individual organs and systems, the investigation of systemic autoimmune diseases has increasingly focused on the physiological activity and impact of NETs in SSC. Apart from RA and SLE, substantial levels of anti-NET antibodies (ANETA) and fragments of NETs have also been identified in the serum of SSC patients. Notably, a gradual reduction in NETs formation was observed during the early stages of the disease, whereas patients with severe and chronic vascular complications exhibited persistently elevated levels of NETs throughout the course of the disease. Additionally, DNA filaments have been detected in skin biopsies of SSC patients, potentially representing NETs. These NETs are thought to undergo modifications by CXCL4, leading to the formation of CXCL4-DNA complexes that contribute to immune activation and the production of IFN-I in individuals with early active SSC [101]. The interaction between platelets and neutrophils plays a crucial role in SSC, enhancing the ability of white blood cells to extravasate from blood vessels and migrate to inflamed tissues, while also promoting the generation of NETs. When phagocytosis is impaired, activated platelets evade clearance mechanisms, leading to the buildup of microparticles (µPs), triggering NETs formation and contributing to vascular damage. The substantial decrease in P-selectin (PSGL-1) expression observed in SSC increases the likelihood of phagocytes being unable to recognize and engulf platelets, thus resulting in sustained µPs release. Therefore, the elimination of activated platelets via PSGL-1 may serve to restrict µPs accumulation and NETs production, ultimately reducing the progression of the disease [102].
The impact of NETs on APS is also considered impossible to ignore. APS is an autoimmune thromboinflammatory disease, which is characterized by the formation of arteriovenous or microvascular thrombosis and poor pregnancy. There are a variety of autoantibodies, such as an-tiphospholipid antibody (aPL), an-ticardiolipin antibody (aCL). APL makes the normal steady-state mechanism on the surface of neutrophils destroyed and directly triggers the release of NETs [103]. High levels of anti-NETs IgG can damage the clearance of NETs, which is also closely related to the formation of venous thrombosis [104]. Pregnancy complications are also the main clinical manifestation when APS occurs. Yuan Lu et al. found that the markers of NETs increased in pregnant women with APS, and NETs may lead to placental defects. This direction may provide a new target for the treatment of the disease [105].
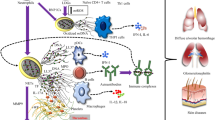
Other autoimmune diseases, such as autoimmune diseases in the kidney, can also see the accumulation of NETs. At the same time, studies have shown that NETs can lead to a vicious circle [106]. It can be seen that NETs are closely related to autoimmune diseases, and the treatment of autoimmune diseases for NETs is worthy of in-depth study (Fig. 2).

The role of NETs in various autoimmune diseases. NETs are closely related to a variety of autoimmune diseases, but the role they play in the pathogenesis of various autoimmune diseases is variable. In SLE, immune complexes such as LL37 and HNP protect the DNA in NETs from nuclease degradation, which induces anti-dsDNA autoantibodies and releases I IFN with pDC to trigger SLE. In AAV, NETs are released by ANCA-stimulated neutrophils and contain targeted autoantigens PR3 and MPO. After ANCA binds to autoantigens such as PR3 and MPO, NETs and MPO-DNA complexes are deposited in inflammatory sites and promote the autoimmune response in patients with vasculitis. The formation of NETs is catalyzed by PADs, in which the arginine residues of the protein are deaminated to citrullinated proteins. Therefore, the production of NETs promotes the production of ACPAs and leads to the production of inflammatory factors and adhesion proteins in RA. If NETs are the chief culprit in SLE, NETs are prey and danger maker in AAV and RA
Neutrophil necroptosis: another potential mechanism contributing to autoimmune pathogenesis
Neutrophil necroptosis is a form of regulated necroptosis that is triggered by RIPK3-MLKL-dependent necroptosis when cells are stimulated by TNF, adhesion receptors, monosodium urate (MSU) crystals or staphylococcus aureus [107]. Necroptosis is similar to other forms of death such as NETs, which are capable of causing inflammation and immune damage. RIPK3-MLKL signalling can work with p38 MAPK and RIPK3-MLKL signalling can activate NOX together with p38 MAPK and PI3K, resulting in ROS production and non-apoptotic cell death [107]. Neutrophil necroptosis has been implicated as a mode of death that triggers inflammation because this mode of cell death results in the release of toxic cellular contents, namely DAMP, which cause tissue damage and induce an inflammatory response [107, 108]. Recently, neutrophil necroptosis has been found in autoimmune diseases such as psoriasis and RA [109]. This suggests that the study of neutrophil necroptosis may provide new ideas for the treatment of autoimmune diseases. The following section will focus on the contribution of neutrophil necroptosis to inflammation in patients with RA.
Neutrophil necroptosis triggers RA through different pathways
Neutrophil necroptosis is essentially a form of programmed death mediated by RIPK1, RIPK3, and MLKL that involves the release of intracellular toxic contents that mediate the onset of inflammation in RA [110].
In RA patients, neutrophil exposure to GM-CSF activates RIPK1, RIPK3, and MLKL results in necroptosis [111], followed by attachment of adhesion receptors [112]. It was found that CD44 attached to GM-CSF-stimulated neutrophils induced necroptosis, and interestingly, CD44 attached to neutrophils isolated from the synovial fluid of RA patients in the absence of GM-CSF stimulation also triggered necroptosis, which could indicate that CD44 can trigger necroptosis in neutrophils in the presence of inflammation. granulocyte necroptosis under inflammatory conditions [113].
Fibroblast-activated protein-a (FAP-a) also plays an important role in the development of necroptosis in neutrophils from RA patients [114]. Neutrophils isolated in the joints of RA patients undergo GM-CSF non-dependent necroptosis after CD44 clearance and this effect can be blocked by the FAP-a inhibitor, MLKL. It was found that FAP-a was activated in parallel with RIPK3-MLKKL-p38 MAPK signalling and that its activity was required for CD44 to trigger ROS production and subsequent cellular necroptosis. Thus, FAP-a is potentially a key target for the inhibition of neutrophil necroptosis in RA [108].
Indeed, in addition to RA, neutrophil necroptosis has been identified in patients with a variety of autoimmune diseases, and because neutrophil adhesion molecules can trigger necrosis under inflammatory conditions, vacuolisation of the neutrophil cytoplasm has been found in inflamed tissue from patients with psoriasis [112]. In AAV, ANCA induces necroptosis in neutrophils, and necroptosis inhibitors can also prevent AAV altogether [115]. Currently, RIPK1 inhibitors are in phase II clinical trials and are being investigated for the treatment of autoimmune diseases such as psoriasis, UC and RA [116, 117].
Neutrophil necroptosis and NETs: intertwined patterns of neutrophil death
Though necroptosis and NETs are non-apoptotic deaths which lead to the release of toxic contents from neutrophils into the extracellular space causing damage to surrounding tissues, the mechanisms of necroptosis and NETs are not the same. However, there appears to be an intrinsic link between them.
Studies have shown that PMA or MSU crystals can trigger the production of NETs in the presence of neutrophil necroptosis, while the absence or inhibition of necroptosis by PIPK3 also inhibits the induction of NETs by MSU crystals [118, 119], and that PIPK3-dependent necroptosis is an upstream activator of NETs. However, necroptosis does not inhibit all NETs, it may be able to control the formation of NETs induced by MSU crystals, but it remains controversial whether there is a role for NETs formed by PMA induction [115]. Outside of this phenomenon, however, NETs have been identified through studies independent of RIPK3 and MLKL, and one possible explanation is that the so-called NETs that occur during necroptosis are actually passive releases of chromatin associated with necrosis, whereas the formation of true NETs is not dependent on the occurrence of necroptosis [107]. It is undeniable that there is an underlying link between NETs and necroptosis, and it is the intertwining and balancing of these death pathways that determines the ultimate fate of neutrophils. Perhaps a deeper understanding of the love-hate relationship between neutrophil death modalities may also provide a novel way to explore the mechanisms of autoimmune disease development.
Neutrophil pyroptosis: a novel pathological mechanism in autoimmune disorders
Pyroptosis is a programmed death process associated with inflammation and has recently been extensively studied by researchers. Cells undergoing pyroptosis form pyroptosis vesicles similar in size to apoptotic vesicles and are accompanied by chromatin sequestration and damage and the formation of numerous voids in the cell membrane, ultimately causing lysis of the cell membrane and the release of intracellular contents leading to inflammation [13, 120]. NLRP3 inflammasome, caspase-1, GSDMD, IL-1β and IL-18 play key roles in the development of pyroptosis [121]. NLRP3 inflammasome are sensors that activate caspase-1, which can be activated by factors such as ROS, and when caspase-1 is activated by inflammatory vesicles, the caspase-1 then cleaves pro-IL-1β and pro-IL-18 to produce IL-1β and IL-18, which in turn cause inflammation to occur [122, 123].
Pyroptosis was firstly detected in macrophages and related diseases, followed by numerous reports confirming that neutrophils can also undergo pyroptosis, which is closely related to the immunity of body. In the early stages of sepsis, neutrophil pyroptosis can act as an amplifier of inflammatory signals and thus recruit immune cells, which may have a positive impact on the treatment of sepsis. During infection, neutrophil pyroptosis is particularly important for bacterial clearance [120]. In but some studies have indicated that neutrophils have a specific resistance to pyroptosis and that the pathway for IL-1β production is somewhat different from the classical pyroptosis pathway. While macrophages, for example, undergo pyroptosis mainly by rupture of the plasma membrane (PM) via GSDMD, the cleavage of N-GSDMD during NLRP3 inflammasome signalling in neutrophils is mainly associated with abundant intracellular organelle membranes. In neutrophils activated by NLRP3, instead of being transported to the plasma membrane, N-GSDMD protein is transported to azurophilic granules and autophagosomes, which ultimately release IL-1β via an autophagy-dependent pathway [124]. Therefore, the mechanism of pyroptosis in neutrophils is not clear, which provides an important direction for research. Instead, it has recently become clear that neutrophils can undergo this form of death when lacking the functional NADPH oxidase Nox2 [121, 123]. However, recent studies have found that methicillin-resistant staphylococcus aureus (MRSA) uses neutrophil necroptosis with pyroptosis to trigger IL-1β production. Neutrophil pyroptosis may not act alone [13].
The role of inflammasome activation in the pathogenesis of rheumatic autoimmune diseases
A variety of typical inflammatory vesicles such as NLRP1, NLRP3, AIM2 and NLRC4 have been identified in a variety of rheumatic autoimmune diseases. In RA, NLRP3 inflammasome mediate the activation of IL-1β production, which leads to inflammation [125]. In SLE, anti-U1-snRNP autoantibodies are able to activate NLRP3 inflammasome in human monocytes, leading to IL-1β production [126, 127]. In macrophages from SLE patients, NETs-mediated activation of NLRP3 inflammasomes is significantly enhanced [128]. Also, in ankylosing spondylitis (AS), IL-1β production was highly induced, suggesting that inflammatory vesicles may be activated and induce IL-1β production in AS [129]. It is not difficult to speculate on the important role of pyroptosis in autoimmune disease in these studies[121], it is also worthwhile to explore in depth the role of pyroptosis as a neutrophil critical for inflammation in the development of autoimmune disease.
Pyroptosis-related elements in other modes of neutrophil death
It has been found that GSDMD, NLRP3, which plays an important role in pyroptosis, also contributes to inflammation and autoimmune diseases by participating in, or being activated by, multiple modes of neutrophil death [125, 130]. In adult Still's disease (AOSD), DNA released by NETs activates NLRP3 inflammasome and increases the expression levels of IL-1β and IL-18 [131].
GSDMD brings together two death pathways, pyroptosis and NETs, and GSDMD is cleaved by NE to drive NETs [124, 132]. the formation of NETs requires GSDMD, while in pyroptosis GSDMD is cleaved, resulting in the formation of membrane pores [133]. In summary, GSDMD is the pro-inflammatory cell pivotal to death. Also, GSDMD is thought to promote neutrophil death and GSDMD deficiency may enhance host defence by delaying neutrophil death [134]. In fact, neutrophil pyroptosis has been associated with autoimmune diseases. Clinical experiments showed that the expressions of NLRP3 and apoptosis associated speck-like protein containing a CARD (ASC) and pro-caspase-1 in neutrophils of RA patients were significantly decreased. The expression of activated caspase-1 protein was significantly increased. Overactivation of caspase-1 in RA neutrophils may mediate IL-18 activation, thereby contributing to RA progression in an NLRP3 inflammasome-independent manner [135].
Currently, death modalities such as pyroptosis and necroptosis are considered to be inflammation-associated death modalities. Perhaps, by inhibiting these inflammation-related death modalities or converting these death pathways to non-inflammatory death, such as apoptosis, may also be a strategy to treat autoimmune diseases. It is therefore particularly important to explore the mechanisms that trigger the various modes of death of neutrophils in autoimmune diseases.
Neutrophil ferroptosis in autoimmune diseases: a new type of neutrophil death mode has emerged
Ferroptosis is a recently discovered form of non-apoptotic cell death that causes disease through iron-catalyzed oxidation of unsaturated fatty acids and thus the accumulation of ROS [136]. Several studies have found a close association between ferroptosis and the development of tumors and other degenerative diseases [137,138,139,140,141], as well as in autoimmune diseases [142]. Abnormalities in cellular ferroptosis have been seen in autoimmune-mediated hepatitis (AIH), but the exact mechanism of occurrence remains unclear. In the GPX4± mouse high glucose and high fat diet model, INOS is significantly more upregulated than in WT mice [143], which indirectly suggests that ferroptosis promotes the production of INOS and thus contributes to AIH [144]. Further, neutrophil ferroptosis is also closely associated with autoimmune diseases. IFN-α and SLE IgG inhibit the transcription of GPX4 by promoting the binding of CREM to the GPX4 promoter, thereby increasing the production of lipid peroxides and inducing the development of neutrophil ferroptosis, thus contributing to the development of SLE. the development of SLE. This places neutrophil ferroptosis at the centre of SLE pathogenesis and highlights the importance of targeting GPX4 transcription and neutrophil ferroptosis in the treatment of SLE [145, 146].
At present, research on ferroptosis in autoimmune diseases is uncommon and in-depth, and the role of neutrophil ferroptosis in autoimmune diseases is not yet clear. However, from the above-mentioned article on neutrophil ferroptosis in SLE published in Nature, it appears that this newly discovered mode of death of neutrophils, which play an important immune role in autoimmune diseases, may indeed exist. This clearly provides us with new research and therapeutic ideas, and the investigation of neutrophil death patterns in autoimmune diseases continues.
Autophagy: the entanglement between survival and death mechanisms
In addition to various forms of cell death, investigating the normal physiological function and survival mecihansms of neutrophils is crucial for understanding neutrophil death. Autophagy is a pivotal mechanism that plays a role in multiple aspects of neutrophil biology and pathophysiology. Not only does it contribute to cellular homeostasis, but its interplay with different modes of neutrophil death also influences neutrophil survival and its potential association with autoimmune diseases.
Autophagy is a process in which eukaryotic cells use lysosomes to degrade cytoplasmic proteins and damaged organelles under the regulation of autophagy related gene (Atg) [13]. Autophagy exhibits significant upregulation in response to oxidative stress, nutritional deficiencies, and diverse antigenic stimuli, including pathogens, in order to ensure cellular survival. While autophagy provides energy and supports cell survival, dysregulated or uncontrolled autophagy can also result in autophagy-dependent cell death, leading to pathological conditions. In summary, autophagy plays a critical role in cellular survival [147]. Autophagy is a mechanism of survival whose function is momentous in the birth and death of neutrophils. It has been found that autophagy can dynamically regulate different stages of neutrophil differentiation in vivo. The accumulation of immature neutrophils in mice with conditionally knocked out Atg7 in hematopoietic stem cells (HSCs) and progenitor cells demonstrated that inhibition of autophagy delayed neutrophilic differentiation [148]. At the same time, in the Atg7 deficient neutrophil precursors, the glycolytic activity increased, but the mitochondrial function impaired, ATP production decreased, and lipid droplets accumulate, suggesting that free fatty acids provided by autophagy are particularly important for the differentiation of neutrophil [149]. However, autophagy can only regulate the number of neutrophils at the production level, but not by regulating the apoptosis of mature neutrophils.
In addition to the growth and development of neutrophils, autophagy is also essential for major neutrophil functions, including degranulation, reactive oxygen species production and release of neutrophil extracellular traps. Degranulation is a process in which neutrophils regulate the secretion of granulocytes, and it is one of the main mechanisms of neutrophilic inflammatory response. It was found that mice with myeloid specific autophagy deficiency showed reduced degranulation both in vivo and in vitro, and showed reduced severity of neutrophil-mediated inflammation and autoimmune diseases. At the same time, NADPH oxidation-mediated ROS production is also reduced in autophagy deficient neutrophils, and inhibition of NADPH oxidase reduces neutrophils degranulation, which not only suggests that autophagy can control neutrophils degranulation, but also proves that NADPH oxidase is the intersection of autophagy and degranulation [150].
In addition to its role in neutrophil degranulation, autophagy exhibits a profound interplay with NETs, forming a complex relationship that contributes to the development of various autoimmune diseases, acting as a critical tipping point. Notably, ROS, serving as a trigger for NETs formation, displays an intricate and reciprocal dependency on autophagy. ROS induction can stimulate autophagy, while autophagy, in turn, becomes a crucial prerequisite for ROS generation. Consequently, inhibition of either autophagy or NADPH oxidase effectively hampers chromatin condensation, ultimately impeding the formation of NETs [147]. The interconnection between autophagy and NETs is vividly exemplified by their convergence. It has been observed that activated platelets exhibit a robust capacity to transfer potent HMGB1 molecules to neutrophils, thereby acting as formidable inducers of NETs. Moreover, this potent NETs inducer simultaneously triggers the activation of autophagy, further highlighting the intricate relationship between these two processes [151]. This evidence not only proves that platelets can affect the formation of NETs, but also reveals the inevitable relationship between NETs and autophagy. Interestingly, heparin as a conventional anticoagulant and antithrombotic agent interferes with the interaction between neutrophils and platelets, thereby inhibiting their eventual activation. low molecular weight heparins (LMWH) in prophylactic dose can inhibit the autophagy of peripheral neutrophils and NETs produced under inflammatory stimulation [152]. Meanwhile, chloroquine (CQ), a traditional treatment for malaria, is also a classic autophagy inhibitor, which can impair autophagosome lysosome fusion and degradation of autophagosome contents. It was found that CQ significantly reduced serum free DNA and Cit-H3 in mice with pancreatitis as well as significantly inhibited the formation of NETs [153]. Autophagy inhibitor 5′-(4-fluorosulfonylbenzoyl) and knockout of Atg7 inhibit LC3 autophagy formation and also significantly reduce the production of NETs in promyelocytes [154]. The interplay between autophagy and NETs offers novel therapeutic prospects for addressing inflammation and autoimmune diseases, thereby underscoring the potential involvement of autophagy in other forms of neutrophil death, thereby showcasing its impact on autoimmune diseases. Initially, autophagy, serving as a survival mechanism, is inherently linked with apoptosis, recognized as an essential cellular pathway. Polymorphonuclear neutrophils (PMN), although short-lived, exhibit evidence of prolonged survival of Staphylococcus aureus within their confines, thereby facilitating pathogen persistence during infection. This survival mechanism employed by the bacterium within PMN primarily relies on the inhibition of neutrophil apoptosis through autophagy promotion and the upregulation of anti-apoptotic genes (mcl-1, bcl-2). Conversely, restoration of normal apoptosis levels in infected PMN is achieved through the inhibition of autophagy. Thus, a compelling association between autophagy and apoptosis in PMN undoubtedly exists [155]. At the same time, autophagy inhibitors 3-methyl-adenine (MA) and CQ can significantly delay TNF-α-induced neutrophils apoptosis and significantly down-regulate the expression of mcl-1 [156]. The relationship between granulocyte autophagy and pyroptosis seems to be more interesting and special. In fact, the pyroptosis of neutrophils is different from that of canonical and non-canonical macrophages, and autophagy is involved in the pyroptosis of neutrophils. In neutrophils, N-GSDMD does not accumulate in large quantities on the plasma membrane, but is transported to azurophilic (primary) granules and autophagosomes. At the same time, it was found that the secretion of IL-1β was impaired in Atg7−/− mouse neutrophils, and the inhibition of autophagy by VPS34-IN1 and the blocking of autophagy by BAFA1 significantly reduced the release of IL-1β, suggesting that neutrophils release IL-1β through an autophagy dependent pathway [124]. This process may be that IL-1β enters lumen of a vesicle and then becomes autophagosome, and finally autophagosome containing IL-1β can directly fuse with the plasma membrane and be secreted into the extracellular [157].
The primary objective of investigating the correlation between autophagy and neutrophil death lies in the quest for novel targets and therapeutic interventions for various diseases. The intricate interplay between autophagy and alternative forms of neutrophil death has been implicated in the pathogenesis of numerous autoimmune disorders. Notably, investigations have revealed elevated levels of autophagy in neutrophils of SLE patients. Among the pivotal players in this context, the REDD1 protein assumes a critical role in maintaining energy homeostasis and orchestrating multiple inflammatory pathways. Furthermore, endothelin-1 (ET-1) and hypoxia-inducible factor-1α (HIF-1α) act as inducers of autophagy, culminating in the release of NETs through the upregulation of REDD1, thus serving as inflammatory triggers that exacerbate the progression of SLE [158]. Similarly, REDD1 was highly expressed in intestinal neutrophils of UC patients and IL-1β and tissue factor (TF) modified NETs were formed in an autophagy dependent manner [159]. In AAV, the lysosomal membrane protein-2 (LAMP-2) ANCA-induced NET formation is involved in initiation of autophagy mechanism. Anti-lamp-2 antibody can inhibit neutrophil spontaneous apoptosis, activate neutrophil autophagy and activate NETs. Apoptosis and necrosis inhibitors did not interfere with the formation of NETs induced by anti-LAMP-2 antibodies [160]. In SSC, activated platelets exhibit a robust release of SSC particles and HMGB1, thereby inducing neutrophils to undergo autophagy. Subsequently, the redistribution of vesicle contents triggers the generation of NETs, consequently exacerbating vascular pathology and tissue fibrosis associated with the disease [161]. Furthermore, the IL-23/17 axis plays a pivotal role not only in T cell differentiation but also in modulating neutrophil homeostasis and migration in psoriasis. Researches demonstrate a reduction in the activities of NETs, NE, and ROS in Wdfy3-deficient neutrophils, with Wdfy3 acting as a key regulator of macroautophagy. Hence, the interplay between autophagy and other forms of neutrophil death, including NETs, warrants significant attention in the context of autoimmune diseases [162].
Conclusion
Neutrophil death is a complex and multifaceted phenomenon that has significant implications for the immune system. While the normal non-lytic morphology of neutrophils is essential for maintaining the body homeostasis, the emergence of abnormal non-lytic morphology and lytic morphology that causes inflammation can lead to the development of autoimmune diseases that pose troublesome problems for the immune system. Unlike other cells, neutrophils exhibit unique mechanisms of death that involve the production of reactive oxygen and nitrogen species. These species play a critical role in the development of infections and autoimmune diseases, highlighting the importance of understanding the various modes of neutrophil death.
Furthermore, it is not uncommon for multiple modes of neutrophil death to be present in a given autoimmune disease, and these modes of death often interact or overlap with each other. This interplay between different modes of neutrophil death underscores the pivotal role played by these cells in the immune system and highlights the potential importance of their death in the development of autoimmune diseases. As such, continued research into the various modes of neutrophil death may provide new insights into the treatment of autoimmune diseases, and this area of study represents an exciting frontier for pharmacological research.
Data availability
Not applicable.
Abbreviations
- ANCA:
-
Anti-neutrophil cytoplasmic antibodies
- RA:
-
Rheumatoid arthritis
- SS:
-
Sjogren's syndrome
- UC:
-
Ulcerative colitis
- MS:
-
Multiple sclerosis
- NE:
-
Neutrophil elastase
- GM-CSF:
-
Granulocyte colony-stimulating factor
- NETs:
-
Neutrophil extracellular traps
- DAMPs:
-
Damage-associated molecular patterns
- TNF-α:
-
Tumor necrosis factor-α
- GPA:
-
Granulomatosis with polyangiitis
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- LRP:
-
Lipoprotein receptor-related protein
- HA:
-
Hyaluronic acid
- PD-L1:
-
Programmed cell death-ligand 1
- PBMC:
-
Peripheral blood mononuclear cell
- ANA:
-
Antinuclear antibodies
- HNP:
-
Human neutrophil peptide
- TLR9:
-
Toll-like receptor 9
- IC:
-
Immune complexes
- MPA:
-
Microscopic poly-vasculitis
- HMGB1:
-
High mobility group protein 1
- ACPAs:
-
Anti-citrullinated peptide antibodies
- SF:
-
Synovial fluid
- TEN:
-
Toxic epidermal necrolysis
- LPMC:
-
Lamina propria mononuclear cells
- TNBS:
-
2,4,6-Trinitrobenzene sulfonic acid
- pfCD:
-
Perianal fistulising Crohn's disease
- μPs:
-
Microparticles
- aPL:
-
An-tiphospholipid antibody
- FAP-a:
-
Fibroblast-activated protein-a
- MRSA:
-
Methicillin-resistant staphylococcus aureus
- AS:
-
Ankylosing spondylitis
- AIH:
-
Autoimmune-mediated hepatitis
- HSCs:
-
Hematopoietic stem cells
- LMWH:
-
Low molecular weight heparins
- PMN:
-
Polymorphonuclear neutrophils
- EGPA:
-
Eosinophilic granuloma with poly-vasculitis
- Hif-1α:
-
Hypoxia-inducible factor-1α
- LAMP-2:
-
Lysosomal membrane protein-2
- RNP IC:
-
Immunocomplexes containing ribonucleoproteins
- TIM-3:
-
T cell immunoglobulin and mucin domain-containing protein 3
- MCPIP-1:
-
Monocyte chemotactic protein-inducible protein-1
- AAV:
-
ANCA-associated vasculitis
- SLE:
-
Systemic lupus erythematosus
- SSC:
-
Systemic sclerosis
- CD:
-
Crohn's disease
- MPO:
-
Myeloperoxidase
- ROS:
-
Reactive oxygen species
- Cyt-C:
-
Cytochrome C
- CTL:
-
Cytotoxic T lymphocytes
- NK:
-
Natural killer cells
- PR3:
-
Proteinase 3
- CRT:
-
Calreticulin
- APS:
-
Antiphospholipid syndrome
- ECM:
-
Extracellular cell matrix
- IFN-I:
-
Type I interferon
- MIF:
-
Migration inhibitory factor
- PAD4:
-
Peptidylarginine deiminase 4
- LDG:
-
Low-density granulocytes
- pDC:
-
Plasmacytoid dendritic cells
- MMP-9:
-
Matrix metalloproteinase-9
- EC:
-
Endothelial cells
- mDCs:
-
Myeloid dendritic cells
- TF:
-
Tissue factor
- IL-17A:
-
Interleukin-17A
- SJS:
-
Stevens Johnson Syndrome
- FPR1:
-
Rormyl peptide receptor 1
- HOCl:
-
Oxidant hypochlorous acid
- RRMS:
-
Relapsing–remitting MS
- ANETA:
-
Anti-NET antibodies
- PSGL-1:
-
P-selectin
- aCL:
-
An-ticardiolipin antibody
- MSU:
-
Monosodium urate
- PM:
-
Plasma membrane
- AOSD:
-
Adult Still's disease (AOSD)
- Atg:
-
Autophagy related gene
- CQ:
-
Chloroquine
- MA:
-
3-Methyl-adenine
- ET-1:
-
Endothelin-1
References
Wang L, Wang FS, Gershwin ME (2015) Human autoimmune diseases: a comprehensive update. J Intern Med 278:369–395. https://doi.org/10.1111/joim.12395
Lesage S, Goodnow CC (2001) Organ-specific autoimmune disease: a deficiency of tolerogenic stimulation. J Exp Med 194:F31-36. https://doi.org/10.1084/jem.194.5.f31
Wahren-Herlenius M, Dörner T (2013) Immunopathogenic mechanisms of systemic autoimmune disease. Lancet 382:819–831. https://doi.org/10.1016/s0140-6736(13)60954-x
Ma WT, Gao F, Gu K, Chen DK (2019) The role of monocytes and macrophages in autoimmune diseases: a comprehensive review. Front Immunol 10:1140. https://doi.org/10.3389/fimmu.2019.01140
Gianchecchi E, Delfino DV, Fierabracci A (2018) NK cells in autoimmune diseases: linking innate and adaptive immune responses. Autoimmun Rev 17:142–154. https://doi.org/10.1016/j.autrev.2017.11.018
Burn GL, Foti A, Marsman G, Patel DF, Zychlinsky A (2021) The neutrophil. Immunity 54:1377–1391. https://doi.org/10.1016/j.immuni.2021.06.006
de Oliveira S, Rosowski EE, Huttenlocher A (2016) Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol 16:378–391. https://doi.org/10.1038/nri.2016.49
Liew PX, Kubes P (2019) The neutrophil’s role during health and disease. Physiol Rev 99:1223–1248. https://doi.org/10.1152/physrev.00012.2018
Németh T, Sperandio M, Mócsai A (2020) Neutrophils as emerging therapeutic targets. Nat Rev Drug Discov 19:253–275. https://doi.org/10.1038/s41573-019-0054-z
Kolaczkowska E, Kubes P (2013) Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13:159–175. https://doi.org/10.1038/nri3399
Vicar T, Raudenska M, Gumulec J, Balvan J (2020) The quantitative-phase dynamics of apoptosis and lytic cell death. Sci Rep 10:1566. https://doi.org/10.1038/s41598-020-58474-w
Jorgensen I, Rayamajhi M, Miao EA (2017) Programmed cell death as a defence against infection. Nat Rev Immunol 17:151–164. https://doi.org/10.1038/nri.2016.147
Lawrence SM, Corriden R, Nizet V (2020) How neutrophils meet their end. Trends Immunol 41:531–544. https://doi.org/10.1016/j.it.2020.03.008
Silvestre-Roig C, Braster Q, Wichapong K, Lee EY, Teulon JM, Berrebeh N, Winter J, Adrover JM, Santos GS, Froese A, Lemnitzer P, Ortega-Gómez A, Chevre R, Marschner J, Schumski A, Winter C, Perez-Olivares L, Pan C, Paulin N, Schoufour T, Hartwig H, González-Ramos S, Kamp F, Megens RTA, Mowen KA, Gunzer M, Maegdefessel L, Hackeng T, Lutgens E, Daemen M, von Blume J, Anders HJ, Nikolaev VO, Pellequer JL, Weber C, Hidalgo A, Nicolaes GAF, Wong GCL, Soehnlein O (2019) Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 569:236–240. https://doi.org/10.1038/s41586-019-1167-6
Obeng E (2021) Apoptosis (programmed cell death) and its signals - a review. Braz J Biol 81:1133–1143. https://doi.org/10.1590/1519-6984.228437
Nagata S (2018) Apoptosis and clearance of apoptotic cells. Annu Rev Immunol 36:489–517. https://doi.org/10.1146/annurev-immunol-042617-053010
Zhang Q, Liu J, Zhang M, Wei S, Li R, Gao Y, Peng W, Wu C (2019) Apoptosis induction of fibroblast-like synoviocytes is an important molecular-mechanism for herbal medicine along with its active components in treating rheumatoid arthritis. Biomolecules. https://doi.org/10.3390/biom9120795
Fox S, Leitch AE, Duffin R, Haslett C, Rossi AG (2010) Neutrophil apoptosis: relevance to the innate immune response and inflammatory disease. J Innate Immun 2:216–227. https://doi.org/10.1159/000284367
Millet A, Martin KR, Bonnefoy F, Saas P, Mocek J, Alkan M, Terrier B, Kerstein A, Tamassia N, Satyanarayanan SK, Ariel A, Ribeil JA, Guillevin L, Cassatella MA, Mueller A, Thieblemont N, Lamprecht P, Mouthon L, Perruche S, Witko-Sarsat V (2015) Proteinase 3 on apoptotic cells disrupts immune silencing in autoimmune vasculitis. J Clin Invest 125:4107–4121. https://doi.org/10.1172/jci78182
Jerke U, Eulenberg-Gustavus C, Rousselle A, Nicklin P, Kreideweiss S, Grundl MA, Eickholz P, Nickles K, Schreiber A, Korkmaz B, Kettritz R (2022) Targeting cathepsin c in PR3-ANCA vasculitis. J Am Soc Nephrol. https://doi.org/10.1681/asn.2021081112
Everts-Graber J, Martin KR, Thieblemont N, Mocek J, Roccabianca A, Chafey P, Le Gall M, Tacnet-Delorme P, Reutelingsperger CP, Naccache JM, Bonnotte B, Karras A, Puéchal X, Guillevin L, Terrier B, Frachet P, Perretti M, Mouthon L, Witko-Sarsat V (2019) Proteomic analysis of neutrophils in ANCA-associated vasculitis reveals a dysregulation in proteinase 3-associated proteins such as annexin-A1 involved in apoptotic cell clearance. Kidney Int 96:397–408. https://doi.org/10.1016/j.kint.2019.02.017
Kitching AR, Anders HJ, Basu N, Brouwer E, Gordon J, Jayne DR, Kullman J, Lyons PA, Merkel PA, Savage COS, Specks U, Kain R (2020) ANCA-associated vasculitis. Nat Rev Dis Primers 6:71. https://doi.org/10.1038/s41572-020-0204-y
Gabillet J, Millet A, Pederzoli-Ribeil M, Tacnet-Delorme P, Guillevin L, Mouthon L, Frachet P, Witko-Sarsat V (2012) Proteinase 3, the autoantigen in granulomatosis with polyangiitis, associates with calreticulin on apoptotic neutrophils, impairs macrophage phagocytosis, and promotes inflammation. J Immunol 189:2574–2583. https://doi.org/10.4049/jimmunol.1200600
Müller A, Krause B, Kerstein-Stähle A, Comdühr S, Klapa S, Ullrich S, Holl-Ulrich K, Lamprecht P (2021) Granulomatous inflammation in ANCA-associated vasculitis. Int J Mol Sci. https://doi.org/10.3390/ijms22126474
Greenlee-Wacker MC (2016) Clearance of apoptotic neutrophils and resolution of inflammation. Immunol Rev 273:357–370. https://doi.org/10.1111/imr.12453
van Rossum AP, Limburg PC, Kallenberg CG (2005) Activation, apoptosis, and clearance of neutrophils in Wegener’s granulomatosis. Ann N Y Acad Sci 1051:1–11. https://doi.org/10.1196/annals.1361.041
Moosig F, Csernok E, Kumanovics G, Gross WL (2000) Opsonization of apoptotic neutrophils by anti-neutrophil cytoplasmic antibodies (ANCA) leads to enhanced uptake by macrophages and increased release of tumour necrosis factor-alpha (TNF-alpha). Clin Exp Immunol 122:499–503. https://doi.org/10.1046/j.1365-2249.2000.01410.x
Casciola-Rosen LA, Anhalt G, Rosen A (1994) Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med 179:1317–1330. https://doi.org/10.1084/jem.179.4.1317
Sandin C, Eriksson P, Segelmark M, Skogh T, Kastbom A (2016) IgA- and SIgA anti-PR3 antibodies in serum versus organ involvement and disease activity in PR3-ANCA-associated vasculitis. Clin Exp Immunol 184:208–215. https://doi.org/10.1111/cei.12769
Defendi F, Thielens NM, Clavarino G, Cesbron JY, Dumestre-Pérard C (2020) The immunopathology of complement proteins and innate immunity in autoimmune disease. Clin Rev Allergy Immunol 58:229–251. https://doi.org/10.1007/s12016-019-08774-5
Donnelly S, Roake W, Brown S, Young P, Naik H, Wordsworth P, Isenberg DA, Reid KB, Eggleton P (2006) Impaired recognition of apoptotic neutrophils by the C1q/calreticulin and CD91 pathway in systemic lupus erythematosus. Arthritis Rheum 54:1543–1556. https://doi.org/10.1002/art.21783
Tacnet-Delorme P, Gabillet J, Chatfield S, Thieblemont N, Frachet P, Witko-Sarsat V (2018) Proteinase 3 interferes with C1q-mediated clearance of apoptotic cells. Front Immunol 9:818. https://doi.org/10.3389/fimmu.2018.00818
Trouw LA, Daha N, Kurreeman FA, Böhringer S, Goulielmos GN, Westra HJ, Zhernakova A, Franke L, Stahl EA, Levarht EW, Stoeken-Rijsbergen G, Verduijn W, Roos A, Li Y, Houwing-Duistermaat JJ, Huizinga TW, Toes RE (2013) Genetic variants in the region of the C1q genes are associated with rheumatoid arthritis. Clin Exp Immunol 173:76–83. https://doi.org/10.1111/cei.12097
Wight TN, Kang I, Evanko SP, Harten IA, Chang MY, Pearce OMT, Allen CE, Frevert CW (2020) Versican-A critical extracellular matrix regulator of immunity and inflammation. Front Immunol 11:512. https://doi.org/10.3389/fimmu.2020.00512
Gee K, Kryworuchko M, Kumar A (2004) Recent advances in the regulation of CD44 expression and its role in inflammation and autoimmune diseases. Arch Immunol Ther Exp (Warsz) 52:13–26
Nagy N, Kuipers HF, Marshall PL, Wang E, Kaber G, Bollyky PL (2019) Hyaluronan in immune dysregulation and autoimmune diseases. Matrix Biol 78–79:292–313. https://doi.org/10.1016/j.matbio.2018.03.022
Wittig B, Seiter S, Schmidt DS, Zuber M, Neurath M, Zöller M (1999) CD44 variant isoforms on blood leukocytes in chronic inflammatory bowel disease and other systemic autoimmune diseases. Lab Invest 79:747–759
Kitano A, Oshitani N, Matsumoto T, Kobayashi K (1996) CD44 variants in ulcerative colitis and Crohn’s disease. Lancet 348:266–267. https://doi.org/10.1016/s0140-6736(05)65573-0
Takazoe K, Tesch GH, Hill PA, Hurst LA, Jun Z, Lan HY, Atkins RC, Nikolic-Paterson DJ (2000) CD44-mediated neutrophil apoptosis in the rat. Kidney Int 58:1920–1930. https://doi.org/10.1111/j.1523-1755.2000.00364.x
Cairns AP, Crockard AD, McConnell JR, Courtney PA, Bell AL (2001) Reduced expression of CD44 on monocytes and neutrophils in systemic lupus erythematosus: relations with apoptotic neutrophils and disease activity. Ann Rheum Dis 60:950–955. https://doi.org/10.1136/ard.60.10.950
Lessard CJ, Adrianto I, Kelly JA, Kaufman KM, Grundahl KM, Adler A, Williams AH, Gallant CJ, Anaya JM, Bae SC, Boackle SA, Brown EE, Chang DM, Criswell LA, Edberg JC, Freedman BI, Gregersen PK, Gilkeson GS, Jacob CO, James JA, Kamen DL, Kimberly RP, Martin J, Merrill JT, Niewold TB, Park SY, Petri MA, Pons-Estel BA, Ramsey-Goldman R, Reveille JD, Song YW, Stevens AM, Tsao BP, Vila LM, Vyse TJ, Yu CY, Guthridge JM, Bruner GR, Langefeld CD, Montgomery C, Harley JB, Scofield RH, Gaffney PM, Moser KL (2011) Identification of a systemic lupus erythematosus susceptibility locus at 11p13 between PDHX and CD44 in a multiethnic study. Am J Hum Genet 88:83–91. https://doi.org/10.1016/j.ajhg.2010.11.014
Karmakar U, Chu JY, Sundaram K, Astier AL, Garside H, Hansen CG, Dransfield I, Vermeren S (2021) Immune complex-induced apoptosis and concurrent immune complex clearance are anti-inflammatory neutrophil functions. Cell Death Dis 12:296. https://doi.org/10.1038/s41419-021-03528-8
Zhu M, Yuan K, Lu Q, Zhu Q, Zhang S, Li X, Zhao L, Wang H, Luo G, Wang T, Huang G, Xu A (2019) Emodin ameliorates rheumatoid arthritis by promoting neutrophil apoptosis and inhibiting neutrophil extracellular trap formation. Mol Immunol 112:188–197. https://doi.org/10.1016/j.molimm.2019.05.010
Andrews LP, Yano H, Vignali DAA (2019) Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: breakthroughs or backups. Nat Immunol 20:1425–1434. https://doi.org/10.1038/s41590-019-0512-0
Zamani MR, Aslani S, Salmaninejad A, Javan MR, Rezaei N (2016) PD-1/PD-L and autoimmunity: a growing relationship. Cell Immunol 310:27–41. https://doi.org/10.1016/j.cellimm.2016.09.009
Wang JF, Wang YP, Xie J, Zhao ZZ, Gupta S, Guo Y, Jia SH, Parodo J, Marshall JC, Deng XM (2021) Upregulated PD-L1 delays human neutrophil apoptosis and promotes lung injury in an experimental mouse model of sepsis. Blood 138:806–810. https://doi.org/10.1182/blood.2020009417
Tanaka H, Arima Y, Kamimura D, Tanaka Y, Takahashi N, Uehata T, Maeda K, Satoh T, Murakami M, Akira S (2019) Phosphorylation-dependent Regnase-1 release from endoplasmic reticulum is critical in IL-17 response. J Exp Med 216:1431–1449. https://doi.org/10.1084/jem.20181078
Dobosz E, Wadowska M, Kaminska M, Wilamowski M, Honarpisheh M, Bryzek D, Potempa J, Jura J, Lech M, Koziel J (2021) MCPIP-1 restricts inflammation via promoting apoptosis of neutrophils. Front Immunol 12:627922. https://doi.org/10.3389/fimmu.2021.627922
Qu B, Cao J, Zhang F, Cui H, Teng J, Li J, Liu Z, Morehouse C, Jallal B, Tang Y, Guo Q, Yao Y, Shen N (2015) Type I interferon inhibition of microRNA-146a maturation through Up-regulation of monocyte chemotactic protein-induced protein 1 in systemic lupus erythematosus. Arthritis Rheumatol 67:3209–3218. https://doi.org/10.1002/art.39398
Schindler L, Zwissler L, Krammer C, Hendgen-Cotta U, Rassaf T, Hampton MB, Dickerhof N, Bernhagen J (2021) Macrophage migration inhibitory factor inhibits neutrophil apoptosis by inducing cytokine release from mononuclear cells. J Leukoc Biol 110:893–905. https://doi.org/10.1002/jlb.3a0420-242rrr
El Kebir D, József L, Khreiss T, Filep JG (2006) Inhibition of K+ efflux prevents mitochondrial dysfunction, and suppresses caspase-3-, apoptosis-inducing factor-, and endonuclease G-mediated constitutive apoptosis in human neutrophils. Cell Signal 18:2302–2313. https://doi.org/10.1016/j.cellsig.2006.05.013
Ottonello L, Cutolo M, Frumento G, Arduino N, Bertolotto M, Mancini M, Sottofattori E, Dallegri F (2002) Synovial fluid from patients with rheumatoid arthritis inhibits neutrophil apoptosis: role of adenosine and proinflammatory cytokines. Rheumatology (Oxford) 41:1249–1260. https://doi.org/10.1093/rheumatology/41.11.1249
Papayannopoulos V (2018) Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 18:134–147. https://doi.org/10.1038/nri.2017.105
Sørensen OE, Borregaard N (2016) Neutrophil extracellular traps - the dark side of neutrophils. J Clin Investig 126:1612–1620. https://doi.org/10.1172/jci84538
Yipp BG, Kubes P (2013) NETosis: how vital is it? Blood 122:2784–2794. https://doi.org/10.1182/blood-2013-04-457671
Jariwala MP, Laxer RM (2021) NETosis in rheumatic diseases. Current Rheumatol Rep 23:9. https://doi.org/10.1007/s11926-020-00977-6
Lou H, Wojciak-Stothard B, Ruseva MM, Cook HT, Kelleher P, Pickering MC, Mongkolsapaya J, Screaton GR, Xu XN (2020) Autoantibody-dependent amplification of inflammation in SLE. Cell Death Dis 11:729. https://doi.org/10.1038/s41419-020-02928-6
Bai Y, Tong Y, Liu Y, Hu H (2018) Self-dsDNA in the pathogenesis of systemic lupus erythematosus. Clin Exp Immunol 191:1–10. https://doi.org/10.1111/cei.13041
Knight JS, Subramanian V, O’Dell AA, Yalavarthi S, Zhao W, Smith CK, Hodgin JB, Thompson PR, Kaplan MJ (2015) Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann Rheum Dis 74:2199–2206. https://doi.org/10.1136/annrheumdis-2014-205365
Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, Kaplan MJ (2016) Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med 22:146–153. https://doi.org/10.1038/nm.4027
Dörner T (2012) SLE in 2011: deciphering the role of NETs and networks in SLE. Nat Rev Rheumatol 8:68–70. https://doi.org/10.1038/nrrheum.2011.200
Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL, Barrat FJ, Banchereau J, Pascual V (2011) Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med 3:73ra20. https://doi.org/10.1126/scitranslmed.3001201
Martinelli S, Urosevic M, Daryadel A, Oberholzer PA, Baumann C, Fey MF, Dummer R, Simon HU, Yousefi S (2004) Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil differentiation. J Biol Chem 279:44123–44132. https://doi.org/10.1074/jbc.M405883200
Pieterse E, Hofstra J, Berden J, Herrmann M, Dieker J, van der Vlag J (2015) Acetylated histones contribute to the immunostimulatory potential of neutrophil extracellular traps in systemic lupus erythematosus. Clin Exp Immunol 179:68–74. https://doi.org/10.1111/cei.12359
Carmona-Rivera C, Zhao W, Yalavarthi S, Kaplan MJ (2015) Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann Rheum Dis 74:1417–1424. https://doi.org/10.1136/annrheumdis-2013-204837
Blanco LP, Wang X, Carlucci PM, Torres-Ruiz JJ, Romo-Tena J, Sun HW, Hafner M, Kaplan MJ (2021) RNA externalized by neutrophil extracellular traps promotes inflammatory pathways in endothelial cells. Arthritis Rheumatol 73:2282–2292. https://doi.org/10.1002/art.41796
Yang B, Huang X, Xu S, Li L, Wu W, Dai Y, Ge MX, Yuan L, Cao W, Yang M, Wu Y, Deng D (2021) Decreased miR-4512 levels in monocytes and macrophages of individuals with systemic lupus erythematosus contribute to innate immune activation and neutrsophil NETosis by targeting TLR4 and CXCL2. Front Immunol 12:756825. https://doi.org/10.3389/fimmu.2021.756825
Salvador F (2020) ANCA associated vasculitis. Eur J Intern Med 74:18–28. https://doi.org/10.1016/j.ejim.2020.01.011
Hunter RW, Welsh N, Farrah TE, Gallacher PJ, Dhaun N (2020) ANCA associated vasculitis. Bmj 369:m1070. https://doi.org/10.1136/bmj.m1070
Fousert E, Toes R, Desai J (2020) Neutrophil extracellular traps (NETs) take the central stage in driving autoimmune responses. Cells. https://doi.org/10.3390/cells9040915
Masuda S, Nonokawa M, Futamata E, Nishibata Y, Iwasaki S, Tsuji T, Hatanaka Y, Nakazawa D, Tanaka S, Tomaru U, Kawakami T, Atsumi T, Ishizu A (2019) Formation and disordered degradation of neutrophil extracellular traps in necrotizing lesions of anti-neutrophil cytoplasmic antibody-associated vasculitis. Am J Pathol 189:839–846. https://doi.org/10.1016/j.ajpath.2019.01.007
Wang H, Wang C, Zhao MH, Chen M (2015) Neutrophil extracellular traps can activate alternative complement pathways. Clin Exp Immunol 181:518–527. https://doi.org/10.1111/cei.12654
Kessenbrock K, Krumbholz M, Schönermarck U, Back W, Gross WL, Werb Z, Gröne HJ, Brinkmann V, Jenne DE (2009) Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med 15:623–625. https://doi.org/10.1038/nm.1959
Panda R, Krieger T, Hopf L, Renné T, Haag F, Röber N, Conrad K, Csernok E, Fuchs TA (2017) Neutrophil extracellular traps contain selected antigens of anti-neutrophil cytoplasmic antibodies. Front Immunol 8:439. https://doi.org/10.3389/fimmu.2017.00439
Kraaij T, Kamerling SWA, van Dam LS, Bakker JA, Bajema IM, Page T, Brunini F, Pusey CD, Toes REM, Scherer HU, Rabelink TJ, van Kooten C, Teng YKO (2018) Excessive neutrophil extracellular trap formation in ANCA-associated vasculitis is independent of ANCA. Kidney Int 94:139–149. https://doi.org/10.1016/j.kint.2018.01.013
Kimura H, Mii A, Shoji J, Arakawa Y, Shimizu A (2021) Immunohistochemical detection of citrullinated histone H3-positive neutrophils is useful for identifying active glomerular and interstitial lesions in antineutrophil cytoplasmic antibody-associated vasculitis. Histopathology 78:520–531. https://doi.org/10.1111/his.14247
Frangou E, Vassilopoulos D, Boletis J, Boumpas DT (2019) An emerging role of neutrophils and NETosis in chronic inflammation and fibrosis in systemic lupus erythematosus (SLE) and ANCA-associated vasculitides (AAV): Implications for the pathogenesis and treatment. Autoimmun Rev 18:751–760. https://doi.org/10.1016/j.autrev.2019.06.011
Nakazawa D, Tomaru U, Suzuki A, Masuda S, Hasegawa R, Kobayashi T, Nishio S, Kasahara M, Ishizu A (2012) Abnormal conformation and impaired degradation of propylthiouracil-induced neutrophil extracellular traps: implications of disordered neutrophil extracellular traps in a rat model of myeloperoxidase antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 64:3779–3787. https://doi.org/10.1002/art.34619
Huang YM, Wang H, Wang C, Chen M, Zhao MH (2015) Promotion of hypercoagulability in antineutrophil cytoplasmic antibody-associated vasculitis by C5a-induced tissue factor-expressing microparticles and neutrophil extracellular traps. Arthritis Rheumatol 67:2780–2790. https://doi.org/10.1002/art.39239
Su B, Mao X, Yin B, Chen C, Zhang M, Cui T, Hao Y (2021) TIM-3 regulates the NETs-mediated dendritic cell activation in myeloperoxidase-ANCA-associated vasculitis. Clin Exp Rheumatol 39(2):13–20. https://doi.org/10.55563/clinexprheumatol/6y0bjb
Matsumoto K, Yasuoka H, Yoshimoto K, Suzuki K, Takeuchi T (2021) Platelet CXCL4 mediates neutrophil extracellular traps formation in ANCA-associated vasculitis. Sci Rep 11:222. https://doi.org/10.1038/s41598-020-80685-4
Söderberg D, Segelmark M (2016) Neutrophil extracellular traps in ANCA-associated vasculitis. Front Immunol 7:256. https://doi.org/10.3389/fimmu.2016.00256
Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, Friday S, Li S, Patel RM, Subramanian V, Thompson P, Chen P, Fox DA, Pennathur S, Kaplan MJ (2013) NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med 5:178140. https://doi.org/10.1126/scitranslmed.3005580
Wright HL, Lyon M, Chapman EA, Moots RJ, Edwards SW (2020) Rheumatoid arthritis synovial fluid neutrophils drive inflammation through production of chemokines, reactive oxygen species, and neutrophil extracellular traps. Front Immunol 11:584116. https://doi.org/10.3389/fimmu.2020.584116
Clarke J (2020) NETs directly injure cartilage in RA. Nat Rev Rheumatol 16:410. https://doi.org/10.1038/s41584-020-0459-4
Chapman EA, Lyon M, Simpson D, Mason D, Beynon RJ, Moots RJ, Wright HL (2019) Caught in a Trap? proteomic analysis of neutrophil extracellular traps in rheumatoid arthritis and systemic lupus erythematosus. Front Immunol 10:423. https://doi.org/10.3389/fimmu.2019.00423
Yamamoto T (2020) Psoriasis and connective tissue diseases. Int J Mol Sci. https://doi.org/10.3390/ijms21165803
Shao S, Fang H, Dang E, Xue K, Zhang J, Li B, Qiao H, Cao T, Zhuang Y, Shen S, Zhang T, Qiao P, Li C, Gudjonsson JE, Wang G (2019) Neutrophil extracellular traps promote inflammatory responses in psoriasis via activating epidermal TLR4/IL-36R crosstalk. Front Immunol 10:746. https://doi.org/10.3389/fimmu.2019.00746
Herster F, Bittner Z, Archer NK, Dickhöfer S, Eisel D, Eigenbrod T, Knorpp T, Schneiderhan-Marra N, Löffler MW, Kalbacher H, Vierbuchen T, Heine H, Miller LS, Hartl D, Freund L, Schäkel K, Heister M, Ghoreschi K, Weber ANR (2020) Neutrophil extracellular trap-associated RNA and LL37 enable self-amplifying inflammation in psoriasis. Nat Commun 11:105. https://doi.org/10.1038/s41467-019-13756-4
Kinoshita M, Ogawa Y, Hama N, Ujiie I, Hasegawa A, Nakajima S, Nomura T, Adachi J, Sato T, Koizumi S, Shimada S, Fujita Y, Takahashi H, Mizukawa Y, Tomonaga T, Nagao K, Abe R, Kawamura T (2021) Neutrophils initiate and exacerbate Stevens-Johnson syndrome and toxic epidermal necrolysis. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aax2398
Li T, Wang C, Liu Y, Li B, Zhang W, Wang L, Yu M, Zhao X, Du J, Zhang J, Dong Z, Jiang T, Xie R, Ma R, Fang S, Zhou J, Shi J (2020) Neutrophil extracellular traps induce intestinal damage and thrombotic tendency in inflammatory bowel disease. J Crohns Colitis 14:240–253. https://doi.org/10.1093/ecco-jcc/jjz132
Lai HJ, Doan HT, Lin EY, Chiu YL, Cheng YK, Lin YH, Chiang HS (2023) Histones of neutrophil extracellular traps directly disrupt the permeability and integrity of the intestinal epithelial barrier. Inflamm Bowel Dis 29:783–797. https://doi.org/10.1093/ibd/izac256
Dinallo V, Marafini I, Di Fusco D, Laudisi F, Franzè E, Di Grazia A, Figliuzzi MM, Caprioli F, Stolfi C, Monteleone I, Monteleone G (2019) Neutrophil extracellular traps sustain inflammatory signals in ulcerative colitis. J Crohns Colitis 13:772–784. https://doi.org/10.1093/ecco-jcc/jjy215
Wang P, Liu D, Zhou Z, Liu F, Shen Y, You Q, Lu S, Wu J (2023) The role of protein arginine deiminase 4-dependent neutrophil extracellular traps formation in ulcerative colitis. Front Immunol 14:1144976. https://doi.org/10.3389/fimmu.2023.1144976
Leppkes M, Lindemann A, Gößwein S, Paulus S, Roth D, Hartung A, Liebing E, Zundler S, Gonzalez-Acera M, Patankar JV, Mascia F, Scheibe K, Hoffmann M, Uderhardt S, Schauer C, Foersch S, Neufert C, Vieth M, Schett G, Atreya R, Kühl AA, Bleich A, Becker C, Herrmann M, Neurath MF (2022) Neutrophils prevent rectal bleeding in ulcerative colitis by peptidyl-arginine deiminase-4-dependent immunothrombosis. Gut 71:2414–2429. https://doi.org/10.1136/gutjnl-2021-324725
Zhang T, Mei Y, Dong W, Wang J, Huang F, Wu J (2020) Evaluation of protein arginine deiminase-4 inhibitor in TNBS- induced colitis in mice. Int Immunopharmacol 84:106583. https://doi.org/10.1016/j.intimp.2020.106583
Cao D, Qian K, Zhao Y, Hong J, Chen H, Wang X, Yang N, Zhang C, Cao J, Jia K, Wu G, Zhu M, Shen J, Zhang Y, Cui Z, Wang Z (2023) Association of neutrophil extracellular traps with fistula healing in patients with complex perianal fistulizing crohn’s disease. J Crohns Colitis 17:580–592. https://doi.org/10.1093/ecco-jcc/jjac171
Pierson ER, Wagner CA, Goverman JM (2018) The contribution of neutrophils to CNS autoimmunity. Clin Immunol 189:23–28. https://doi.org/10.1016/j.clim.2016.06.017
Woodberry T, Bouffler SE, Wilson AS, Buckland RL, Brüstle A (2018) The emerging role of neutrophil granulocytes in multiple sclerosis. J Clin Med. https://doi.org/10.3390/jcm7120511
Tillack K, Naegele M, Haueis C, Schippling S, Wandinger KP, Martin R, Sospedra M (2013) Gender differences in circulating levels of neutrophil extracellular traps in serum of multiple sclerosis patients. J Neuroimmunol 261:108–119. https://doi.org/10.1016/j.jneuroim.2013.05.004
Didier K, Giusti D, Le Jan S, Terryn C, Muller C, Pham BN, Le Naour R, Antonicelli FD, Servettaz A (2020) Neutrophil Extracellular Traps Generation Relates with Early Stage and Vascular Complications in Systemic Sclerosis. J Clin Med. https://doi.org/10.3390/jcm9072136
Manfredi AA, Ramirez GA, Godino C, Capobianco A, Monno A, Franchini S, Tombetti E, Corradetti S, Distler JHW, Bianchi ME, Rovere-Querini P, Maugeri N (2022) Platelet phagocytosis via P-selectin glycoprotein ligand 1 and accumulation of microparticles in systemic sclerosis. Arthritis Rheumatol 74:318–328. https://doi.org/10.1002/art.41926
Knight JS, Kanthi Y (2022) Mechanisms of immunothrombosis and vasculopathy in antiphospholipid syndrome. Semin Immunopathol. https://doi.org/10.1007/s00281-022-00916-w
Zuo Y, Yalavarthi S, Gockman K, Madison JA, Gudjonsson JE, Kahlenberg JM, Joseph McCune W, Bockenstedt PL, Karp DR, Knight JS (2020) Anti-neutrophil extracellular trap antibodies and impaired neutrophil extracellular trap degradation in antiphospholipid syndrome. Arthritis Rheumatol 72:2130–2135. https://doi.org/10.1002/art.41460
Lu Y, Dong Y, Zhang Y, Shen D, Wang X, Ge R, Zhang M, Xia Y, Wang X (2020) Antiphospholipid antibody-activated NETs exacerbate trophoblast and endothelial cell injury in obstetric antiphospholipid syndrome. J Cell Mol Med 24:6690–6703. https://doi.org/10.1111/jcmm.15321
Arneth B (2021) Defects in ubiquitination and NETosis and their associations with human diseases. Pathology 53:439–445. https://doi.org/10.1016/j.pathol.2020.10.014
Wang X, Yousefi S, Simon HU (2018) Necroptosis and neutrophil-associated disorders. Cell Death Dis 9:111. https://doi.org/10.1038/s41419-017-0058-8
Wang X, Gessier F, Perozzo R, Stojkov D, Hosseini A, Amirshahrokhi K, Kuchen S, Yousefi S, Lötscher P, Simon HU (2020) RIPK3-MLKL-Mediated Neutrophil Death Requires Concurrent Activation of Fibroblast Activation Protein-α. Journal of immunology 205:1653–1663. https://doi.org/10.4049/jimmunol.2000113
Honda T, Yamamoto O, Sawada Y, Egawa G, Kitoh A, Otsuka A, Dainichi T, Nakajima S, Miyachi Y, Kabashima K (2017) Receptor-interacting protein kinase 3 controls keratinocyte activation in a necroptosis-independent manner and promotes psoriatic dermatitis in mice. J Allergy Clin Immunol 140:619-622.e616. https://doi.org/10.1016/j.jaci.2017.02.027
Kril I, Havrylyuk A, Potomkina H, Chopyak V (2020) Apoptosis and secondary necrosis of neutrophils and monocytes in the immunopathogenesis of rheumatoid arthritis: a cohort study. Rheumatol Int 40:1449–1454. https://doi.org/10.1007/s00296-020-04642-0
Zhao J, Jiang P, Guo S, Schrodi SJ, He D (2021) Apoptosis, Autophagy, NETosis, necroptosis, and pyroptosis mediated programmed cell death as targets for innovative therapy in rheumatoid arthritis. Front Immunology 12:809806. https://doi.org/10.3389/fimmu.2021.809806
Wang X, He Z, Liu H, Yousefi S, Simon HU (2016) Neutrophil necroptosis is triggered by ligation of adhesion molecules following GM-CSF priming. J Immunol 197:4090–4100. https://doi.org/10.4049/jimmunol.1600051
Mihalache CC, Yousefi S, Conus S, Villiger PM, Schneider EM, Simon HU (2011) Inflammation-associated autophagy-related programmed necrotic death of human neutrophils characterized by organelle fusion events. J Immunol 186:6532–6542. https://doi.org/10.4049/jimmunol.1004055
Mousavi MJ, Farhadi E, Vodjgani M, Karami J, Tahmasebi MN, Sharafat Vaziri A, Asgari M, Rezaei N, Mostafaei S, Jamshidi A, Mahmoudi M (2021) Role of Fibroblast activation protein alpha in fibroblast-like synoviocytes of rheumatoid arthritis. Iran J Allergy Asthma Immunol 20:338–349. https://doi.org/10.18502/ijaai.v20i3.6335
Schreiber A, Rousselle A, Becker JU, von Mässenhausen A, Linkermann A, Kettritz R (2017) Necroptosis controls NET generation and mediates complement activation, endothelial damage, and autoimmune vasculitis. Proc Natl Acad Sci USA 114:E9618-e9625. https://doi.org/10.1073/pnas.1708247114
Weisel K, Berger S, Thorn K, Taylor PC, Peterfy C, Siddall H, Tompson D, Wang S, Quattrocchi E, Burriss SW, Walter J, Tak PP (2021) A randomized, placebo-controlled experimental medicine study of RIPK1 inhibitor GSK2982772 in patients with moderate to severe rheumatoid arthritis. Arthritis Res Ther 23:85. https://doi.org/10.1186/s13075-021-02468-0
Tompson DJ, Davies C, Scott NE, Cannons EP, Kostapanos M, Gross AS, Powell M, Ino H, Shimamura R, Ogura H, Nagakubo T, Igarashi H, Nakano A (2021) Comparison of the Pharmacokinetics of RIPK1 Inhibitor GSK2982772 in Healthy Western and Japanese Subjects. Eur J Drug Metab Pharmacokinet 46:71–83. https://doi.org/10.1007/s13318-020-00652-2
Desai J, Mulay SR, Nakazawa D, Anders HJ (2016) Matters of life and death. How neutrophils die or survive along NET release and is “NETosis” = necroptosis? Cell Mol Life Sci 73:2211–2219. https://doi.org/10.1007/s00018-016-2195-0
Desai J, Kumar SV, Mulay SR, Konrad L, Romoli S, Schauer C, Herrmann M, Bilyy R, Müller S, Popper B, Nakazawa D, Weidenbusch M, Thomasova D, Krautwald S, Linkermann A, Anders HJ (2016) PMA and crystal-induced neutrophil extracellular trap formation involves RIPK1-RIPK3-MLKL signaling. Eur J Immunol 46:223–229. https://doi.org/10.1002/eji.201545605
Chen L, Zhao Y, Lai D, Zhang P, Yang Y, Li Y, Fei K, Jiang G, Fan J (2018) Neutrophil extracellular traps promote macrophage pyroptosis in sepsis. Cell Death Dis 9:597. https://doi.org/10.1038/s41419-018-0538-5
Pérez-Figueroa E, Álvarez-Carrasco P, Ortega E, Maldonado-Bernal C (2021) Neutrophils: many ways to die. Front Immunol 12:631821. https://doi.org/10.3389/fimmu.2021.631821
Li Z, Guo J, Bi L (2020) Role of the NLRP3 inflammasome in autoimmune diseases. Biomed PharmacotheR 130:110542. https://doi.org/10.1016/j.biopha.2020.110542
Ryu JC, Kim MJ, Kwon Y, Oh JH, Yoon SS, Shin SJ, Yoon JH, Ryu JH (2017) Neutrophil pyroptosis mediates pathology of P. aeruginosa lung infection in the absence of the NADPH oxidase NOX2. Mucosal Immunol 10:757–774. https://doi.org/10.1038/mi.2016.73
Karmakar M, Minns M, Greenberg EN, Diaz-Aponte J, Pestonjamasp K, Johnson JL, Rathkey JK, Abbott DW, Wang K, Shao F, Catz SD, Dubyak GR, Pearlman E (2020) N-GSDMD trafficking to neutrophil organelles facilitates IL-1β release independently of plasma membrane pores and pyroptosis. Nat Commun 11:2212. https://doi.org/10.1038/s41467-020-16043-9
Yi YS (2018) Role of inflammasomes in inflammatory autoimmune rheumatic diseases. Korean J Physiol Pharmacol 22:1–15. https://doi.org/10.4196/kjpp.2018.22.1.1
Kattah NH, Kattah MG, Utz PJ (2010) The U1-snRNP complex: structural properties relating to autoimmune pathogenesis in rheumatic diseases. Immunol Rev 233:126–145. https://doi.org/10.1111/j.0105-2896.2009.00863.x
Shin MS, Kang Y, Lee N, Kim SH, Kang KS, Lazova R, Kang I (2012) U1-small nuclear ribonucleoprotein activates the NLRP3 inflammasome in human monocytes. J Immunol 188:4769–4775. https://doi.org/10.4049/jimmunol.1103355
Huang W, Jiao J, Liu J, Huang M, Hu Y, Ran W, Yan L, Xiong Y, Li M, Quan Z, Rao Y, Chen J, Huang Y, Zhang D (2020) MFG-E8 accelerates wound healing in diabetes by regulating “NLRP3 inflammasome-neutrophil extracellular traps” axis. Cell Death Discov 6:84. https://doi.org/10.1038/s41420-020-00318-7
Tan AL, Marzo-Ortega H, O’Connor P, Fraser A, Emery P, McGonagle D (2004) Efficacy of anakinra in active ankylosing spondylitis: a clinical and magnetic resonance imaging study. Ann Rheum Dis 63:1041–1045. https://doi.org/10.1136/ard.2004.020800
Sasaki Y, Otsuka K, Arimochi H, Tsukumo SI, Yasutomo K (2020) Distinct Roles of IL-1β and IL-18 in NLRC4-Induced Autoinflammation. Front Immunol 11:591713. https://doi.org/10.3389/fimmu.2020.591713
Hu Q, Shi H, Zeng T, Liu H, Su Y, Cheng X, Ye J, Yin Y, Liu M, Zheng H, Wu X, Chi H, Zhou Z, Jia J, Sun Y, Teng J, Yang C (2019) Increased neutrophil extracellular traps activate NLRP3 and inflammatory macrophages in adult-onset Still’s disease. Arthritis Res Ther 21:9. https://doi.org/10.1186/s13075-018-1800-z
Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, von Pein JB, Broz P, Sweet MJ, Schroder K (2018) Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol. https://doi.org/10.1126/sciimmunol.aar6676
Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, Menninger S, Eickhoff J, Nussbaumer P, Klebl B, Krüger R, Herzig A, Zychlinsky A (2018) Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. https://doi.org/10.1126/sciimmunol.aar6689
Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, Zhao L, Zhou S, Yu H, Zhou W, Silberstein LE, Cheng T, Han M, Xu Y, Luo HR (2018) Gasdermin D exerts anti-inflammatory effects by promoting neutrophil death. Cell Rep 22:2924–2936. https://doi.org/10.1016/j.celrep.2018.02.067
Yang Z, Cao J, Yu C, Yang Q, Zhang Y, Han L (2016) Caspase-1 mediated interleukin-18 activation in neutrophils promotes the activity of rheumatoid arthritis in a NLRP3 inflammasome independent manner. Joint Bone Spine 83:282–289. https://doi.org/10.1016/j.jbspin.2015.07.006
Xie Z, Hou H, Luo D, An R, Zhao Y, Qiu C (2021) ROS-Dependent Lipid peroxidation and reliant antioxidant ferroptosis-suppressor-protein 1 in rheumatoid arthritis: a covert clue for potential therapy. Inflammation 44:35–47. https://doi.org/10.1007/s10753-020-01338-2
Qiu Y, Cao Y, Cao W, Jia Y, Lu N (2020) The Application of Ferroptosis in Diseases. Pharmacol Res 159:104919. https://doi.org/10.1016/j.phrs.2020.104919
Jakaria M, Belaidi AA, Bush AI, Ayton S (2021) Ferroptosis as a mechanism of neurodegeneration in Alzheimer’s disease. J Neurochem 159:804–825. https://doi.org/10.1111/jnc.15519
Zhang G, Zhang Y, Shen Y, Wang Y, Zhao M, Sun L (2021) The potential role of ferroptosis in alzheimer’s disease. J Alzheimers Dis 80:907–925. https://doi.org/10.3233/jad-201369
Xiong R, He R, Liu B, Jiang W, Wang B, Li N, Geng Q (2021) Ferroptosis: a new promising target for lung cancer therapy. Oxid Med Cell Longev 2021:8457521. https://doi.org/10.1155/2021/8457521
Battaglia AM, Chirillo R, Aversa I, Sacco A, Costanzo F, Biamonte F (2020) Ferroptosis and cancer: mitochondria meet the “iron maiden” cell death. Cells. https://doi.org/10.3390/cells9061505
Chen X, Kang R, Kroemer G, Tang D (2021) Ferroptosis in infection, inflammation, and immunity. J Exp Med. https://doi.org/10.1084/jem.20210518
Seibt TM, Proneth B, Conrad M (2019) Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med 133:144–152. https://doi.org/10.1016/j.freeradbiomed.2018.09.014
Deng G, Li Y, Ma S, Gao Z, Zeng T, Chen L, Ye H, Yang M, Shi H, Yao X, Zeng Z, Chen Y, Song Y, Liu B, Gao L (2020) Caveolin-1 dictates ferroptosis in the execution of acute immune-mediated hepatic damage by attenuating nitrogen stress. Free Radic Biol Med 148:151–161. https://doi.org/10.1016/j.freeradbiomed.2019.12.026
Li P, Jiang M, Li K, Li H, Zhou Y, Xiao X, Xu Y, Krishfield S, Lipsky PE, Tsokos GC, Zhang X (2021) Glutathione peroxidase 4-regulated neutrophil ferroptosis induces systemic autoimmunity. Nat Immunol 22:1107–1117. https://doi.org/10.1038/s41590-021-00993-3
Ohl K, Rauen T, Tenbrock K (2021) Dysregulated neutrophilic cell death in SLE: a spotlight on ferroptosis. Signal Transduct Target Ther 6:392. https://doi.org/10.1038/s41392-021-00804-z
Skendros P, Mitroulis I, Ritis K (2018) Autophagy in neutrophils: from granulopoiesis to neutrophil extracellular traps. Front Cell Dev Biol 6:109. https://doi.org/10.3389/fcell.2018.00109
Sidaway P (2017) Neutrophils: neutrophil differentiation is autophagy dependent. Nat Rev Immunol 17:662. https://doi.org/10.1038/nri.2017.122
Riffelmacher T, Clarke A, Richter FC, Stranks A, Pandey S, Danielli S, Hublitz P, Yu Z, Johnson E, Schwerd T, McCullagh J, Uhlig H, Jacobsen SEW, Simon AK (2017) Autophagy-dependent generation of free fatty acids is critical for normal neutrophil differentiation. Immunity 47:466-480.e465. https://doi.org/10.1016/j.immuni.2017.08.005
Bhattacharya A, Wei Q, Shin JN, Abdel Fattah E, Bonilla DL, Xiang Q, Eissa NT (2015) Autophagy is required for neutrophil-mediated inflammation. Cell Rep 12:1731–1739. https://doi.org/10.1016/j.celrep.2015.08.019
Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, Maiuri L, Maseri A, D’Angelo A, Bianchi ME, Rovere-Querini P, Manfredi AA (2014) Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost 12:2074–2088. https://doi.org/10.1111/jth.12710
Manfredi AA, Rovere-Querini P, D’Angelo A, Maugeri N (2017) Low molecular weight heparins prevent the induction of autophagy of activated neutrophils and the formation of neutrophil extracellular traps. Pharmacol Res 123:146–156. https://doi.org/10.1016/j.phrs.2016.08.008
Murthy P, Singhi AD, Ross MA, Loughran P, Paragomi P, Papachristou GI, Whitcomb DC, Zureikat AH, Lotze MT, Zeh Iii HJ, Boone BA (2019) Enhanced neutrophil extracellular trap formation in acute pancreatitis contributes to disease severity and is reduced by chloroquine. Front Immunol 10:28. https://doi.org/10.3389/fimmu.2019.00028
Ma R, Li T, Cao M, Si Y, Wu X, Zhao L, Yao Z, Zhang Y, Fang S, Deng R, Novakovic VA, Bi Y, Kou J, Yu B, Yang S, Wang J, Zhou J, Shi J (2016) Extracellular DNA traps released by acute promyelocytic leukemia cells through autophagy. Cell Death Dis 7:e2283. https://doi.org/10.1038/cddis.2016.186
Mulcahy ME, O’Brien EC, O’Keeffe KM, Vozza EG, Leddy N, McLoughlin RM (2020) Manipulation of Autophagy and Apoptosis Facilitates Intracellular Survival of Staphylococcus aureus in Human Neutrophils. Front Immunol 11:565545. https://doi.org/10.3389/fimmu.2020.565545
Pliyev BK, Menshikov M (2012) Differential effects of the autophagy inhibitors 3-methyladenine and chloroquine on spontaneous and TNF-α-induced neutrophil apoptosis. Apoptosis 17:1050–1065. https://doi.org/10.1007/s10495-012-0738-x
Iula L, Keitelman IA, Sabbione F, Fuentes F, Guzman M, Galletti JG, Gerber PP, Ostrowski M, Geffner JR, Jancic CC, Trevani AS (2018) Autophagy mediates interleukin-1β secretion in human neutrophils. Front Immunol 9:269. https://doi.org/10.3389/fimmu.2018.00269
Frangou E, Chrysanthopoulou A, Mitsios A, Kambas K, Arelaki S, Angelidou I, Arampatzioglou A, Gakiopoulou H, Bertsias GK, Verginis P, Ritis K, Boumpas DT (2019) REDD1/autophagy pathway promotes thromboinflammation and fibrosis in human systemic lupus erythematosus (SLE) through NETs decorated with tissue factor (TF) and interleukin-17A (IL-17A). Ann Rheum Dis 78:238–248. https://doi.org/10.1136/annrheumdis-2018-213181
Angelidou I, Chrysanthopoulou A, Mitsios A, Arelaki S, Arampatzioglou A, Kambas K, Ritis D, Tsironidou V, Moschos I, Dalla V, Stakos D, Kouklakis G, Mitroulis I, Ritis K, Skendros P (2018) REDD1/autophagy pathway is associated with neutrophil-driven IL-1β inflammatory response in active ulcerative colitis. J Immunol 200:3950–3961. https://doi.org/10.4049/jimmunol.1701643
Tang S, Zhang Y, Yin SW, Gao XJ, Shi WW, Wang Y, Huang X, Wang L, Zou LY, Zhao JH, Huang YJ, Shan LY, Gounni AS, Wu YZ, Zhang JB (2015) Neutrophil extracellular trap formation is associated with autophagy-related signalling in ANCA-associated vasculitis. Clin Exp Immunol 180:408–418. https://doi.org/10.1111/cei.12589
Maugeri N, Capobianco A, Rovere-Querini P, Ramirez GA, Tombetti E, Valle PD, Monno A, D’Alberti V, Gasparri AM, Franchini S, D’Angelo A, Bianchi ME, Manfredi AA (2018) Platelet microparticles sustain autophagy-associated activation of neutrophils in systemic sclerosis. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aao3089
Suzuki E, Maverakis E, Sarin R, Bouchareychas L, Kuchroo VK, Nestle FO, Adamopoulos IE (2016) T Cell-independent mechanisms associated with neutrophil extracellular trap formation and selective autophagy in IL-17A-Mediated epidermal hyperplasia. J Immunol 197:4403–4412. https://doi.org/10.4049/jimmunol.1600383
Speir M, Nowell CJ, Chen AA, O’Donnell JA, Shamie IS, Lakin PR, D’Cruz AA, Braun RO, Babon JJ, Lewis RS, Bliss-Moreau M, Shlomovitz I, Wang S, Cengia LH, Stoica AI, Hakem R, Kelliher MA, O’Reilly LA, Patsiouras H, Lawlor KE, Weller E, Lewis NE, Roberts AW, Gerlic M, Croker BA (2020) Ptpn6 inhibits caspase-8- and Ripk3/Mlkl-dependent inflammation. Nat Immunol 21:54–64. https://doi.org/10.1038/s41590-019-0550-7
Soni C, Reizis B (2019) Self-DNA at the Epicenter of SLE: Immunogenic Forms, Regulation, and Effects. Front Immunol 10:1601. https://doi.org/10.3389/fimmu.2019.01601
Mahajan A, Herrmann M, Muñoz LE (2016) Clearance deficiency and cell death pathways: a model for the pathogenesis of SLE. Front Immunol 7:35. https://doi.org/10.3389/fimmu.2016.00035
Tsokos GC, Lo MS, Costa Reis P, Sullivan KE (2016) New insights into the immunopathogenesis of systemic lupus erythematosus. Nat Rev Rheumatol 12:716–730. https://doi.org/10.1038/nrrheum.2016.186
Huang W, Wu J, Yang H, Xiong Y, Jiang R, Cui T, Ye D (2017) Milk fat globule-EGF factor 8 suppresses the aberrant immune response of systemic lupus erythematosus-derived neutrophils and associated tissue damage. Cell Death Differ 24:263–275. https://doi.org/10.1038/cdd.2016.115
Lauber K, Keppeler H, Munoz LE, Koppe U, Schröder K, Yamaguchi H, Krönke G, Uderhardt S, Wesselborg S, Belka C, Nagata S, Herrmann M (2013) Milk fat globule-EGF factor 8 mediates the enhancement of apoptotic cell clearance by glucocorticoids. Cell Death Differ 20:1230–1240. https://doi.org/10.1038/cdd.2013.82
Stefanescu M, Matache C, Onu A, Tanaseanu S, Dragomir C, Constantinescu I, Schönlau F, Rohdewald P, Szegli G (2001) Pycnogenol efficacy in the treatment of systemic lupus erythematosus patients. Phytother Res 15:698–704. https://doi.org/10.1002/ptr.915
Wehrli M, Schneider C, Cortinas-Elizondo F, Verschoor D, Frias Boligan K, Adams OJ, Hlushchuk R, Engelmann C, Daudel F, Villiger PM, Seibold F, Yawalkar N, Vonarburg C, Miescher S, Lötscher M, Kaufmann T, Münz C, Mueller C, Djonov V, Simon HU, von Gunten S (2020) IgA triggers cell death of neutrophils when primed by inflammatory mediators. J Immunol 205:2640–2648. https://doi.org/10.4049/jimmunol.1900883
Graeter S, Schneider C, Verschoor D, von Däniken S, Seibold F, Yawalkar N, Villiger P, Dimitrov JD, Smith DF, Cummings RD, Simon HU, Vassilev T, von Gunten S (2020) Enhanced pro-apoptotic effects of Fe(II)-modified IVIG on human neutrophils. Front Immunol 11:973. https://doi.org/10.3389/fimmu.2020.00973
Watanabe-Kusunoki K, Nakazawa D, Kusunoki Y, Kudo T, Hattanda F, Nishio S, Masuda S, Tomaru U, Kondo T, Atsumi T, Ishizu A (2020) Recombinant thrombomodulin ameliorates autoimmune vasculitis via immune response regulation and tissue injury protection. J Autoimmun 108:102390. https://doi.org/10.1016/j.jaut.2019.102390
Uozumi R, Iguchi R, Masuda S, Nishibata Y, Nakazawa D, Tomaru U, Ishizu A (2020) Pharmaceutical immunoglobulins reduce neutrophil extracellular trap formation and ameliorate the development of MPO-ANCA-associated vasculitis. Modern Rheumatol 30:544–550. https://doi.org/10.1080/14397595.2019.1602292
Fortner KA, Blanco LP, Buskiewicz I, Huang N, Gibson PC, Cook DL, Pedersen HL, Yuen PST, Murphy MP, Perl A, Kaplan MJ, Budd RC (2020) Targeting mitochondrial oxidative stress with MitoQ reduces NET formation and kidney disease in lupus-prone MRL-lpr mice. Lupus Sci Med. https://doi.org/10.1136/lupus-2020-000387
Hair PS, Enos AI, Krishna NK, Cunnion KM (2018) Inhibition of immune complex complement activation and neutrophil extracellular trap formation by peptide inhibitor of complement C1. Front Immunol 9:558. https://doi.org/10.3389/fimmu.2018.00558
Yu Y, Koehn CD, Yue Y, Li S, Thiele GM, Hearth-Holmes MP, Mikuls TR, O’Dell JR, Klassen LW, Zhang Z, Su K (2015) Celastrol inhibits inflammatory stimuli-induced neutrophil extracellular trap formation. Curr Mol Med 15:401–410. https://doi.org/10.2174/1566524015666150505160743
Liao P, He Y, Yang F, Luo G, Zhuang J, Zhai Z, Zhuang L, Lin Z, Zheng J, Sun E (2018) Polydatin effectively attenuates disease activity in lupus-prone mouse models by blocking ROS-mediated NET formation. Arthritis Res Ther 20:254. https://doi.org/10.1186/s13075-018-1749-y
Yang F, Luo X, Luo G, Zhai Z, Zhuang J, He J, Han J, Zhang Y, Zhuang L, Sun E, He Y (2019) Inhibition of NET formation by polydatin protects against collagen-induced arthritis. Int Immunopharmacol 77:105919. https://doi.org/10.1016/j.intimp.2019.105919
Furumoto Y, Smith CK, Blanco L, Zhao W, Brooks SR, Thacker SG, Abdalrahman Z, Sciumè G, Tsai WL, Trier AM, Nunez L, Mast L, Hoffmann V, Remaley AT, O’Shea JJ, Kaplan MJ, Gadina M (2017) Tofacitinib ameliorates murine lupus and its associated vascular dysfunction. Arthritis Rheumatol 69:148–160. https://doi.org/10.1002/art.39818
Kraaij T, Kamerling SWA, de Rooij ENM, van Daele PLA, Bredewold OW, Bakker JA, Bajema IM, Scherer HU, Toes REM, Huizinga TJW, Rabelink TJ, van Kooten C, Teng YKO (2018) The NET-effect of combining rituximab with belimumab in severe systemic lupus erythematosus. J Autoimmun 91:45–54. https://doi.org/10.1016/j.jaut.2018.03.003
Pérez-Sánchez C, Ruiz-Limón P, Aguirre MA, Jiménez-Gómez Y, Arias-de la Rosa I, Ábalos-Aguilera MC, Rodriguez-Ariza A, Castro-Villegas MC, Ortega-Castro R, Segui P, Martinez C, Gonzalez-Conejero R, Rodríguez-López S, Gonzalez-Reyes JA, Villalba JM, Collantes-Estévez E, Escudero A, Barbarroja N, López-Pedrera C (2017) Diagnostic potential of NETosis-derived products for disease activity, atherosclerosis and therapeutic effectiveness in rheumatoid arthritis patients. J Autoimmun 82:31–40. https://doi.org/10.1016/j.jaut.2017.04.007
Yuan K, Zhu Q, Lu Q, Jiang H, Zhu M, Li X, Huang G, Xu A (2020) Quercetin alleviates rheumatoid arthritis by inhibiting neutrophil inflammatory activities. J Nutr Biochem 84:108454. https://doi.org/10.1016/j.jnutbio.2020.108454
Zhang S, Huang G, Yuan K, Zhu Q, Sheng H, Yu R, Luo G, Xu A (2017) Tanshinone IIA ameliorates chronic arthritis in mice by modulating neutrophil activities. Clin Exp Immunol 190:29–39. https://doi.org/10.1111/cei.12993
Fukui S, Gutch S, Fukui S, Chu L, Wagner DD (2022) Anti-inflammatory protective effect of ADAMTS-13 in murine arthritis models. J Thromb Haemost 20:2386–2393. https://doi.org/10.1111/jth.15828
Funding
This work was funded by the National Natural Science Foundation of China (No.82204403); China Postdoctoral Science Foundation (No.2022M72199); Key Research and Development Plan of Anhui Province in 2021-Population Health Project (202104j07020032) and open project of Key Laboratory of Anti-inflammatory Immune Drugs of Ministry of Education (KFJJ-2021-07).
Author information
Authors and Affiliations
Contributions
ZW ZHANG and L JIN: preparing the original manuscript; LH LIU and MQ ZHOU: preparing the figures; XZ ZHANG and LL ZHANG: performing manuscript review and revision. All authors have read and approved the final draft of the paper.
Corresponding authors
Ethics declarations
Conflict of interest
Authors have no relevant financial or non-financial interests to disclose.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
All authors approve the submission of the manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, Z., Jin, L., Liu, L. et al. The intricate relationship between autoimmunity disease and neutrophils death patterns: a love-hate story. Apoptosis 28, 1259–1284 (2023). https://doi.org/10.1007/s10495-023-01874-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10495-023-01874-w